Co-reporter:Catherine Kanimozhi, Gerald J. Brady, Matthew J. Shea, Peishen Huang, Yongho Joo, Michael S. Arnold, and Padma Gopalan
ACS Applied Materials & Interfaces November 22, 2017 Volume 9(Issue 46) pp:40734-40734
Publication Date(Web):October 25, 2017
DOI:10.1021/acsami.7b14115
Separation of electronically pure, narrowly dispersed, pristine, semiconducting single-walled carbon nanotubes (CNTs) from a heterogeneous as-synthesized mixture is essential for various semiconducting technologies and biomedical applications. Although conjugated polymer wrappers are often utilized to facilitate electronic-type sorting, it is highly desirable to remove organic residues from the resulting devices. We report here the design and synthesis of a mild acid-degradable π-conjugated polyimine polymer, poly[(9,9-di-n-octyl-2,7-fluoren-dinitrilomethine)-alt-co-(6,6′-{2,2′-bipyridyl-dimethine})] (PFO-N-BPy), that is structurally analogous to the commonly used and commercially available poly[(9,9-dioctylfluorenyl-2,7-diyl)-alt-co-(6,6′-(2,2′-bipyridine))] (PFO-BPy). An acid cleavable imine link (−HC═N−) was introduced in the PFO-N-BPy backbone to impart degradability, which is absent in PFO-BPy. PFO-N-BPy was synthesized via a metal catalyst-free aza-Wittig reaction in high yields. PFO-N-BPy with a degree of polymerization of just ∼10 showed excellent (>99% electronic purity) selectivity for both large-diameter (1.3–1.7 nm) arc-discharge semiconducting CNTs (S-CNTs) and smaller diameter (0.8–1.2 nm) high-pressure carbon monoxide disproportionation reaction S-CNTs. Overall, the selectivity for the semiconducting species is similar to that of PFO-BPy but with an advantage of complete depolymerization under mild acidic conditions into recyclable monomers. We further show by ultraviolet–visible spectroscopy, X-ray photoelectron spectroscopy, and scanning electron microscopy that the PFO-N-BPy-wrapped S-CNTs can be aligned into a monolayer array on gate dielectrics using a floating evaporative self-assembly process from which the polymer can be completely removed. Short channel field effect transistors were fabricated from the polymer-stripped aligned S-CNT arrays, which further confirmed the semiconducting purity on the order of 99.9% or higher.Keywords: aligned arrays; degradable conjugated polymers; evaporative self-assembly; nanotube electronics; selective sorting;
Co-reporter:Kyle M. McElhinny, Peishen Huang, Yongho Joo, Catherine Kanimozhi, Arunee Lakkham, Kenji Sakurai, Paul G. Evans, and Padma Gopalan
Langmuir 2017 Volume 33(Issue 9) pp:
Publication Date(Web):February 7, 2017
DOI:10.1021/acs.langmuir.6b04585
The structural configuration of molecules assembled at organic–inorganic interfaces within electronic materials strongly influences the functional electronic and vibrational properties relevant to applications ranging from energy storage to photovoltaics. Controlling and characterizing the structural state of an interface and its evolution under external stimuli is crucial both for the fundamental understanding of the factors influenced by molecular structure and for the development of methods for material synthesis. It has been challenging to create complete molecular monolayers that exhibit external reversible control of the structure and electronic configuration. We report a monolayer/inorganic interface consisting of an organic monolayer assembled on an oxide surface, exhibiting structural and electronic reconfiguration under ultraviolet illumination. The molecular monolayer is linked to the surface through a carboxylate link, with the backbone bearing an azobenzene functional group and the head group consisting of a rhenium–bipyridine group. Optical spectroscopy, X-ray photoelectron spectroscopy, atomic force microscopy, and X-ray reflectivity show that closely packed monolayers are formed from these molecules via the Langmuir–Blodgett technique. Reversible photoisomerization is observed in solution and in monolayers assembled on Si and quartz substrates. The reconfiguration of these monolayers provides additional means to control excitation and charge transfer processes that are important in applications in catalysis, molecular electronics, and solar energy conversion.
Co-reporter:Wei Wei, Leith Samad, Jonathan W. Choi, Yongho Joo, Austin Way, Michael S. Arnold, Song Jin, and Padma Gopalan
Chemistry of Materials 2016 Volume 28(Issue 11) pp:4017
Publication Date(Web):May 9, 2016
DOI:10.1021/acs.chemmater.6b01453
A simple route for the synthesis of arrays of sub-20-nm-wide molybdenum disulfide (MoS2) nanowires using a self-assembled cylinder forming poly(styrene-b-2-vinylpyridine) thin films is demonstrated. The protonated 2-vinylpyridine selectively seeds molybdenum precursors in the aqueous solution, and the precursors are converted to molybdenum sulfide during a sulfur annealing process. An ultraviolet cross-linking step is introduced to ensure the successful transfer of the morphologies of the block copolymer templates to the MoS2 nanowires. The nanowires transition from an amorphous to a crystalline MoS2 phase upon thermal annealing in the presence of sulfur, as confirmed by X-ray photoelectron spectroscopy, Raman spectroscopy, X-ray diffraction, and transmission electron microscopy. This work provides a pathway to large area, dense, spatially localized arrays of transition metal dichalcogenide nanowires for catalytic and sensing applications.
