Co-reporter: Dr. Thorsten Berg
Angewandte Chemie 2017 Volume 129(Issue 40) pp:12214-12216
Publication Date(Web):2017/09/25
DOI:10.1002/ange.201706479
Wacht auf! Die Schlafkrankheit und die Chagas-Krankheit werden durch Infektionen mit Trypanosomen verursacht. Die Entwicklung niedermolekularer Hemmstoffe der für die Parasiten essenziellen PEX14/PEX5-Wechselwirkung hat großes Potenzial für die Wirkstoffentwicklung.
Co-reporter: Dr. Thorsten Berg
Angewandte Chemie International Edition 2017 Volume 56(Issue 40) pp:12048-12050
Publication Date(Web):2017/09/25
DOI:10.1002/anie.201706479
Wake up! Sleeping sickness and Chagas disease are neglected tropical diseases caused by trypanosome infections. Small molecules that disrupt a crucial protein–protein interaction in the parasites offer a new approach to drug development for these diseases.
Co-reporter:Angela Berg, Thorsten Berg
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmcl.2017.06.012
The transcription factor STAT5a is a potential target for tumor therapy. We present a fluorescence polarization-based, high-throughput screen of chemical libraries containing natural products and known bioactive molecules, for the identification of small-molecule inhibitors of the STAT5a SH2 domain. This screen identified suramin, a drug used to treat African trypanosomiasis, and its analogues NF023 and NF449 as inhibitors of the SH2 domains of STAT5a/b. Our data extend the known in vitro targets of suramin and analogues to include members of the STAT protein family.Download high-res image (93KB)Download full-size image
Co-reporter:Nagarajan Elumalai, Kalaiselvi Natarajan, Thorsten Berg
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 14(Issue 14) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.bmc.2017.05.039
The transcription factor STAT5b is an antitumor target. Recently, we presented the small molecules Stafib-1 and Stafib-2 as potent, selective inhibitors of the STAT5b SH2 domain. Here we report that halogen substitutions on the terminal phenyl ring of Stafib-1 and a close derivative are tolerated and specificity over the STAT5a SH2 domain is maintained, albeit with a slight reduction in activity. Our data demonstrate that the synthetic methodology used for generating Stafib-1 and Stafib-2 can be utilized to synthesize a small library of halogen-substituted derivatives, and extend the panel of catechol bisphosphate-based submicromolar and selective STAT5b inhibitors.Download high-res image (59KB)Download full-size image
Co-reporter:Dr. Andrej Scharow;Dr. Daniel Knappe;Dr. Wolfgang Reindl;Dr. Ralf Hoffmann;Dr. Thorsten Berg
ChemBioChem 2016 Volume 17( Issue 8) pp:759-767
Publication Date(Web):
DOI:10.1002/cbic.201500535
Abstract
Polo-like kinase 1 (Plk1), a validated cancer target, harbors a protein–protein interaction domain referred to as the polo-box domain (PBD), in addition to its enzymatic domain. Although functional inhibition either of the enzymatic domain or of the PBD has been shown to inhibit Plk1, so far there have been no reports of bifunctional agents with the potential to target both protein domains. Here we report the development of Plk1 inhibitors that incorporate both an ATP-competitive ligand of the enzymatic domain, derived from BI 2536, and a functional inhibitor of the PBD, based either on the small molecule poloxin-2 or on a PBD-binding peptide. Although these bifunctional agents do not seem to bind both protein domains simultaneously, the most potent compound displays low-nanomolar activity against the Plk1 PBD, with excellent selectivity over the PBDs of Plk2 and Plk3. Our data provide insights into challenges and opportunities relating to the optimization of Plk1 PBD ligands as potent Plk1 inhibitors.
Co-reporter:Angela Berg ;Dr. Thorsten Berg
ChemBioChem 2016 Volume 17( Issue 8) pp:650-656
Publication Date(Web):
DOI:10.1002/cbic.201500580
Abstract
Polo-like kinase 1 (Plk1), a key player in mitosis, is overexpressed in a wide range of tumor types and has been validated as a target for tumor therapy. In addition to its N-terminal kinase domain, Plk1 harbors a C-terminal protein–protein interaction domain, referred to as the polo-box domain (PBD). Because the PBD is unique to the five-member family of polo-like kinases, and its inhibition is sufficient to inhibit the enzyme, the Plk1 PBD is an attractive target for the inhibition of Plk1 function. Although peptide-based inhibitors are invaluable tools for elucidating the nature of the binding interface, small molecules are better suited for the induction of mitotic arrest and apoptosis in tumor cells by Plk1 inhibition. This review describes the considerable progress that has been made in developing small-molecule and peptide-based inhibitors of the Plk1 PBD.
