Co-reporter:Yuriy Slutskyy, Christopher R. Jamison, Peng Zhao, Juyeol Lee, Young Ho Rhee, and Larry E. Overman
Journal of the American Chemical Society May 31, 2017 Volume 139(Issue 21) pp:7192-7192
Publication Date(Web):May 17, 2017
DOI:10.1021/jacs.7b04265
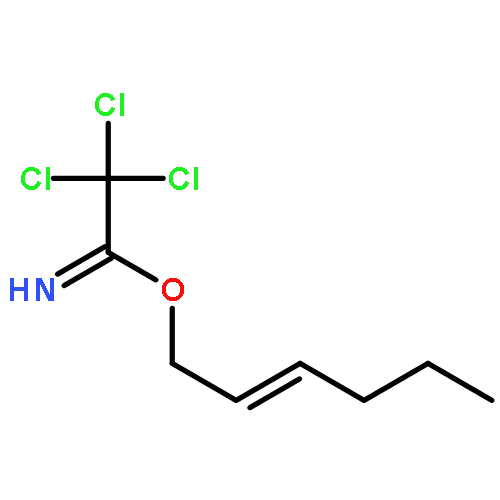
A short enantioselective synthesis of 6-substituted cis-2,8-dioxabicyclo[3.3.0]octan-3-ones is described. The pivotal step is coupling of a tertiary radical generated directly from a tertiary alcohol with a 3-chloro-5-alkoxybutenolide. This strategy is applied toward scalable 14–15 step syntheses of three rearranged spongian diterpenoids: cheloviolenes A and B and dendrillolide C.
Co-reporter:Christopher R. Jamison and Larry E. Overman
Accounts of Chemical Research 2016 Volume 49(Issue 8) pp:1578
Publication Date(Web):August 4, 2016
DOI:10.1021/acs.accounts.6b00284
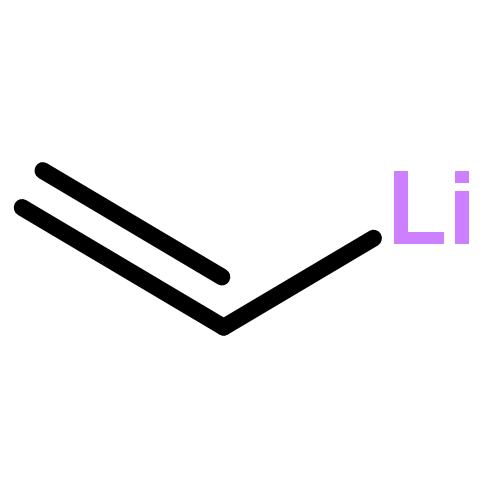
ConspectusConvergent synthesis strategies in which a target molecule is prepared by a branched approach wherein two or more complex fragments are combined at a late stage are almost always preferred over a linear approach in which the overall yield of the target molecule is eroded by the efficiency of each successive step in the sequence. As a result, bimolecular reactions that are able to combine complex fragments in good yield and, where important, with high stereocontrol are essential for implementing convergent synthetic strategies. Although intramolecular reactions of carbon radicals have long been exploited to assemble polycyclic ring systems, bimolecular coupling reactions of structurally complex carbon radicals have rarely been employed to combine elaborate fragments in the synthesis of structurally intricate molecules. We highlight in this Account recent discoveries from our laboratories that demonstrate that bimolecular reactions of structurally elaborate tertiary carbon radicals and electron-deficient alkenes can unite complex fragments in high yield using nearly equimolar amounts of the two coupling partners.Our discussion begins by considering several aspects of the bimolecular addition of tertiary carbon radicals to electron-deficient alkenes that commend these transformations for the union of structurally complex, sterically bulky fragments. We then discuss how in the context of synthesizing rearranged spongian diterpenoids we became aware of the exceptional utility of coupling reactions of alkenes and tertiary carbon radicals to unite structurally complex synthetic intermediates. Our initial investigations exploit the early report of Okada that N-(acyloxy)phthalimides reductively fragment at room temperature in the presence of visible light and catalytic amounts of the photocatalyst Ru(bpy)3Cl2 to form carbon radicals that react with alkenes. We show that this reaction of a tertiary radical precursor and an enone can combine two elaborate enantioenriched fragments in good yield with the formation of new quaternary and secondary stereocenters. As a result of the ready availability of tertiary alcohols, we describe two methods that were developed, one in collaboration with the MacMillan group, to generate tertiary radicals from tertiary alcohols. In the method that will be preferred in most instances, the tertiary alcohol is esterified in high yield to give a tert-alkyl hemioxalate salt, which—without purification—reacts with electron-deficient alkenes in the presence of visible light and an Ir(III) photocatalyst to give coupled products having a newly formed quaternary carbon in high yield. Hemioxalate salts containing Li, Na, K, and Cs countercations can be employed in this reaction, whose only other product is CO2. These reactions are carried out using nearly equimolar amounts of the addends, making them ideal for coupling of complex fragments at the late stage in a synthetic sequence. The attractive attributes of the fragment-coupling chemistry that we discuss in this Account are illustrated by an enantioselective total synthesis of a tricyclic trans-clerodane diterpenoid in eight steps and 34% overall yield from commercially available precursors. We anticipate that bimolecular reactions of carbon radicals will be increasingly used for fragment coupling in the future.