Co-reporter:Samantha K. Schmitt, David J. Trebatoski, John D. Krutty, Angela W. Xie, Benjamin Rollins, William L. Murphy, and Padma Gopalan
Biomacromolecules 2016 Volume 17(Issue 3) pp:
Publication Date(Web):February 2, 2016
DOI:10.1021/acs.biomac.5b01682
Conjugation of biomolecules for stable presentation is an essential step toward reliable chemically defined platforms for cell culture studies. In this work, we describe the formation of a stable and site-specific amide bond via the coupling of a cysteine terminated peptide at low concentration to an azlactone containing copolymer coating. A copolymer of polyethylene glycol methyl ether methacrylate-ran-vinyl azlactone-ran-glycidyl methacrylate P(PEGMEMA-r-VDM-r-GMA) was used to form a thin coating (20–30 nm) on silicon and polycarbonate substrates. The formation and stability of coating-peptide bonds for peptides containing free thiols and amines were quantified by X-ray photoelectron spectroscopy (XPS) after exposure to cell culture conditions. Peptides containing a thiol as the only nucleophile coupled via a thioester bond; however, the bond was labile under cell culture conditions and almost all the bound peptides were displaced from the surface over a period of 2 days. Coupling with N-terminal primary amine peptides resulted in the formation of an amide bond with low efficiency (<20%). In contrast, peptides containing an N-terminal cysteine, which contain both nucleophiles (free thiol and amine) in close proximity, bound with 67% efficiency under neutral pH, and were stable under the same conditions for 2 weeks. Control studies confirm that the stable amide formation was a result of an intramolecular rearrangement through a N-acyl intermediate that resembles native chemical ligation. Through a combination of XPS and cell culture studies, we show that the cysteine terminated peptides undergo a native chemical ligation process at low peptide concentration in aqueous media, short reaction time, and at room temperature resulting in the stable presentation of peptides beyond 2 weeks for cell culture studies.
Co-reporter:Yongho Joo
The Journal of Physical Chemistry C 2016 Volume 120(Issue 25) pp:13815-13824
Publication Date(Web):June 8, 2016
DOI:10.1021/acs.jpcc.6b04098
Graphene-enhanced Raman scattering (GERS) is a promising characterization technique which uses a single layer of graphene. As the electronic coupling of adsorbates with graphene leads to enhancement in the Raman signal, it is of immense interest to explore the factors that affect the coupling of the adsorbates with graphene. To probe this effect we have designed and synthesized a series of dipolar molecules with the general structure, N-ethyl-N-(2-ethyl(1-pyrenebutyrate)-4-(4-R-phenylazo)aniline) where the R-groups are varied from methoxy (−OCH3), methyl (−CH3), hydrogen (−H), nitrile (−CN), nitro (−NO2) to tricyanofuran (TCF) groups. This systematically changes the dipole moments and electronic/optical band gap of the molecules. By noncovalently interfacing these molecules on graphene, the Raman signal is enhanced by a factor of 40–90 at the excitation wavelength of 532 nm. Measurements of the Raman enhancement factor and Raman cross section are complemented with DFT calculations to correlate the dipole moment and the energy level of the hybrid to the Raman scattering efficiency. These studies highlight the relevance of the dipolar nature of chromophores, which determines their dipole moment and the band gap, and the resulting electronic coupling to graphene which simultaneously alters the energy level of the orbitals in the molecule and the Fermi level in graphene, resulting in efficient Raman excitations and GERS.
Co-reporter:Samantha K. Schmitt;Angela W. Xie;Raha M. Ghassemi;David J. Trebatoski;William L. Murphy
Advanced Healthcare Materials 2015 Volume 4( Issue 10) pp:1555-1564
Publication Date(Web):
DOI:10.1002/adhm.201500191
Human mesenchymal stem cells (hMSCs) are a widely available and clinically relevant cell type with a host of applications in regenerative medicine. Current clinical expansion methods can lead to selective changes in hMSC phenotype potentially resulting from relatively undefined cell culture surfaces. Chemically defined synthetic surfaces can aid in understanding the influence of cell–material interactions on stem cell behavior. Here, a thin copolymer coating for hMSC culture on plastic substrates is developed. The random copolymer is synthesized by living free radical polymerization and characterized in solution before application to the substrate, ensuring a homogeneous coating and limiting the sample-to-sample variations. The ability to coat multiple substrate types and cover large surface areas is reported. Arg–Gly–Asp-containing peptides are incorporated into the coating under aqueous conditions via their lysine or cysteine side chains, resulting in amide and thioester linkages, respectively. Stability studies show amide linkages to be stable and thioester linkages to be labile under standard serum-containing culture conditions. In addition, chemically defined passaging of hMSCs using only ethylenediaminetetraacetic acid on polystyrene dishes is shown. After passage, the hMSCs can be seeded back onto the same plate, indicating potential reusability of the coating.
Co-reporter:Yongho Joo, Gerald J. Brady, Matthew J. Shea, M. Belén Oviedo, Catherine Kanimozhi, Samantha K. Schmitt, Bryan M. Wong, Michael S. Arnold, and Padma Gopalan
ACS Nano 2015 Volume 9(Issue 10) pp:10203
Publication Date(Web):September 8, 2015
DOI:10.1021/acsnano.5b03835
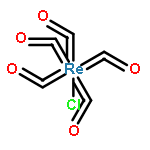
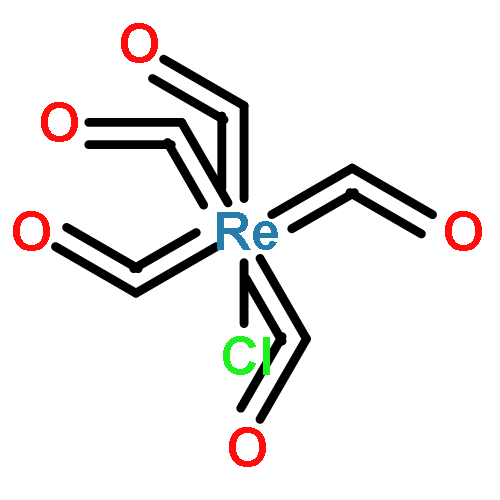