Co-reporter:Andrej Scharow, Monika Raab, Krishna Saxena, Sridhar Sreeramulu, Denis Kudlinzki, Santosh Gande, Christina Dötsch, Elisabeth Kurunci-Csacsko, Susan Klaeger, Bernhard Kuster, Harald Schwalbe, Klaus Strebhardt, and Thorsten Berg
ACS Chemical Biology 2015 Volume 10(Issue 11) pp:2570
Publication Date(Web):August 17, 2015
DOI:10.1021/acschembio.5b00565
Polo-like kinase 1 (Plk1) is a central regulator of mitosis and has been validated as a target for antitumor therapy. The polo-box domain (PBD) of Plk1 regulates its kinase activity and mediates the subcellular localization of Plk1 and its interactions with a subset of its substrates. Functional inhibition of the Plk1 PBD by low-molecular weight inhibitors has been shown to represent a viable strategy by which to inhibit the enzyme, while avoiding selectivity issues caused by the conserved nature of the ATP binding site. Here, we report structure–activity relationships and mechanistic analysis for the first reported Plk1 PBD inhibitor, Poloxin. We present the identification of the optimized analog Poloxin-2, displaying significantly improved potency and selectivity over Poloxin. Poloxin-2 induces mitotic arrest and apoptosis in cultured human tumor cells at low micromolar concentrations, highlighting it as a valuable tool compound for exploring the function of the Plk1 PBD in living cells.
Co-reporter:Nagarajan Elumalai, Angela Berg, Stefan Rubner, and Thorsten Berg
ACS Chemical Biology 2015 Volume 10(Issue 12) pp:2884
Publication Date(Web):October 15, 2015
DOI:10.1021/acschembio.5b00817
Design approaches for inhibitors of protein–protein interactions are rare, but highly sought after. Here, we report that O-phosphorylation of simple derivatives of the natural products dihydrocapsaicin and N-vanillylnonanamide leads to inhibitors of the SH2 domain of the transcription factor STAT5b. The most potent molecule is obtained from dihydrocapsaicin in only three synthetic steps. It has submicromolar affinity for the SH2 domain of STAT5b (Ki = 0.34 μM), while displaying 35-fold selectivity over the highly homologous STAT5a (Ki = 13.0 μM). The corresponding pivaloyloxymethyl ester inhibits STAT5b with selectivity over STAT5a in human tumor cells. Importantly, it inhibits cell viability and induces apoptosis in human tumor cells in a STAT5-dependent manner. Our data validate O-phosphorylation of appropriately preselected natural products or natural product derivatives as a semirational design approach for small molecules that selectively inhibit phosphorylation-dependent protein–protein interaction domains in cultured human tumor cells.
Co-reporter:Corinna Gröst and Thorsten Berg
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 13) pp:3866-3870
Publication Date(Web):25 Feb 2015
DOI:10.1039/C5OB00212E
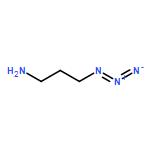
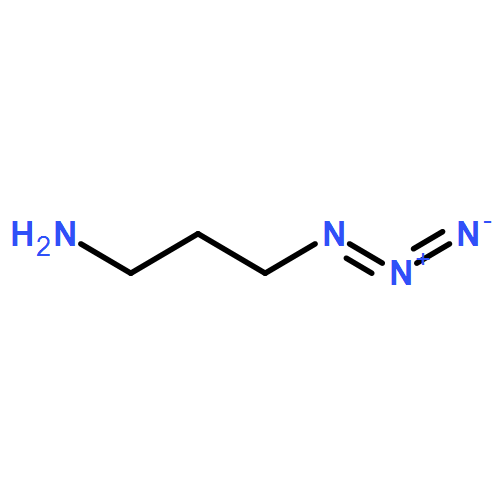
We present the concept, synthesis, and kinetic characterization of PYRROC as the first functionalized cycloalkyne which cannot form isomers in the reaction with azides. In aqueous buffer, PYRROC displays unprecedented rate accelerations in SPAAC of three to four orders of magnitude, leading to rate constants exceeding 400 M−1 s−1.