Co-reporter:Jeffrey S. Cannon and Larry E. Overman
Accounts of Chemical Research 2016 Volume 49(Issue 10) pp:2220
Publication Date(Web):September 30, 2016
DOI:10.1021/acs.accounts.6b00398
Allylic amides, amines, and esters are key synthetic building blocks. Their enantioselective syntheses under mild conditions is a continuing pursuit of organic synthesis methods development. One opportunity for the synthesis of these building blocks is by functionalization of prochiral double bonds using palladium(II) catalysis. In these reactions, nucleopalladation mediated by a chiral palladium(II) catalyst generates a new heteroatom-substituted chiral center. However, reactions where nucleopalladation occurs with antarafacial stereoselectivity are difficult to render enantioselective because of the challenge of transferring chiral ligand information across the square-planar palladium complex to the incoming nucleophile.In this Account, we describe the development and use of enantiopure palladium(II) catalysts of the COP (chiral cobalt oxazoline palladacyclic) family for the synthesis of enantioenriched products from starting materials derived from prochiral allylic alcohols. We begin with initial studies aimed at rendering catalyzed [3,3]-sigmatropic rearrangements of allylic imidates enantioselective, which ultimately led to the identification of the significant utility of the COP family of Pd(II) catalysts. The first use of an enantioselective COP catalyst was reported by Richards’ and our laboratories in 2003 for the enantioselective rearrangement of allylic N-arylimidates. Shortly thereafter, we discovered that the chloride-bridged COP dimer, [COP-Cl]2, was an excellent enantioselective catalyst for the rearrangement of (E)-allylic trichloroacetimidates to enantioenriched allylic trichloroacetamides, this conversion being the most widely used of the allylic imidate rearrangements. We then turn to discuss SN2′ reactions catalyzed by the acetate-bridged COP dimer, [COP-OAc]2, which proceed by a unique mechanism to provide branched allylic esters and allylic phenyl ethers in high enantioselectivity. Furthermore, because of the unique nucleopalladation/deoxypalladation mechanism of these SN2′ reactions, they provide exclusively the branched allylic product. Importantly, both enantiomers of the [COP-Cl]2 and [COP-OAc]2 catalysts are commercially available. We also briefly consider several other enantioselective reactions catalyzed by COP complexes.The mechanism of enantioselective COP-catalyzed allylic rearrangements and allylic substitutions is discussed in some detail. In both reactions, nucleopalladation is found to be the enantiodetermining step. The cyclobutadienyl “floor” of the COP catalyst is critical for transmitting chiral information across the palladium square plane in these reactions. This structural feature enables high enantioselection to be realized in spite of the nearly 180° angle between the catalyst, electrophile and nucleophile in the enantiodetermining step. Our discussion concludes by considering several uses of the COP family of catalysts by other researchers for the enantioselective synthesis of biologically active chiral molecules. We anticipate that additional uses for COP catalysts will emerge in the future. In addition, the structural features of these catalysts that we have identified as important for achieving high enantioselection should be useful in the future development of improved enantioselective Pd(II) catalysts.
Co-reporter:Daniel J. Tao; Yuriy Slutskyy
Journal of the American Chemical Society 2016 Volume 138(Issue 7) pp:2186-2189
Publication Date(Web):February 16, 2016
DOI:10.1021/jacs.6b00541
The first total synthesis of a chromodorolide diterpenoid is described. The synthesis features a bimolecular radical addition/cyclization/fragmentation cascade that unites butenolide and trans-hydrindane fragments while fashioning two C–C bonds and stereoselectively forming three of the ten contiguous stereocenters of chromodorolide B.
Co-reporter:Yuriy Slutskyy and Larry E. Overman
Organic Letters 2016 Volume 18(Issue 11) pp:2564-2567
Publication Date(Web):May 17, 2016
DOI:10.1021/acs.orglett.6b00895
Visible-light photoredox-catalyzed fragmentation of methyl N-phthalimidoyl oxalate allows the direct construction of a 1,4-dicarbonyl structural motif by a conjugate addition of the methoxycarbonyl radical to reactive Michael acceptors. The regioselectivity of the addition of this alkoxyacyl radical species to electron-deficient olefins is heavily influenced by the electronic nature of the acceptor, behavior similar to that exhibited by nucleophilic alkyl radicals.
Co-reporter:Yuriy Slutskyy, Christopher R. Jamison, Gregory L. Lackner, Daniel S. Müller, André P. Dieskau, Nicholas L. Untiedt, and Larry E. Overman
The Journal of Organic Chemistry 2016 Volume 81(Issue 16) pp:7029-7035
Publication Date(Web):June 2, 2016
DOI:10.1021/acs.joc.6b00697
The evolution of a convergent fragment-coupling strategy for the enantioselective total synthesis of trans-clerodane diterpenoids is described. The key bond construction is accomplished by 1,6-addition of a trans-decalin tertiary radical with 4-vinylfuran-2-one. The tertiary radical is optimally generated from the hemioxalate salt of the corresponding tertiary alcohol upon activation by visible light and an Ir(III) photoredox catalyst. The enantioselective total synthesis of trans-clerodane diterpenoid 1 reported here was accomplished in seven steps from 3-methyl-2-cyclohexenone. The synthetic strategy described in this report allows a number of trans-clerodane diterpenoids to be synthesized in enantioselective fashion by synthetic sequences of 10 steps or less. This study illustrates a powerful tactic in organic synthesis in which a structurally complex target structure is disconnected at a quaternary carbon stereocenter to fragments of comparable complexity, which are united in the synthetic pathway by conjugate addition of a nucleophilic tertiary radical to a fragment harboring an electron-deficient C–C double bond.
Co-reporter:Christopher C. Nawrat; Christopher R. Jamison; Yuriy Slutskyy; David W. C. MacMillan
Journal of the American Chemical Society 2015 Volume 137(Issue 35) pp:11270-11273
Publication Date(Web):August 31, 2015
DOI:10.1021/jacs.5b07678
Alkyl oxalates are new bench-stable alcohol-activating groups for radical generation under visible light photoredox conditions. Using these precursors, the first net redox-neutral coupling of tertiary and secondary alcohols with electron-deficient alkenes is achieved.
Co-reporter:Daniel S. Müller; Nicholas L. Untiedt; André P. Dieskau; Gregory L. Lackner
Journal of the American Chemical Society 2015 Volume 137(Issue 2) pp:660-663
Publication Date(Web):January 7, 2015
DOI:10.1021/ja512527s
A new concise construction of trans-clerodane diterpenoids is reported in which oxacyclic and trans-hydronaphthalene fragments are coupled, and the critical C9-quaternary carbon stereocenter formed stereoselectively, by 1,6-addition of a tertiary cuprate or a tertiary carbon radical to β-vinylbutenolide. This strategy is specifically illustrated by total syntheses of (−)-solidagolactone (4), (−)-16-hydroxycleroda-3,13-dien-15,16-olide (5, PL3), and (−)-annonene (6).