Conjugated polymers are among the most selective carbon nanotube sorting agents discovered and enable the isolation of ultrahigh purity semiconducting singled-walled carbon nanotubes (s-SWCNTs) from heterogeneous mixtures that contain problematic metallic nanotubes. The strong selectivity though highly desirable for sorting, also leads to irreversible adsorption of the polymer on the s-SWCNTs, limiting their electronic and optoelectronic properties. We demonstrate how changes in polymer backbone rigidity can trigger its release from the nanotube surface. To do so, we choose a model polymer, namely poly[(9,9-dioctylfluorenyl-2,7-diyl)-alt-co-(6,60-(2,20-bipyridine))] (PFO-BPy), which provides ultrahigh selectivity for s-SWCNTs, which are useful specifically for FETs, and has the chemical functionality (BPy) to alter the rigidity using mild chemistry. Upon addition of Re(CO)5Cl to the solution of PFO-BPy wrapped s-SWCNTs, selective chelation with the BPy unit in the copolymer leads to the unwrapping of PFO-BPy. UV–vis, XPS, and Raman spectroscopy studies show that binding of the metal ligand complex to BPy triggers up to 85% removal of the PFO-BPy from arc-discharge s-SWCNTs (diameter = 1.3–1.7 nm) and up to 72% from CoMoCAT s-SWCNTs (diameter = 0.7–0.8 nm). Importantly, Raman studies show that the electronic structure of the s-SWCNTs is preserved through this process. The generalizability of this method is demonstrated with two other transition metal salts. Molecular dynamics simulations support our experimental findings that the complexation of BPy with Re(CO)5Cl in the PFO-BPy backbone induces a dramatic conformational change that leads to a dynamic unwrapping of the polymer off the nanotube yielding pristine s-SWCNTs.Keywords: conjugated polymers; polymer backbone rigidity; s- SWCNTs;
Co-reporter:Yongho Joo, Gerald J. Brady, Michael S. Arnold, and Padma Gopalan
Langmuir 2014 Volume 30(Issue 12) pp:3460-3466
Publication Date(Web):2017-2-22
DOI:10.1021/la500162x
Arrays of aligned semiconducting single-walled carbon nanotubes (s-SWCNTs) with exceptional electronic-type purity were deposited at high deposition velocity of 5 mm min–1 by a novel “dose-controlled, floating evaporative self-assembly” process with excellent control over the placement of stripes and quantity of s-SWCNTs deposited. This approach uses the diffusion of organic solvent on the water–air interface to deposit aligned s-SWCNT (99.9%) tubes on a partially submerged hydrophobic substrate, which is withdrawn vertically from the surface of water. By decoupling the s-SWCNT stripe formation from the evaporation of the bulk solution and by iteratively applying the s-SWCNTs in controlled “doses”, we show through polarized Raman studies that the s-SWCNTs are aligned within ±14°, are packed at a density of ∼50 s-SWCNTs μm–1, and constitute primarily a well-ordered monodispersed layer. The resulting field-effect transistor devices show high performance with a mobility of 38 cm2 V–1 s–1 and on/off ratio of 2.2 × 106 at 9 μm channel length.
Co-reporter:Yongho Joo, Josef W. Spalenka, Kyle M. McElhinny, Samantha K. Schmitt, Paul G. Evans, and Padma Gopalan
Langmuir 2014 Volume 30(Issue 21) pp:6104-6113
Publication Date(Web):2017-2-22
DOI:10.1021/la5006133
We demonstrate the Langmuir–Blodgett assembly of two rhenium–bipyridine complexes containing a flexible or an aromatic bridge, and transfer of the monolayer to SiO2 and single crystal TiO2 substrates. Both of the complexes (ReEC and Re2TC) have a hydrophilic carboxylic acid group, which preferentially anchors into the water subphase, and forms stable monolayers at surface pressures up to 40 mN/m. The optimum conditions for the formation of complete monolayers of both ReEC and Re2TC were identified through characterization of the morphology by atomic force microscopy (AFM), the thickness by ellipsometry, and the surface coverage by X-ray photoelectron spectroscopy (XPS). X-ray reflectivity measurements (XRR) are consistent with the orientation of the molecules normal to the substrate, and their extension to close to their calculated maximum length. Parameters derived from XRR analysis show that there is a higher packing density for Re2TC monolayers than for ReEC monolayers, attributable to the more rigid bridge in the Re2TC molecule.
Co-reporter:Samantha K. Schmitt, William L. Murphy and Padma Gopalan
Journal of Materials Chemistry A 2013 vol. 1(Issue 9) pp:1349-1360
Publication Date(Web):07 Jan 2013
DOI:10.1039/C2TB00253A
We have designed a lightly crosslinked PEG based copolymer coating with compositional flexibility as well as extended stability for studying human mesenchymal stem cells (hMSCs). Copolymers contain a majority of poly(ethylene glycol) methyl ether methacrylate (PEGMEMA) as a cytophobic background with poly(ethylene glycol) methacrylate (PEGMA) for peptide coupling, and less than 10% glycidyl methacrylate (GMA) for crosslinking. Copolymer thin films were crosslinked into 30 nm thick mats by either thermal treatment or ultraviolet light and were stable for 35 days in water at 37 °C. The amount of PEGMA in the copolymer was optimized to ∼11% to minimize non-specific cell–protein interactions while maximizing the amount of total bound peptides. Following the binding of RGDSP to the mat, hMSCs were seeded. The hMSC adhesion, spreading and focal adhesion complex formation were promoted in a concentration dependent manner. Mats coupled with a non-adhesive scramble (RDGSP) maintained their cytophobicity. Competitive detachment experiments further demonstrated that cell adhesion was mediated by receptor binding to the RGDSP peptide. Cell culture experiments performed at 1 and 2 weeks show that mats can still resist cell adhesion after incubation in a serum containing medium. X-ray photoelectron spectroscopy (XPS) was effectively used to quantify the average total peptide concentration as 12.6 ± 6.14 pmol cm−2. A square 2.2 mm N (1s) element map shows an average value of 17.9 pmol cm−2 of RGDSP, which correlates well with the multipoint high resolution data. The stability of the copolymer, compositional flexibility, ease of application and the ability to precisely quantify bound peptides on the mats make these materials ideal for the study of cellular processes, where stability, functionality and topography of the biointerface are relevant.