Co-reporter:M.Sc. Nagarajan Elumalai;M.Sc. Angela Berg;M.Sc. Kalaiselvi Natarajan;M.Sc. Andrej Scharow ;Dr. Thorsten Berg
Angewandte Chemie International Edition 2015 Volume 54( Issue 16) pp:4758-4763
Publication Date(Web):
DOI:10.1002/anie.201410672
Abstract
Src homology 2 (SH2) domains play a central role in signal transduction. Although many SH2 domains have been validated as drug targets, their structural similarity makes development of specific inhibitors difficult. The cancer-relevant transcription factors STAT5a and STAT5b are particularly challenging small-molecule targets because their SH2 domains are 93 % identical on the amino acid level. Here we present the natural product-inspired development of the low-nanomolar inhibitor Stafib-1, as the first small molecule which inhibits the STAT5b SH2 domain (Ki=44 nM) with more than 50-fold selectivity over STAT5a. The binding site of the core moiety of Stafib-1 was validated by functional analysis of point mutants. A prodrug of Stafib-1 was shown to inhibit STAT5b with high selectivity over STAT5a in tumor cells. Stafib-1 provides the first demonstration that naturally occurring SH2 domains with more than 90 % sequence identity can be selectively targeted with small organic molecules.
Co-reporter:M.Sc. Nagarajan Elumalai;M.Sc. Angela Berg;M.Sc. Kalaiselvi Natarajan;M.Sc. Andrej Scharow ;Dr. Thorsten Berg
Angewandte Chemie 2015 Volume 127( Issue 16) pp:4840-4845
Publication Date(Web):
DOI:10.1002/ange.201410672
Abstract
Src homology 2 (SH2) domains play a central role in signal transduction. Although many SH2 domains have been validated as drug targets, their structural similarity makes development of specific inhibitors difficult. The cancer-relevant transcription factors STAT5a and STAT5b are particularly challenging small-molecule targets because their SH2 domains are 93 % identical on the amino acid level. Here we present the natural product-inspired development of the low-nanomolar inhibitor Stafib-1, as the first small molecule which inhibits the STAT5b SH2 domain (Ki=44 nM) with more than 50-fold selectivity over STAT5a. The binding site of the core moiety of Stafib-1 was validated by functional analysis of point mutants. A prodrug of Stafib-1 was shown to inhibit STAT5b with high selectivity over STAT5a in tumor cells. Stafib-1 provides the first demonstration that naturally occurring SH2 domains with more than 90 % sequence identity can be selectively targeted with small organic molecules.
Co-reporter:Corinna Gröst, Martin Gräber, Michael Hell, Thorsten Berg
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 23) pp:7357-7363
Publication Date(Web):1 December 2013
DOI:10.1016/j.bmc.2013.09.057
Alexidine is in everyday human use as oral disinfectant and contact lens disinfectant. It is used as a mixture of stereoisomers. Since all of alexidine’s known biological targets are chiral, the biological activity of any of its chiral stereoisomers could be significantly higher than that of the mixture of stereoisomers. This makes a synthetic methodology for obtaining the individual enantiomers of the chiral diastereoisomer highly desirable. Here, we describe the first synthesis of both enantiomers of alexidine in high enantiomeric purity, and demonstrate their activity against the protein–protein interaction between the anti-apoptotic protein Bcl-xL and the pro-apoptotic protein Bak.