Co-reporter:Marcus Baumann, André P. Dieskau, Brad M. Loertscher, Mary C. Walton, Sangkil Nam, Jun Xie, David Horne and Larry E. Overman
Chemical Science 2015 vol. 6(Issue 8) pp:4451-4457
Publication Date(Web):26 May 2015
DOI:10.1039/C5SC01536G
Epipolythiodioxopiperazine (ETP) alkaloids are structurally elaborate alkaloids that show potent antitumor activity. However, their high toxicity and demonstrated interactions with various biological receptors compromises their therapeutic potential. In an effort to mitigate these disadvantages, a short stereocontrolled construction of tricyclic analogues of epidithiodioxopiperazine alkaloids was developed. Evaluation of a small library of such structures against two invasive cancer cell lines defined initial structure–activity relationships (SAR), which identified 1,4-dioxohexahydro-6H-3,8a-epidithiopyrrolo[1,2-a]pyrazine 3c and related structures as particularly promising antitumor agents. ETP alkaloid analogue 3c exhibits low nanomolar activity against both solid and blood tumors in vitro. In addition, 3c significantly suppresses tumor growth in mouse xenograft models of melanoma and lung cancer, without obvious signs of toxicity, following either intraperitoneal (IP) or oral administration. The short synthesis of molecules in this series will enable future mechanistic and translational studies of these structurally novel and highly promising clinical antitumor candidates.
Co-reporter:Mary C. Walton, Yun-Fang Yang, Xin Hong, K. N. Houk, and Larry E. Overman
Organic Letters 2015 Volume 17(Issue 24) pp:6166-6169
Publication Date(Web):November 25, 2015
DOI:10.1021/acs.orglett.5b03171
Copper-catalyzed reactions of glycine ester arylimines and methacrylonitrile provide selective access to either the endo or exo pyrrolidine cycloadducts. DFT calculations have elucidated the origins of ligand-controlled diastereoselectivity.
Co-reporter:Stephen M. Canham, Benjamin D. Hafensteiner, Alec D. Lebsack, Tricia L. May-Dracka, Sangkil Nam, Brian A. Stearns, Larry E. Overman
Tetrahedron 2015 Volume 71(Issue 37) pp:6424-6436
Publication Date(Web):16 September 2015
DOI:10.1016/j.tet.2015.02.080
A unified strategy for enantioselective total synthesis of all stereoisomers of the [2+2] family of quadrigemine alkaloids is reported. In this approach, two enantioselective intramolecular Heck reactions are carried out at the same time on precursors fashioned in four steps from either meso- or (+)-chimonanthine to form the two critical quaternary carbons of the peripheral cyclotryptamine rings of these products. Useful levels of catalyst control are realized in either desymmetrizing a meso precursor or controlling diastereoselectivity in elaborating C2-symmetric intermediates. None of the synthetic quadrigemines are identical with alkaloids isolated previously and referred to as quadrigemines A and E. In addition, we report improvements in our previous total syntheses of (+)- or (−)-quadrigemine C that shortened the synthetic sequence to 10 steps and provided these products in 2.2% overall yield from tryptamine.
Co-reporter:Gerald Pratsch, Gregory L. Lackner, and Larry E. Overman
The Journal of Organic Chemistry 2015 Volume 80(Issue 12) pp:6025-6036
Publication Date(Web):June 1, 2015
DOI:10.1021/acs.joc.5b00795
Tertiary carbon radicals have notable utility for uniting complex carbon fragments with concomitant formation of new quaternary carbons. This article explores the scope, limitations, and certain mechanistic aspects of Okada’s method for forming tertiary carbon radicals from N-(acyloxy)phthalimides by visible-light photocatalysis. Optimized conditions for generating tertiary radicals from N-(acyloxy)phthalimide derivatives of tertiary carboxylic acids by visible-light irradiation in the presence of 1 mol % of commercially available Ru(bpy)3(PF6)2, diethyl 1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (8), and i-Pr2NEt and their coupling in dichloromethane at room temperature with alkene acceptors were developed. Four representative tertiary N-(acyloxy)phthalimides and 15 alkene radical acceptors were examined. Both reductive couplings with electron-deficient alkenes and radical substitution reactions with allylic and vinylic bromides and chlorides were examined with many such reactions occurring in good yield using only a slight excess (typically 1.5 equiv) of the alkene. In general, the yields of these photocatalytic reactions were higher than the analogous transformations of the corresponding N-phthalimidoyl oxalates. Deuterium labeling and competition experiments reveal that the reductive radical coupling of tertiary N-(acyloxy)phthalimides with electron-deficient alkenes can be terminated by both hydrogen-atom transfer and single-electron reduction followed by protonation, and that this mechanistic duality is controlled by the presence or absence of i-Pr2NEt.
Co-reporter:Gregory L. Lackner, Kyle W. Quasdorf, Gerald Pratsch, and Larry E. Overman
The Journal of Organic Chemistry 2015 Volume 80(Issue 12) pp:6012-6024
Publication Date(Web):June 1, 2015
DOI:10.1021/acs.joc.5b00794
The coupling of tertiary carbon radicals with alkene acceptors is an underdeveloped strategy for uniting complex carbon fragments and forming new quaternary carbons. The scope and limitations of a new approach for generating nucleophilic tertiary radicals from tertiary alcohols and utilizing these intermediates in fragment coupling reactions is described. In this method, the tertiary alcohol is first acylated to give the tert-alkyl N-phthalimidoyl oxalate, which in the presence of visible-light, catalytic Ru(bpy)3(PF6)2, and a reductant fragments to form the corresponding tertiary carbon radical. In addition to reductive coupling with alkenes, substitution reactions of tertiary radicals with allylic and vinylic halides is described. A mechanism for the generation of tertiary carbon radicals from tert-alkyl N-phthalimidoyl oxalates is proposed that is based on earlier pioneering investigations of Okada and Barton. Deuterium labeling and competition experiments reveal that the reductive radical coupling of tert-alkyl N-phthalimidoyl oxalates with electron-deficient alkenes is terminated by hydrogen-atom transfer.