Co-reporter:Yuanchun Zhao, Changshui Huang, Myungwoong Kim, Bryan M. Wong, François Léonard, Padma Gopalan, and Mark A. Eriksson
ACS Applied Materials & Interfaces 2013 Volume 5(Issue 19) pp:9355
Publication Date(Web):September 23, 2013
DOI:10.1021/am4024753
We report the functionalization of carbon nanotubes with two azobenzene-based chromophores with large internal dipole moments and opposite dipole orientations. The molecules are attached to the nanotubes noncovalently via a pyrene tether. A combination of characterization techniques shows uniform molecular coverage on the nanotubes, with minimal aggregation of excess chromophores on the substrate. The large on/off ratios and the subthreshold swings of the nanotube-based field-effect transistors (FETs) are preserved after functionalization, and different shifts in threshold voltage are observed for each chromophore. Ab initio calculations verify the properties of the synthesized chromophores and indicate very small charge transfer, confirming a strong, noncovalent functionalization.Keywords: chromophore; DFT calculations; dipole orientation; field-effect transistor; functionalization; single-wall carbon nanotube;
Co-reporter:Peerasak Paoprasert, Srikanth Kandala, Daniel P. Sweat, Rose Ruther and Padma Gopalan
Journal of Materials Chemistry A 2012 vol. 22(Issue 3) pp:1046-1053
Publication Date(Web):14 Nov 2011
DOI:10.1039/C1JM13293H
We demonstrate that hydroxyl containing bipyridine-based molecules can be efficiently grafted onto SiO2 and TiO2 substrates to create a stable monolayer. These surface-grafted ligands are relevant to several optoelectronic applications as well as catalysis since many metal–bipyridine complexes have been used as dye sensitizers, light emitters, and catalysts. Control experiments were carried out to infer that the grafting occurs through the formation of an ether link with available hydroxyl groups on the surface. The coverage was found to be a function of annealing temperature, annealing time, and availability of surface hydroxyl groups. Further complexation with rhenium salts resulted in a rhenium–bipyridine complex, a common electron injector in dye-sensitized solar cells. All samples were characterized by X-ray photoelectron spectroscopy (XPS) and infrared reflection absorption spectroscopy (IRRAS). This simple grafting method leads to monolayer coverage on the surface, effectively avoiding crosslinking, and shows good stability in acetonitrile and aqueous solutions for at least four weeks. We further expand the scope of this approach by grafting an electroactive ferrocene unit onto indium tin oxide. Based on these results, the formation of an ether bond via thermal annealing of small molecules offers a versatile strategy for preparing stable organic layers on a variety of oxide surfaces, and therefore, expands the tool box for functionalizing organic/inorganic interfaces.
Co-reporter:Eungnak Han;Huiman Kang;Chi-Chun Liu;Paul F. Nealey
Advanced Materials 2010 Volume 22( Issue 38) pp:4325-4329
Publication Date(Web):
DOI:10.1002/adma.201001669
Co-reporter:Peerasak Paoprasert, Josef W. Spalenka, Dane L. Peterson, Rose E. Ruther, Robert J. Hamers, Paul G. Evans and Padma Gopalan
Journal of Materials Chemistry A 2010 vol. 20(Issue 13) pp:2651-2658
Publication Date(Web):15 Dec 2009
DOI:10.1039/B920233A
Poly(3-hexylthiophene) (P3HT) brushes on silicon dioxide (SiO2) were prepared using a click reaction between ethynyl-terminated P3HT and an azide self-assembled monolayer (SAM). Regioregular ethynyl-terminated P3HT with molecular weight of 5900 g mol−1 and polydispersity of 1.2 was synthesized by catalyst-transfer polycondensation using Grignard metathesis mediated by a nickel-based catalyst. The azide SAM was prepared from bifunctional molecules containing azide and siloxane as click reaction precursor and surface linker, respectively. The P3HT brushes were characterized by atomic force microscopy, ellipsometry, X-ray photoelectron spectroscopy, infrared reflection absorption spectroscopy, and UV-visible spectroscopy. The grafting of P3HT brushes was studied as a function of click reaction time and the growth of the brushes is governed by a diffusion-controlled process. P3HT brushes were prepared on pre-fabricated field-effect transistor structures in order to probe the electrical properties of the brushes. The versatile synthetic methodology developed in this work can be generalized to prepare a wide variety of semiconducting conjugated polymer brushes on oxide surfaces relevant to organic electronic devices.
Co-reporter:Eungnak Han, Melvina Leolukman, Myungwoong Kim, and Padma Gopalan
ACS Nano 2010 Volume 4(Issue 11) pp:6527
Publication Date(Web):October 19, 2010
DOI:10.1021/nn101616d
We report the design of a direct electron beam patternable buffer layer to spatially control the orientation of the microdomains in an overlaying polystyrene-block-poly(methyl methacrylate) (PS-b-PMMA) block copolymer (BCP) film. The buffer layer consists of a surface anchored low molecular weight PS-b-PMMA, with the PMMA segment anchored to the surface and a short PS block at the buffer layer/BCP interface. The block architecture of the buffer layer combines the essential features of “bottom up” and “top down” approaches as it functions as a nonpreferential layer to dictate perpendicular orientation of BCP domains from the substrate interface and as an e-beam resist to allow top-down lithographic process to spatially define the buffer layer on the substrate. The composition of the buffer layer can be tuned by changing the relative block lengths to create a nonpreferential surface which effectively induces perpendicular orientation of domains in an overlying BCP film. The grafted block copolymer can be locally shaved by e-beam lithography resulting in spatial control of domain orientation in the BCP film. The direct patterning approach reduces the number of steps involved in forming chemical patterns by conventional lithography.Keywords: block copolymer; direct patterning; directed assembly; e-beam lithography; neutral surface
Co-reporter:Peerasak Paoprasert, Jennifer E. Laaser, Wei Xiong, Ryan A. Franking, Robert J. Hamers, Martin T. Zanni, J. R. Schmidt and Padma Gopalan
The Journal of Physical Chemistry C 2010 Volume 114(Issue 21) pp:9898-9907
Publication Date(Web):May 10, 2010
DOI:10.1021/jp102022d
We have measured the electron injection kinetics of four rhenium−bipyridine complexes (Re1C, ReEC, Re1TC, and Re2TC) on TiO2 nanocrystalline films using transient infrared spectroscopy. The self-assembled monolayer formation of these complexes was characterized by UV−visible spectroscopy, infrared reflection absorption spectroscopy, and X-ray photoelectron spectroscopy. These complexes bind to the TiO2 surface through the formation of carboxylate groups, and these self-assembled layers are approximately a monolayer. The kinetics studies address the effect of insulating and conjugated spacers and the length of conjugation on the electron-transfer process. The insulating bridge leads to a slower injection rate and poorer injection yield compared with the conjugated spacers. The electron injection of Re2TC was found to be a fast, high-yielding, and multiple electron injector process. The ground and electronically excited states of the dye complexes were characterized using ground-state and time-dependent density functional theory. We present the role of electronic conjugation in modulating electron injection using a combination of computational and experimental work and find that these metal-based complexes adsorbed on a semiconductor surface can be used to read out the electron injection kinetics through tailored molecular bridges.