Co-reporter:Dr. Martin Gräber;Dipl.-Chem. Michael Hell;M.Sc. Corinna Gröst;Dr. Anders Friberg;Bianca Sperl;Dr. Michael Sattler;Dr. Thorsten Berg
Angewandte Chemie 2013 Volume 125( Issue 16) pp:4583-4588
Publication Date(Web):
DOI:10.1002/ange.201208889
Co-reporter:Dr. Martin Gräber;Dipl.-Chem. Michael Hell;M.Sc. Corinna Gröst;Dr. Anders Friberg;Bianca Sperl;Dr. Michael Sattler;Dr. Thorsten Berg
Angewandte Chemie International Edition 2013 Volume 52( Issue 16) pp:4487-4491
Publication Date(Web):
DOI:10.1002/anie.201208889
Co-reporter:Thorsten Berg
Chemistry & Biology 2012 Volume 19(Issue 10) pp:1215-1216
Publication Date(Web):26 October 2012
DOI:10.1016/j.chembiol.2012.09.007
Co-reporter:Angela Hollis;Bianca Sperl;Dr. Martin Gräber; Dr. Thorsten Berg
ChemBioChem 2012 Volume 13( Issue 2) pp:302-307
Publication Date(Web):
DOI:10.1002/cbic.201100652
Co-reporter:Martin Gräber, Weronika Janczyk, Bianca Sperl, Nagarajan Elumalai, Christian Kozany, Felix Hausch, Tad A. Holak, and Thorsten Berg
ACS Chemical Biology 2011 Volume 6(Issue 10) pp:1008
Publication Date(Web):July 28, 2011
DOI:10.1021/cb2001796
Phosphorylation-dependent protein binding domains are crucially important for intracellular signaling pathways and thus highly relevant targets in chemical biology. By screening of chemical libraries against 12 structurally diverse phosphorylation-dependent protein binding domains, we have identified fosfosal and dexamethasone-21-phosphate as selective inhibitors of two antitumor targets: the SH2 domain of the transcription factor STAT5b and the substrate-binding domain of the peptidyl-prolyl isomerase Pin1, respectively. Both compounds are phosphate prodrugs with documented clinical use as anti-inflammatory agents in humans and were discovered with a high hit rate from a small subgroup within the screening library. Our study indicates O-phosphorylation of appropriately preselected natural products or natural product derivatives as a generally applicable strategy for the identification of non-reactive and non-peptidic ligands of phosphorylation-dependent protein binding domains. Moreover, our data indicate that it would be advisable to monitor the bioactivities of clinically used prodrugs in their uncleaved state against phosphorylation-dependent protein binding domains.
Co-reporter:Corinna Gröst and Thorsten Berg
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 13) pp:NaN3870-3870
Publication Date(Web):2015/02/25
DOI:10.1039/C5OB00212E
We present the concept, synthesis, and kinetic characterization of PYRROC as the first functionalized cycloalkyne which cannot form isomers in the reaction with azides. In aqueous buffer, PYRROC displays unprecedented rate accelerations in SPAAC of three to four orders of magnitude, leading to rate constants exceeding 400 M−1 s−1.



![Piperidine, 1-[(2E,4E)-5-(3,4-dihydroxyphenyl)-1-oxo-2,4-pentadienyl]-](http://img.cochemist.com/ccimg/255000/254974-70-4.png)
![Piperidine, 1-[(2E,4E)-5-(3,4-dihydroxyphenyl)-1-oxo-2,4-pentadienyl]-](http://img.cochemist.com/ccimg/255000/254974-70-4_b.png)


![Nonanamide, N-[(3,4-dihydroxyphenyl)methyl]-](http://img.cochemist.com/ccimg/139500/139446-83-6.png)
![Nonanamide, N-[(3,4-dihydroxyphenyl)methyl]-](http://img.cochemist.com/ccimg/139500/139446-83-6_b.png)
![N-[(9h-fluoren-9-ylmethoxy)carbonyl]-3-hydroxy-l-tyrosine](http://img.cochemist.com/ccimg/137100/137018-93-0.png)
![N-[(9h-fluoren-9-ylmethoxy)carbonyl]-3-hydroxy-l-tyrosine](http://img.cochemist.com/ccimg/137100/137018-93-0_b.png)
![Nonanamide, N-[2-(3,4-dihydroxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/106000/105955-08-6.png)
![Nonanamide, N-[2-(3,4-dihydroxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/106000/105955-08-6_b.png)
![(1R,7AR)-7-{[(2,3-DIHYDROXY-2-ISOPROPYLBUTANOYL)OXY]METHYL}-2,3,5,7A-TETRAHYDRO-1H-PYRROLIZIN-1-YL (2E)-2-METHYL-2-BUTENOATE](http://img.cochemist.com/ccimg/42100/42092-05-7.png)
![(1R,7AR)-7-{[(2,3-DIHYDROXY-2-ISOPROPYLBUTANOYL)OXY]METHYL}-2,3,5,7A-TETRAHYDRO-1H-PYRROLIZIN-1-YL (2E)-2-METHYL-2-BUTENOATE](http://img.cochemist.com/ccimg/42100/42092-05-7_b.png)