Co-reporter:Gerald Pratsch and Larry E. Overman
The Journal of Organic Chemistry 2015 Volume 80(Issue 22) pp:11388-11397
Publication Date(Web):October 30, 2015
DOI:10.1021/acs.joc.5b01962
Visible-light photoreductive coupling of 2-arylallyl bromides in the presence of the photocatalyst Ru(bpy)3(PF6)2, a Hantzsch ester, and i-Pr2NEt gives 2,5-diaryl-1,5-dienes in high yield. This method avoids the use of stoichiometric metal reductants and is compatible with the presence of halogen, alkyl, electron-donating, and electron-withdrawing substituents on the aromatic ring.
Co-reporter:Michael B. Shaghafi, David G. Barrett, Francis S. Willard, Larry E. Overman
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 4) pp:1031-1036
Publication Date(Web):15 February 2014
DOI:10.1016/j.bmcl.2014.01.021
We report the discovery of the glucose-dependent insulin secretogogue activity of a novel class of polycyclic guanidines through phenotypic screening as part of the Lilly Open Innovation Drug Discovery platform. Three compounds from the University of California, Irvine, 1–3, having the 3-arylhexahydropyrrolo[1,2-c]pyrimidin-1-amine scaffold acted as insulin secretagogues under high, but not low, glucose conditions. Exploration of the structure–activity relationship around the scaffold demonstrated the key role of the guanidine moiety, as well as the importance of two lipophilic regions, and led to the identification of 9h, which stimulated insulin secretion in isolated rat pancreatic islets in a glucose-dependent manner.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:John E. DeLorbe ; David Horne ; Richard Jove ; Steven M. Mennen ; Sangkil Nam ; Fang-Li Zhang
Journal of the American Chemical Society 2013 Volume 135(Issue 10) pp:4117-4128
Publication Date(Web):March 1, 2013
DOI:10.1021/ja400315y
A common strategy for preparing tryptophan-derived epidithiodioxopiperazine (ETP) natural products containing a hydroxyl substituent adjacent to a quaternary carbon stereocenter is reported. This strategy is exemplified by enantioselective total syntheses of four heptacyclic ETP natural products—gliocladine C (6), leptosin D (7), T988C (8), and bionectin A (9)—starting with the di-(tert-butoxycarbonyl) derivative 17 of the trioxopiperazine natural product gliocladin C, which is readily available by enantioselective chemical synthesis. In addition, total syntheses of the enantiomer of gliocladine C (ent-6) and gliocladin A (11), the di(methylthio) congener of bionectin A, are reported. These syntheses illustrate a synthetic strategy wherein diversity in the dioxopiperazine unit of ETP natural products is introduced at a late stage in a synthetic sequence. In vitro cytotoxicity of compounds in this series against invasive human prostrate (DU145) and melanoma (A2058) cancer cell lines is described and compared to that of chaetocin A (4).
Co-reporter:Gregory L. Lackner ; Kyle W. Quasdorf
Journal of the American Chemical Society 2013 Volume 135(Issue 41) pp:15342-15345
Publication Date(Web):September 27, 2013
DOI:10.1021/ja408971t
A convenient method for the direct construction of quaternary carbons from tertiary alcohols by visible-light photoredox coupling of tert-alkyl N-phthalimidoyl oxalate intermediates with electron-deficient alkenes is reported.
Co-reporter:Salman Y. Jabri
Journal of the American Chemical Society 2013 Volume 135(Issue 11) pp:4231-4234
Publication Date(Web):March 3, 2013
DOI:10.1021/ja401423j
The first total synthesis of a member of the plectosphaeroic acid family of fungal natural products is reported. Key steps include the late-stage formation of the hindered N6–C9″ bond and stereoselective introduction of the two methylthio substituents.
Co-reporter:Stephen M. Canham, David J. France, and Larry E. Overman
The Journal of Organic Chemistry 2013 Volume 78(Issue 1) pp:9-34
Publication Date(Web):June 26, 2012
DOI:10.1021/jo300872y
This article describes synthetic studies that culminated in the first total synthesis of the Lycopodium alkaloid sieboldine A. During this study, a number of pinacol-terminated cationic cyclizations were examined to form the cis-hydrindanone core of sieboldine A. Of these, a mild Au(I)-promoted 1,6-enyne cyclization that was terminated by a semipinacol rearrangement proved to be most efficient. Fashioning the unprecedented N-hydroxyazacyclononane ring embedded within the bicyclo[5.2.1]decane-N,O-acetal moiety of sieboldine A was a formidable challenge. Ultimately, the enantioselective total synthesis of (+)-sieboldine A was completed by forming this ring in good yield by cyclization of a protected-hydroxylamine thioglycoside precursor.
Co-reporter:Salman Y. Jabri and Larry E. Overman
The Journal of Organic Chemistry 2013 Volume 78(Issue 17) pp:8766-8788
Publication Date(Web):August 27, 2013
DOI:10.1021/jo4015479
Evolution of the synthetic strategy that culminated in the first total syntheses of the structurally unique plectosphaeroic acids B (2) and C (3) is described. The successful enantioselective route to (+)-2 and (+)-3 proceeds in 6 and 11 steps from the known hexahydro-2H-pyrazinopyrrolo[2,3-b]indole-1,4-dione 39, which in turn is available in enantiomerically pure form by chemical synthesis. The central challenge in this synthesis endeavor was uniting the hexahydro-2H-pyrazinopyrrolo[2,3-b]indole-1,4-dione and cinnabarinic acid fragments of these marine alkaloids. Critical for achieving this successful C–N bond formation was the use of an iodocinnabarinic acid diester in which the amino group was masked with two Boc substituents, a Cu(I) carboxylate complex and the weak base KOAc. The highly congested C–N bond generated in this coupling, in conjunction with the delicate nature of the densely functionalized coupling partners, provided a striking testament to the power of modern copper-mediated amination methods. Two approaches, one stereoselective, for introducing the methylthio substituents of (+)-plectosphaeroic acid B were developed. The epitrisulfide ring of (+)-plectosphaeroic acid C was formed by ring expansion of an epidisulfide precursor.