Co-reporter:Eungnak Han and Padma Gopalan
Langmuir 2010 Volume 26(Issue 2) pp:1311-1315
Publication Date(Web):September 30, 2009
DOI:10.1021/la902483m
We report the effect of increasing amounts of glycidyl methacrylate (GMA) in random copolymers of P(S-r-MMA-r-GMA) on the formation of nonpreferential mats for the assembly of P(S-b-MMA) block copolymers. Increasing the GMA concentration in the random copolymer from 1 (PG1) to 4 (PG4) mole % increased the cross-linking efficiency and reduced the effective minimum thickness of the cross-linked mat for perpendicular alignment of P(S-b-MMA) from ∼6 nm to ∼2 nm. The compositional window (so-called perpendicular window) of PG4 was defined for both symmetric and asymmetric P(S-b-MMA). Compared to PG1, incorporation of higher amount of polar comonomer (GMA) in PG4 shifted the perpendicular window toward higher styrene fraction as a result of the increased polarity. The defined perpendicular window for P(S-r-MMA-r-GMA) is equally applicable for random copolymers prepared by both controlled living and classical free-radical polymerizations.
Co-reporter:Eungnak Han, Karl O. Stuen, Melvina Leolukman, Chi-Chun Liu, Paul F. Nealey and Padma Gopalan
Macromolecules 2009 Volume 42(Issue 13) pp:4896-4901
Publication Date(Web):June 11, 2009
DOI:10.1021/ma9002903
We report the induction of perpendicularly oriented cylindrical domains in PS-b-PMMA block copolymer (BCP) films thicker than 100 nm by thermally annealing on a substrate modified with a random copolymer. The effects of annealing temperature, composition of the substrate-modifying random copolymer, and BCP film thickness on the morphology of PMMA cylinder forming PS-b-PMMA were studied. For BCP films thicker than 100 nm, the fabrication of perpendicular PMMA cylinders is highly dependent on both the substrate-modifying random copolymer and the annealing temperature as these two parameters control the interactions of the BCP with the substrate and the free surface, respectively. We found the best perpendicular structures to be created by using a random copolymer brush with a styrene fraction (FSt) near 0.70 and an annealing temperature near 230 °C. Perpendicular cylinder structures were achieved in ∼300 nm thick films using these conditions. When the BCP film was thicker than 300 nm, nucleation and growth of the microdomains proceeded independently from each interface. We present scanning electron microscope (SEM) and cross-sectional transmission electron microscope (TEM) images of these perpendicular structures and explain the results on the basis of previous simulation reports.
Co-reporter:Tomoyasu Hirai, Melvina Leolukman, Sangwoo Jin, Raita Goseki, Yoshihito Ishida, Masa-aki Kakimoto, Teruaki Hayakawa, Moonhor Ree and Padma Gopalan
Macromolecules 2009 Volume 42(Issue 22) pp:8835-8843
Publication Date(Web):October 29, 2009
DOI:10.1021/ma9018944
Two kinds of polyhedral oligomeric silsesquioxane (POSS)-containing block copolymers (BCPs), namely PS-b-PMAPOSS and PMMA-b-PMAPOSS, were synthesized by living anionic polymerization. A wide range of molecular weights were obtained with a very narrow polydispersity index of less than 1.09. The bulk samples prepared by slow evaporation from a polymer solution in chloroform exhibit well-defined microphase-separated structures with long-range order. Thermal annealing induced hierarchical structures consisting of a smaller length scale ordered crystalline POSS domains within the larger microphase-separated structures. We report detailed structural characterization of these hierarchical structures in bulk and thin films by transmission electron microscopy and grazing incidence wide-angle X-ray scattering (GIWAXS). On the basis of this structural analysis, we propose a model for the formation of an orthorhombic lattice structure through the aggregation of POSS segments which formed a helix-like structure.
Co-reporter:Melvina Leolukman;Peerasak Paoprasert;Ian Mel;Solimar J. Diaz;David J. Mcgee
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 19) pp:5017-5026
Publication Date(Web):
DOI:10.1002/pola.23554
Abstract
Linear-dendritic block copolymer hosts were synthesized by end-functionalizing poly(methylmethacrylate) with dendrons that acted as hydrogen-bonding acceptors for nonlinear optical chromophores. Second harmonic generation experiments indicate that the d33 coefficients and maximum chromophore loading are increased in linear-dendritic block copolymer hosts over comparable homopolymer hosts. Transmission electron microscopy shows 5–10 nm chromophore domains, confirming the effective spatial dispersion of the chromophores. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 5017–5026, 2009
Co-reporter:Melvina Leolukman, Young-Hye La, Xuefa Li and Padma Gopalan
Polymer Journal 2008 40(9) pp:825-831
Publication Date(Web):July 16, 2008
DOI:10.1295/polymj.PJ2008014
Morphology dependence of asymmetric poly(styrene-b-tert-butylacrylate) [P(S-b-tBA)] thin film on casting solvent, annealing solvent and annealing time was investigated. Two casting solvents, toluene and propylene glycol monomethyl ester acetate (PGMEA), and two annealing solvents, p-xylene and tert-butylacrylate (tBA), were studied. The casting solvent dominated the initial morphology. P(S-b-tBA) film cast from toluene resulted in inverted phase (PtBA cylinders in PS matrix) and the one cast from PGMEA led to normal phase (PS cylinders in PtBA matrix) morphology. The annealing solvents preference to either the majority or the minority block in combination with the annealing time controlled the orientation of the cylinders. When the annealing solvent preferentially interacted with the majority block in the as-cast film, a metastable highly ordered perpendicular cylinder morphology was trapped. The best conditions to obtain perpendicular PS cylinders were casting P(S-b-tBA) film from PGMEA and annealing in tBA vapor for 20 and 25 min on silicon and NaCl substrates respectively.