Co-reporter:Zachary D. Aron, Tatsuya Ito, Tricia L. May, Larry E. Overman, and Jocelyn Wang
The Journal of Organic Chemistry 2013 Volume 78(Issue 19) pp:9929-9948
Publication Date(Web):September 10, 2013
DOI:10.1021/jo4018099
A new strategy for enantioselective synthesis of azacyclic molecules in which dynamic kinetic equilibration of diastereomeric iminium ions precedes a stereochemistry-determining sigmatropic rearrangement is reported. The method is illustrated by the synthesis, in high enantiomeric purity (generally 95–99% ee), of a variety of 1-azabicyclic molecules containing angular allyl or 3-substituted 2-propenyl side chains adjacent to nitrogen and up to three stereogenic centers. In these products, the size of the carbocyclic ring is varied widely (5–12 membered); however, useful yields are obtained in forming 1-azabicyclic products containing only fused pyrrolidine and piperidine rings. Chirality transfer from substituents at carbons 1 and 2 of the 3-butenylamine fragment of the starting material is investigated, with methyl and phenyl substituents at the allylic position shown to provide exquisite stereocontrol (generally 98–99% chirality transfer). An attractive feature of the method is the ability to carry out the key transformation in the absence of solvent. Illustrated also is the high yielding conversion of four such products to a new family of bicyclic β-amino acids of high enantiomeric purity.
Co-reporter:Hao Wang ; Philipp Kohler ; Larry E. Overman ;K. N. Houk
Journal of the American Chemical Society 2012 Volume 134(Issue 38) pp:16054-16058
Publication Date(Web):September 6, 2012
DOI:10.1021/ja3075538
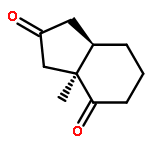
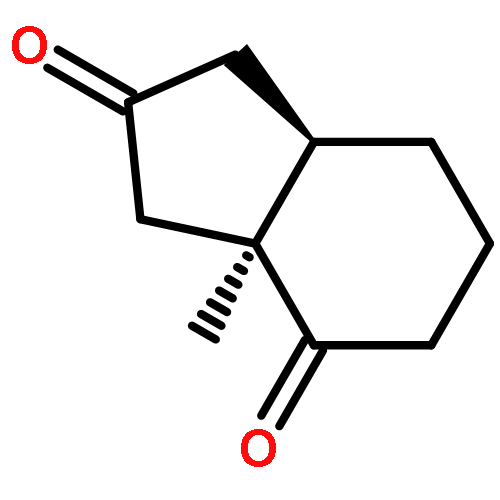
Stereoselectivities of the dihydroxylations of cis-bicyclo[3.3.0]octene intermediates for a projected total synthesis of chromodorolide A have been explored experimentally. The reaction occurs unexpectedly on the apparently more hindered (concave) face; this result has been explained through computational studies using B3LYP and B3LYP-D3 methods. Torsional effects are largely responsible for the stereoselectivity encountered in the chromodorolide A synthesis. Many literature examples have been reported on related cases. QM calculations show that the stereoselectivities of dihydroxylations of fused cyclopentenes are influenced by the conformational rigidity or flexibility of the substrate. Torsional, electrostatic, and steric effects can all influence stereoselectivity, and the rigidity or flexibility of conformations of reactants provides a predictive guide to stereoselectivity.
Co-reporter:Jeffrey S. Cannon, Angela C. Olson, Larry E. Overman, and Nicole S. Solomon
The Journal of Organic Chemistry 2012 Volume 77(Issue 4) pp:1961-1973
Publication Date(Web):February 8, 2012
DOI:10.1021/jo202553a
2-Vinylchromanes (1), 2-vinyl-1,4-benzodioxanes (2), and 2,3-dihydro-2-vinyl-2H-1,4-benzoxazines (3) can be prepared in high yields (90–98%) and excellent enantiomeric purities (87–98% ee) by [COP-OAc]2-catalyzed cyclization of phenolic (E)-allylic trichloroacetimidate precursors. Deuterium-labeling and computational experiments are consistent with these cyclization reactions taking place by an anti-oxypalladation/syn-deoxypalladation mechanism. 2-Vinylchromanes can also be prepared in good yields and high enantiomeric purities from analogous (E)-allylic acetate precursors, which constitutes the first report that acetate is a competent leaving group in COP-catalyzed enantioselective SN2′ substitution reactions.
Co-reporter:Jeffrey S. Cannon ;Dr. Larry E. Overman
Angewandte Chemie 2012 Volume 124( Issue 18) pp:4362-4386
Publication Date(Web):
DOI:10.1002/ange.201107385
Abstract
Von Beginn des 19. Jahrhunderts an bis in die Gegenwart hinein haben sich Chemiker mit dem komplexen Indol-Alkaloid Strychnin beschäftigt. In diesem Aufsatz wollen wir untersuchen, weshalb Strychnin bis zum heutigen Tag eine bedeutende Zielstruktur für direkte Syntheseversuche ist. Wir werden eine Reihe von Syntheserouten zu Strychnin diskutieren, um so letztlich ihren Einfluss auf die Entwicklung von Strategien und Methoden in der organischen Synthese aufzuklären.
Co-reporter:Dr. Martin J. Schnermann ; Larry E. Overman
Angewandte Chemie 2012 Volume 124( Issue 38) pp:9714-9718
Publication Date(Web):
DOI:10.1002/ange.201204977
Co-reporter:Jeffrey S. Cannon ;Dr. Larry E. Overman
Angewandte Chemie International Edition 2012 Volume 51( Issue 18) pp:4288-4311
Publication Date(Web):
DOI:10.1002/anie.201107385
Abstract
From the 19th century to the present, the complex indole alkaloid strychnine has engaged the chemical community. In this Review, we examine why strychnine has been and remains today an important target for directed synthesis efforts. A selection of the diverse syntheses of strychnine is discussed with the aim of identifying their influence on the evolution of the strategy and tactics of organic synthesis.