Co-reporter:Sean P. Cullen;Sangkeun Ha;Max G. Lagally
Journal of Polymer Science Part A: Polymer Chemistry 2008 Volume 46( Issue 17) pp:5826-5838
Publication Date(Web):
DOI:10.1002/pola.22896
Abstract
We report the synthesis and characterization of a photocrosslinkable copolymer containing reactive epoxy groups for binding biomolecules. The epoxide-containing copolymer poly(glycidyl methacrylate-ran-2-(acryloyloxy) ethyl 2-methylacrylate) offers distinct advantages such as ease of application to various substrates, enhanced stability of the bound oligonucleotide, low autofluorescence, and the ability to be photopatterned allowing localization of the linkers. The copolymer uses pendant acryloyl groups to control the crosslinking without sacrificing the epoxide groups. The films were characterized using ellipsometry, atomic force microscopy, and fluorescence microscopy. The films on glass, silicon wafer, and stainless steel showed no appreciable degradation in water, tetrahydrofuran, and acetone for ∼4 months. The surface topography for a given thickness of crosslinked film was dictated by the deposition conditions. A 16mer oligonucleotide was immobilized on the thin films. A linear relationship between the film thickness and amount of oligonucleotide immobilized was observed with a maximum signal-to-background ratio (S/B) of 225 for a 60-nm-thick film, a value 50% higher than the S/B for an epoxide monolayer. The crosslinked films maintained a high fluorescence signal following long aqueous washing which is appealing for biological microarrays, immobilizing proteins, and study of slow differentiating cells where stability of the scaffold is relevant. © 2008 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 46: 5826–5838, 2008
Co-reporter:Melvina Leolukman, Peerasak Paoprasert, Yao Wang, Varun Makhija, David J. McGee and Padma Gopalan
Macromolecules 2008 Volume 41(Issue 13) pp:4651-4660
Publication Date(Web):June 11, 2008
DOI:10.1021/ma800318s
Factors affecting the electric-field-induced poling of nonlinear optical chromophores in block copolymer domains were investigated by encapsulating the chromophores in a linear−diblock copolymer [poly(styrene-b-4-vinylpyridine)] and linear−dendritic (poly(methyl methacrylate)−dendron) block copolymer via hydrogen bonding. Temperature-dependent Fourier transform infrared spectroscopy and morphology evaluation by X-ray scattering and transmission electron microscopy were used with in situ second harmonic generation to correlate domain architectures, processing conditions such as thermal history, and chromophore concentrations with poling efficiency. Poling of chromophores encapsulated in the minority domain (spheres or cylinders) of a linear−diblock copolymer was inhibited by the increasing chromophore concentration within the domain and the chemical nature of the majority domain. Chromophore encapsulation in the majority domain produced the most favorable conditions for poling as measured by in situ second harmonic generation. Thermal annealing of the linear−diblock copolymer/chromophore composites resulted in chromophore aggregation with a corresponding decrease in nonlinear optical activity. The linear−dendron/chromophore system presented the most effective architecture for spatially dispersing chromophores. These findings suggest that while well-ordered phase-separated systems such as block copolymers enhance chromophore isolation over homopolymer systems, a more effective approach is to explore polymer chains end functionalized with chromophores.
Co-reporter:Eungnak Han, Karl O. Stuen, Young-Hye La, Paul F. Nealey and Padma Gopalan
Macromolecules 2008 Volume 41(Issue 23) pp:9090-9097
Publication Date(Web):November 6, 2008
DOI:10.1021/ma8018393
The ability of random copolymer brushes and cross-linked mats to induce the vertical orientation of domains in overlying films of lamellae- and cylinder-forming block copolymers was investigated as a function of the composition. The substrate-modifying layers consisted of styrene and methyl methacrylate random copolymers and contained either a terminal hydroxyl group or a third polar comonomer of 2-hydroxyethyl methacrylate (HEMA) for grafting brushes to silicon oxide surfaces or glycidyl methacrylate (GMA) for cross-linking the random copolymer into a mat. Polystyrene-block-poly(methyl methacrylate) (PS-b-PMMA) lamellae- and cylinder-forming block copolymers (both PS and PMMA minority block copolymers) were deposited and annealed on the modified surfaces. In all cases the vertical orientation of domains was observed for a range of random copolymer composition, but the ranges of composition were different for each combination of surface layer and block copolymer. The cylindrical domains of PS exhibited vertical structures for a very narrow range of compositions compared to cylindrical domains of PMMA or lamellae. As expected, the incorporation of polar HEMA or GMA monomers in the surface layers shifted the composition range for the perpendicular orientation of domains to higher fractions of styrene. The results are discussed in terms of the equilibration of the films in the presence of the chemically modified surfaces.
Co-reporter:Sean P. Cullen, Ian C. Mandel and Padma Gopalan
Langmuir 2008 Volume 24(Issue 23) pp:13701-13709
Publication Date(Web):October 28, 2008
DOI:10.1021/la8024952
We explored surface-anchored poly(2-vinyl-4,4-dimethyl azlactone) (PVDMA) brushes as potential templates for protein immobilization. The brushes were grown using atom transfer radical polymerization from surface-anchored initiators and characterized by a combination of ellipsometry, atomic force microscopy, and X-ray photoelectron spectroscopy. RNase A was immobilized as a model enzyme through the nucleophilic attack of azlactone by the amine groups in the lysines located in the protein. The surface density of RNase A increased linearly from 5 to 50 nm. For 50 nm thick poly(2-vinyl-4,4-dimethyl azlactone) brushes, 7.5 μg/cm2 of RNase A was bound. The kinetics and thermodynamics of RNase A immobilization, the activity relative to surface density, and the pH and temperature dependence were examined. A Langmuir-like model for binding kinetics indicates that the kinetics are controlled by the rate of adsorption of RNase A and has an adsorption rate constant, kads, of 2.8 × 10−8 μg−1 s−1 cm3. A maximum relative activity of ∼0.95, which is near the activity of free RNase A, was reached at 1.2 μg/cm2 (∼3.0 monolayers) of immobilized RNase A. The immobilized RNase A had a similar temperature and pH dependence as free RNase A, indicating no significant change in conformation. The PVDMA template was extended to other biotechnologically relevant enzymes, such as deoxyribonuclease I, glucose oxidase, glucoamylase, and trypsin, with relative activities higher than or comparable to those of enzymes immobilized by other means. PVDMA brushes offer an efficient route to immobilize proteins via the ring opening of azlactone without the need for activation or pretreatment while retaining high relative activities of the bound enzymes.