Co-reporter:Dr. Martin J. Schnermann;Nicholas L. Untiedt;Dr. Gonzalo Jiménez-Osés; Kendall N. Houk; Larry E. Overman
Angewandte Chemie International Edition 2012 Volume 51( Issue 38) pp:9581-9586
Publication Date(Web):
DOI:10.1002/anie.201205001
Co-reporter:Dr. Martin J. Schnermann ; Larry E. Overman
Angewandte Chemie International Edition 2012 Volume 51( Issue 38) pp:9576-9580
Publication Date(Web):
DOI:10.1002/anie.201204977
Co-reporter:Jeffrey S. Cannon, James H. Frederich, and Larry E. Overman
The Journal of Organic Chemistry 2012 Volume 77(Issue 4) pp:1939-1951
Publication Date(Web):January 30, 2012
DOI:10.1021/jo2025724
A new family of air- and moisture-stable enantiopure C,N-palladacycles (PIN-acac complexes) were prepared in good overall yield in three steps from 2-iodo-1-naphthoic acid and enantiopure β-amino alcohols. Three of these PIN complexes were characterized by single-crystal X-ray analysis. As anticipated, the naphthalene and imidazoline rings of PIN-acac complexes 18a and 18b were canted significantly from planarity and projected the imidazoline substituents R1 and R2 on opposite faces of the palladium square plane. Fifteen PIN complexes were evaluated as catalysts for the rearrangement of prochiral (E)-allylic trichloroacetimidate 19 (eq 2) and the SN2′ allylic substitution of acetic acid with prochiral (Z)-allylic trichloroacetimidate 23. Although these complexes were kinetically poor catalysts for the Overman rearrangement, they were good catalysts for the allylic substitution reaction, providing branched allylic esters in high yield. However, enantioselectivities were low to moderate and significantly less than that realized with palladacyclic complexes of the COP family. Computational studies support an anti-acetoxypalladation/syn-deoxypalladation mechanism analogous to that observed with COP catalysts. The computational study further suggests that optimizing steric influence in the vicinity of the carbon ligand of a chiral C,N-palladacycle, rather than near the nitrogen heterocycle, is the direction to pursue in future development of improved enantioselective catalysts of this motif.
Co-reporter:John E. DeLorbe ; Salman Y. Jabri ; Steven M. Mennen ; Larry E. Overman ;Fang-Li Zhang
Journal of the American Chemical Society 2011 Volume 133(Issue 17) pp:6549-6552
Publication Date(Web):April 7, 2011
DOI:10.1021/ja201789v
A concise second-generation total synthesis of the fungal-derived alkaloid (+)-gliocladin C (11) in 10 steps and 11% overall yield from isatin is reported. In addition, the epipolythiodioxopiperazine (ETP) natural product (+)-gliocladine C (6) has been prepared in six steps and 29% yield from the di-(tert-butoxycarbonyl) precursor of 11. The total synthesis of 6 constitutes the first total synthesis of an ETP natural product containing a hydroxyl substituent adjacent to a quaternary carbon stereocenter in the pyrrolidine ring.
Co-reporter:Martin J. Schnermann
Journal of the American Chemical Society 2011 Volume 133(Issue 41) pp:16425-16427
Publication Date(Web):September 21, 2011
DOI:10.1021/ja208018s
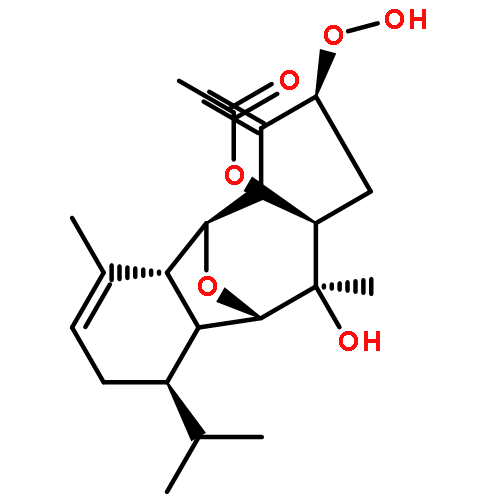
The enantioselective total synthesis of the rearranged spongian diterpene aplyviolene has been completed in 14 steps from the known hydroazulenone 8. The key junction of the hydrocarbon and oxygenated fragments to form the critical C8 quaternary carbon stereocenter and set the stage for elaborating the delicate bicyclic lactone functionality was accomplished in high yield and exquisite stereoselectivity by Michael addition of an enantioenriched hydroazulenone enolate to an enantiopure α-bromocyclopentenone.
Co-reporter:Martin J. Schnermann ; Christopher M. Beaudry ; Nathan E. Genung ; Stephen M. Canham ; Nicholas L. Untiedt ; Breanne D. W. Karanikolas ; Christine Sütterlin
Journal of the American Chemical Society 2011 Volume 133(Issue 43) pp:17494-17503
Publication Date(Web):October 11, 2011
DOI:10.1021/ja207727h
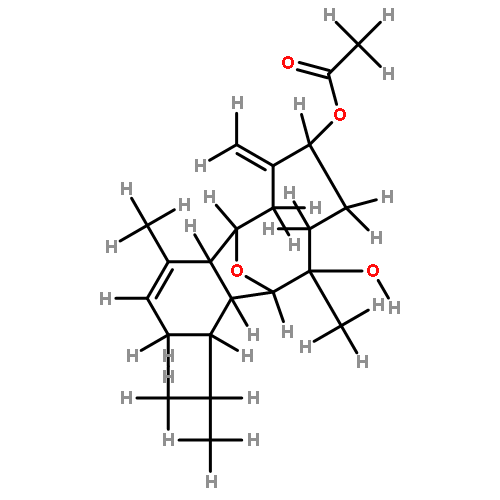
The synthesis and direct comparison of the chemical reactivity of the two highly oxidized bicyclic lactone fragments found in rearranged spongian diterpenes (8-substituted 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one and 6-substituted 7-acetoxy-2,8-dioxabicyclo[3.3.0]octan-3-one) are reported. Details of the first synthesis of the 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one ring system, including an examination of several possibilities for the key bridging cyclization reaction, are described. In addition, the first synthesis of 7-acetoxy-2,8-dioxabicyclo[3.3.0]octanones containing quaternary carbon substituents at C6 is disclosed. Aspects of the chemical reactivity and Golgi-modifying properties of these bicyclic lactone analogs of rearranged spongian diterpenes are also reported. Under both acidic and basic conditions, 8-substituted 2,7-dioxabicyclo[3.2.1]octanones are converted to 6-substituted-2,8-dioxabicyclo[3.3.0]octanones. Moreover, these dioxabicyclic lactones react with primary amines and lysine side chains of lysozyme to form substituted pyrroles, a conjugation that could be responsible for the unique biological properties of these compounds. These studies demonstrate that acetoxylation adjacent to the lactone carbonyl group, in either the bridged or fused series, is required to produce fragmented Golgi membranes in the pericentriolar region that is characteristic of macfarlandin E.