Co-reporter:Victoria E. Campbell;Justin D. Mykietyn;Peerasak Paoprasert;David J. McGee;Insik In
Journal of Polymer Science Part A: Polymer Chemistry 2007 Volume 45(Issue 15) pp:3166-3177
Publication Date(Web):6 JUN 2007
DOI:10.1002/pola.22145
A family of fluorinated azobenzene-based push-pull chromophores with one, two, and three trifluorovinyl ether (TFV) groups in linear and branched architecture was synthesized and utilized as active materials in the low optical loss electro-optic (EO) composites. The fluorinated azobenzene chromophores exhibited increased solubility (30–50 wt %) in semifluorinated polymer host, such as perfluorocyclobutane (PFCB) aromatic ether resin after crosslinking, compared with the commercially available nonfluorinated azobenzene chromophore Disperse Red 1 (1–2 wt %). The impact of this approach on the optical properties on the polymer blends is assessed through optical propagation loss measurements and EO characterization. The resulting fluorinated EO composites showed excellent optical clarity, low birefringence, and low optical loss less than 0.5 dB/cm, while giving EO coefficients of about 3–7 pm/V at 1550 nm. © 2007 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 45: 3166–3177, 2007
Co-reporter:Tomoyasu Hirai ; Melvina Leolukman ; Teruaki Hayakawa ; Masa-aki Kakimoto
Macromolecules () pp:
Publication Date(Web):June 11, 2008
DOI:10.1021/ma800872v
Co-reporter:Peerasak Paoprasert, Srikanth Kandala, Daniel P. Sweat, Rose Ruther and Padma Gopalan
Journal of Materials Chemistry A 2012 - vol. 22(Issue 3) pp:NaN1053-1053
Publication Date(Web):2011/11/14
DOI:10.1039/C1JM13293H
We demonstrate that hydroxyl containing bipyridine-based molecules can be efficiently grafted onto SiO2 and TiO2 substrates to create a stable monolayer. These surface-grafted ligands are relevant to several optoelectronic applications as well as catalysis since many metal–bipyridine complexes have been used as dye sensitizers, light emitters, and catalysts. Control experiments were carried out to infer that the grafting occurs through the formation of an ether link with available hydroxyl groups on the surface. The coverage was found to be a function of annealing temperature, annealing time, and availability of surface hydroxyl groups. Further complexation with rhenium salts resulted in a rhenium–bipyridine complex, a common electron injector in dye-sensitized solar cells. All samples were characterized by X-ray photoelectron spectroscopy (XPS) and infrared reflection absorption spectroscopy (IRRAS). This simple grafting method leads to monolayer coverage on the surface, effectively avoiding crosslinking, and shows good stability in acetonitrile and aqueous solutions for at least four weeks. We further expand the scope of this approach by grafting an electroactive ferrocene unit onto indium tin oxide. Based on these results, the formation of an ether bond via thermal annealing of small molecules offers a versatile strategy for preparing stable organic layers on a variety of oxide surfaces, and therefore, expands the tool box for functionalizing organic/inorganic interfaces.
Co-reporter:Peerasak Paoprasert, Josef W. Spalenka, Dane L. Peterson, Rose E. Ruther, Robert J. Hamers, Paul G. Evans and Padma Gopalan
Journal of Materials Chemistry A 2010 - vol. 20(Issue 13) pp:NaN2658-2658
Publication Date(Web):2009/12/15
DOI:10.1039/B920233A
Poly(3-hexylthiophene) (P3HT) brushes on silicon dioxide (SiO2) were prepared using a click reaction between ethynyl-terminated P3HT and an azide self-assembled monolayer (SAM). Regioregular ethynyl-terminated P3HT with molecular weight of 5900 g mol−1 and polydispersity of 1.2 was synthesized by catalyst-transfer polycondensation using Grignard metathesis mediated by a nickel-based catalyst. The azide SAM was prepared from bifunctional molecules containing azide and siloxane as click reaction precursor and surface linker, respectively. The P3HT brushes were characterized by atomic force microscopy, ellipsometry, X-ray photoelectron spectroscopy, infrared reflection absorption spectroscopy, and UV-visible spectroscopy. The grafting of P3HT brushes was studied as a function of click reaction time and the growth of the brushes is governed by a diffusion-controlled process. P3HT brushes were prepared on pre-fabricated field-effect transistor structures in order to probe the electrical properties of the brushes. The versatile synthetic methodology developed in this work can be generalized to prepare a wide variety of semiconducting conjugated polymer brushes on oxide surfaces relevant to organic electronic devices.
Co-reporter:Samantha K. Schmitt, William L. Murphy and Padma Gopalan
Journal of Materials Chemistry A 2013 - vol. 1(Issue 9) pp:NaN1360-1360
Publication Date(Web):2013/01/07
DOI:10.1039/C2TB00253A
We have designed a lightly crosslinked PEG based copolymer coating with compositional flexibility as well as extended stability for studying human mesenchymal stem cells (hMSCs). Copolymers contain a majority of poly(ethylene glycol) methyl ether methacrylate (PEGMEMA) as a cytophobic background with poly(ethylene glycol) methacrylate (PEGMA) for peptide coupling, and less than 10% glycidyl methacrylate (GMA) for crosslinking. Copolymer thin films were crosslinked into 30 nm thick mats by either thermal treatment or ultraviolet light and were stable for 35 days in water at 37 °C. The amount of PEGMA in the copolymer was optimized to ∼11% to minimize non-specific cell–protein interactions while maximizing the amount of total bound peptides. Following the binding of RGDSP to the mat, hMSCs were seeded. The hMSC adhesion, spreading and focal adhesion complex formation were promoted in a concentration dependent manner. Mats coupled with a non-adhesive scramble (RDGSP) maintained their cytophobicity. Competitive detachment experiments further demonstrated that cell adhesion was mediated by receptor binding to the RGDSP peptide. Cell culture experiments performed at 1 and 2 weeks show that mats can still resist cell adhesion after incubation in a serum containing medium. X-ray photoelectron spectroscopy (XPS) was effectively used to quantify the average total peptide concentration as 12.6 ± 6.14 pmol cm−2. A square 2.2 mm N (1s) element map shows an average value of 17.9 pmol cm−2 of RGDSP, which correlates well with the multipoint high resolution data. The stability of the copolymer, compositional flexibility, ease of application and the ability to precisely quantify bound peptides on the mats make these materials ideal for the study of cellular processes, where stability, functionality and topography of the biointerface are relevant.