Co-reporter:Connor L. Martin, Seiichi Nakamura, Ralf Otte, and Larry E. Overman
Organic Letters 2011 Volume 13(Issue 1) pp:138-141
Publication Date(Web):December 6, 2010
DOI:10.1021/ol102709s
The first enantioselective total syntheses of indole alkaloids of the condylocarpine type are reported. (+)-Condylocarpine, (+)-isocondylocarpine, and (+)-tubotaiwine were prepared in high enantiomeric purity (er > 99:1) from (1S,5R)-hexahydro-1,5-methano-1H-azocino[4,3-b]indole-12-one 7b by way of five or six isolated intermediates.
Co-reporter:Stephen M. Canham, Larry E. Overman, Paul S. Tanis
Tetrahedron 2011 67(51) pp: 9837-9843
Publication Date(Web):
DOI:10.1016/j.tet.2011.09.079
Co-reporter:Stephen M. Canham ; David J. France
Journal of the American Chemical Society 2010 Volume 132(Issue 23) pp:7876-7877
Publication Date(Web):May 19, 2010
DOI:10.1021/ja103666n
The first total synthesis of (+)-sieboldine A was completed in 20 steps from readily available (3aS,6aR)-3,3a,4,6a-tetrahydro-2H-cyclopenta[b]furan-2-one (5). Key steps are as follows: (a) a pinacol-terminated 1,6-enyne cyclization reaction to form the cis-hydrindanone core (11 → 12), (b) formation of the spiro tetrahydrofuran ring by stereoselective DMDO oxidation of tricyclic dihydropyran intermediate 15, and (c) formation of the unprecedented N-hydroxyazacyclononane ring by cyclization of a thioglycoside precursor (18 → 19).
Co-reporter:Tatsuya Ito ; Larry E. Overman ;Jocelyn Wang
Journal of the American Chemical Society 2010 Volume 132(Issue 10) pp:3272-3273
Publication Date(Web):February 22, 2010
DOI:10.1021/ja100607z
A useful enantioselective synthesis of angularly substituted 1-azabicyclic molecules that delivers the bicyclic amine products in good yield and 99% ee is reported. Angularly substituted cis-octahydrocyclopenta[b]pyrroles, cis-octahydroindoles, cis-decahydrocyclohepta[b]pyrroles, and cis-octahydrocyclopenta[b]pyridine are formed in good yields and 99% ee exclusively as the cis stereoisomers. Decahydroquinolines are generated as mixtures of cis and trans stereoisomers in similarly high ee. The starting materials for this cascade transformation are assembled in five steps from cycloalkanones and enantiomerically pure (R)-2-phenyl-3-butenamine. This enantioselective synthesis introduces a new strategy for dynamic kinetic resolution in which a rapid tautomeric equilibration of diastereomeric iminium cations is combined with a diastereoselective sigmatropic rearrangement.
Co-reporter:Connor L. Martin ; Larry E. Overman ;Jason M. Rohde
Journal of the American Chemical Society 2010 Volume 132(Issue 13) pp:4894-4906
Publication Date(Web):March 10, 2010
DOI:10.1021/ja100178u
Development of efficient sequences for the total syntheses of (±)-actinophyllic acid (rac-1) and (−)-actinophyllic acid (1) are described. The central step in these syntheses is the aza-Cope/Mannich reaction, which constructs the previously unknown hexacyclic ring system of actinophyllic acid in one step from much simpler tetracyclic precursors. The tetracyclic hexahydro-1,5-methano-1H-azocino[4,3-b]indole ketone rac-37 is assembled from o-nitrophenylacetic acid in four steps, with oxidative cyclization of a dienolate derivative of tricyclic precursor rac-35 being the central step. In the first-generation synthesis, this intermediate is transformed in two steps to homoallyl amine rac-43, whose formaldiminium derivative undergoes efficient aza-Cope/Mannich reaction to give pentacyclic ketone rac-44. In four additional steps, this intermediate is advanced to (±)-actinophyllic acid. The synthesis is streamlined by elaborating ketone rac-37 to β-hydroxyester intermediate rac-53, which is directly transformed to (±)-actinophyllic acid upon exposure to HCl and paraformaldehyde. This concise second-generation total synthesis of (±)-actinophyllic acid is realized in 22% overall yield from commercially available di-tert-butyl malonate and o-nitrophenylacetic acid by a sequence that proceeds by way of only six isolated intermediates. The first enantioselective total synthesis of (−)-actinophyllic acid (1) is accomplished by this direct sequence from tricyclic keto malonate (S)-35. Catalytic enantioselective reduction of α,β-unsaturated ketone 66 is the key step in the preparation of intermediate (S)-35 from the commercially available Boc-amino acid 65. Discussed also is the possibility that the aza-Cope/Mannich reaction might be involved in the biosynthesis of (−)-actinophyllic acid.