![Propanedinitrile,[4-[2-(4-aminophenyl)ethenyl]-3-cyano-5,5-dimethyl-2(5H)-furanylidene]-](http://img.cochemist.com/ccimg/669000/668984-45-0.png)
![Propanedinitrile,[4-[2-(4-aminophenyl)ethenyl]-3-cyano-5,5-dimethyl-2(5H)-furanylidene]-](http://img.cochemist.com/ccimg/669000/668984-45-0_b.png)







![Ethanol, 2-[ethyl[4-[(4-methylphenyl)azo]phenyl]amino]-](http://img.cochemist.com/ccimg/143100/143057-14-1.png)
![Ethanol, 2-[ethyl[4-[(4-methylphenyl)azo]phenyl]amino]-](http://img.cochemist.com/ccimg/143100/143057-14-1_b.png)

![Ethanol, 2-[ethyl[4-[(4-methoxyphenyl)azo]phenyl]amino]-](http://img.cochemist.com/ccimg/88600/88580-92-1.png)
![Ethanol, 2-[ethyl[4-[(4-methoxyphenyl)azo]phenyl]amino]-](http://img.cochemist.com/ccimg/88600/88580-92-1_b.png)
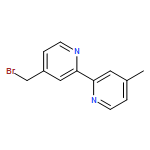
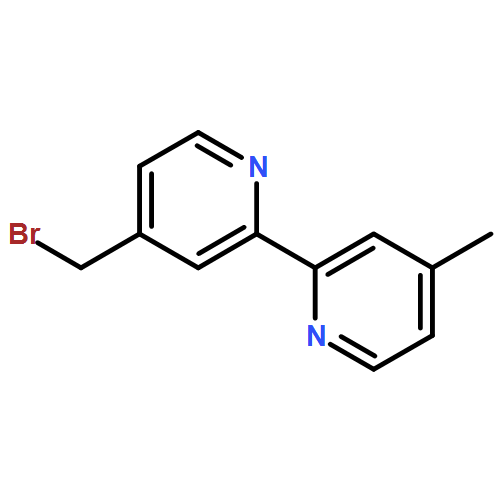
![4-[[4-[ETHYL(2-HYDROXYETHYL)AMINO]PHENYL]DIAZENYL]BENZONITRILE](http://img.cochemist.com/ccimg/79300/79252-67-8.png)
![4-[[4-[ETHYL(2-HYDROXYETHYL)AMINO]PHENYL]DIAZENYL]BENZONITRILE](http://img.cochemist.com/ccimg/79300/79252-67-8_b.png)
![Benzaldehyde, 3-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/34800/34716-73-9.png)
![Benzaldehyde, 3-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/34800/34716-73-9_b.png)
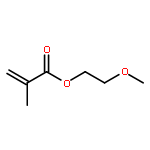
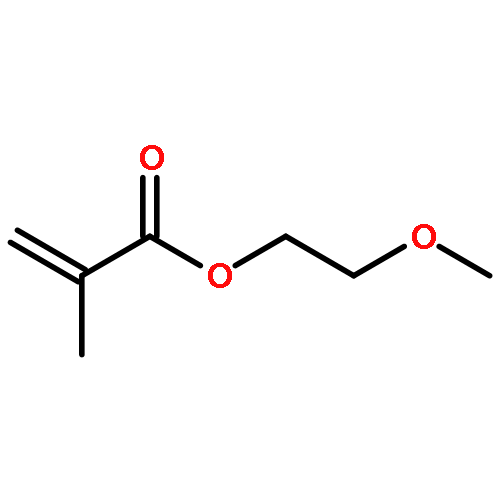
![METHYL 4-[2-(4-OXOCYCLOHEXA-2,5-DIEN-1-YLIDENE)HYDRAZINYL]BENZOATE](http://img.cochemist.com/ccimg/26300/26257-98-7.png)
![METHYL 4-[2-(4-OXOCYCLOHEXA-2,5-DIEN-1-YLIDENE)HYDRAZINYL]BENZOATE](http://img.cochemist.com/ccimg/26300/26257-98-7_b.png)

















![2-Propenoic acid, 2-methyl-, 3-[3,5,7,9,11,13,15-heptakis(2-methylpropyl)pentacyclo[9.5.1.13,9.15,15.17,13]octasiloxan-1-yl]propyl ester, homopolymer](/data/chemimg/109700/425409-08-1.png)
![2-Propenoic acid, 2-methyl-, 3-[3,5,7,9,11,13,15-heptakis(2-methylpropyl)pentacyclo[9.5.1.13,9.15,15.17,13]octasiloxan-1-yl]propyl ester, homopolymer](/data/chemimg/109700/425409-08-1_b.png)






![[2,2'-Bithiophene]-5,5'-dicarbonyl dichloride](http://img.cochemist.com/ccimg/99000/98994-67-3.png)
![[2,2'-Bithiophene]-5,5'-dicarbonyl dichloride](http://img.cochemist.com/ccimg/99000/98994-67-3_b.png)
![[2,2'-Bipyridine]-4-carboxylic acid, methyl ester](/data/chemimg/3721100/98820-73-6.png)
![[2,2'-Bipyridine]-4-carboxylic acid, methyl ester](/data/chemimg/3721100/98820-73-6_b.png)













![Poly[[2,2'-bipyridine]-6,6'-diyl(9,9-dioctyl-9H-fluorene-2,7-diyl)]](/data/chemimg/3726600/1423043-97-3.png)
![Poly[[2,2'-bipyridine]-6,6'-diyl(9,9-dioctyl-9H-fluorene-2,7-diyl)]](/data/chemimg/3726600/1423043-97-3_b.png)