Co-reporter:Jeffrey S. Cannon ; Stefan F. Kirsch ; Larry E. Overman ;Helen F. Sneddon
Journal of the American Chemical Society 2010 Volume 132(Issue 43) pp:15192-15203
Publication Date(Web):October 13, 2010
DOI:10.1021/ja106688j
The catalytic enantioselective SN2′ displacement of (Z)-allylic trichloroacetimidates catalyzed by the palladium(II) complex [COP-OAc]2 is a broadly useful method for the asymmetric synthesis of chiral branched allylic esters. A variety of experiments aimed at elucidating the nature of the catalytic mechanism and its rate- and enantiodetermining steps are reported. Key findings include the following: (a) the demonstration that a variety of bridged-dipalladium complexes are present and constitute resting states of the COP catalyst (however, monomeric palladium(II) complexes are likely involved in the catalytic cycle); (b) labeling experiments establishing that the reaction proceeds in an overall antarafacial fashion; (c) secondary deuterium kinetic isotope effects that suggest substantial rehybridization at both C1 and C3 in the rate-limiting step; and (d) DFT computational studies (B3-LYP/def2-TZVP) that provide evidence for bidentate substrate-bound intermediates and an anti-oxypalladation/syn-deoxypalladation pathway. These results are consistent with a novel mechanism in which chelation of the imidate nitrogen to form a cationic palladium(II) intermediate activates the alkene for attack by external carboxylate in the enantiodetermining step. Computational modeling of the transition-state structure for the acyloxy palladation step provides a model for enantioinduction.
Co-reporter:Jeffrey S. Cannon ; Stefan F. Kirsch
Journal of the American Chemical Society 2010 Volume 132(Issue 43) pp:15185-15191
Publication Date(Web):October 13, 2010
DOI:10.1021/ja106685w
A broadly useful catalytic enantioselective synthesis of branched allylic esters from prochiral (Z)-2-alkene-1-ols has been developed. The starting allylic alcohol is converted to its trichloroacetimidate intermediate by reaction with trichloroacetonitrile, either in situ or in a separate step, and this intermediate undergoes clean enantioselective SN2′ substitution with a variety of carboxylic acids in the presence of the palladium(II) catalyst (Rp,S)-di-μ-acetatobis[(η5-2-(2′-(4′-methylethyl)oxazolinyl)cyclopentadienyl-1-C,3′-N)(η4-tetraphenylcyclobutadiene)cobalt]dipalladium, (Rp,S)-[COP-OAc]2, or its enantiomer. The scope and limitations of this useful catalytic asymmetric allylic esterification are defined.
Co-reporter:Larry E. Overman, Mark D. Rosen
Tetrahedron 2010 66(33) pp: 6514-6525
Publication Date(Web):
DOI:10.1016/j.tet.2010.05.048
Co-reporter:Martin J. Schnermann;Christopher M. Beaudry;Anastasia V. Egorova;Roman S. Polishchuk;Christine Sütterlin;
Proceedings of the National Academy of Sciences 2010 107(14) pp:6158-6163
Publication Date(Web):March 23, 2010
DOI:10.1073/pnas.1001421107
Golgi-modifying properties of the spongian diterpene macfarlandin E (MacE) and a synthetic analog, t-Bu-MacE, containing its 2,7-dioxabicyclo[3.2.1]octan-3-one moiety are reported. Natural product screening efforts identified
MacE as inducing a novel morphological change in Golgi structure defined by ribbon fragmentation with maintenance of the resulting
Golgi fragments in the pericentriolar region. t-Bu-MacE, which possesses the substituted 2,7-dioxabicyclo[3.2.1]octan-3-one but contains a tert-butyl group in place of the hydroazulene subunit of MacE, was prepared by chemical synthesis. Examination of the Golgi-modifying
properties of MacE, t-Bu-MacE, and several related structures revealed that the entire oxygen-rich bridged-bicyclic fragment is required for induction
of this unique Golgi organization phenotype. Further characterization of MacE-induced Golgi modification showed that protein
secretion is inhibited, with no effect on the actin or microtubule cytoskeleton being observed. The conversion of t-Bu-MacE and a structurally related des-acetoxy congener to substituted pyrroles in the presence of primary amines in protic
solvent at ambient temperatures suggests that covalent modification might be involved in the Golgi-altering activity of MacE.
Co-reporter:Travis B. Dunn, J. Michael Ellis, Christiane C. Kofink, James R. Manning and Larry E. Overman
Organic Letters 2009 Volume 11(Issue 24) pp:5658-5661
Publication Date(Web):November 11, 2009
DOI:10.1021/ol902373m
The aza-Cope-Mannich reaction and ring-closing metathesis are key steps in the assembly of intermediates containing rings A−D of Daphniphyllum alkaloids of the daphnicyclidin type such as daphnipaxinin and oldhamine A.
Co-reporter:AngelaC. Olson;LarryE. Overman;HelenF. Sneddon ;JosephW. Ziller
Advanced Synthesis & Catalysis 2009 Volume 351( Issue 18) pp:3186-3192
Publication Date(Web):
DOI:10.1002/adsc.200900678
Abstract
The first di-μ-amidate dipalladium complexes and a new di-μ-carboxylate dipalladium complex of the COP (cobalt oxazoline palladacycle) palladium(II) catalyst family are reported and characterized crystallographically. The di-μ-amidate complex 3 and its enantiomer (ent-3) are the first asymmetric catalysts that allow commercially available, or readily accessible, (E)-2-alken-1-ols to be transformed to enantioenriched branched allylic aryl ethers upon reaction of their trichloroacetimidate derivatives with phenols. The 3-aryloxy-1-alkene products are formed in high enantiomeric purity (typically 90–98% ee) and useful yields (61–88%).
Co-reporter:Larry E. Overman
Tetrahedron 2009 65(33) pp: 6432-6446
Publication Date(Web):
DOI:10.1016/j.tet.2009.05.067
Co-reporter:Olivier Corminboeuf, Larry E. Overman and Lewis D. Pennington
The Journal of Organic Chemistry 2009 Volume 74(Issue 15) pp:5458-5470
Publication Date(Web):June 17, 2009
DOI:10.1021/jo9010156
Enantioselective total syntheses of briarellin E (12) and briarellin F (13), as well as the structure originally proposed for the cladiellin diterpene alcyonin (10), have been realized. Comparison of the spectral data for synthetic 10, natural alcyonin, cladiellisin (33), and cladiellaperoxide (34), as well as chemical transformations of 10 and natural alcyonin, suggest that the structure of this coral metabolite is allylic peroxide 11. The unified approach detailed herein can be used to access both C4-deoxygenated and C4-oxygenated cladiellins and briarellins. The central step in these syntheses is acid-promoted condensation of (Z)-α,β-unsaturated aldehydes 17 with cyclohexadienyl diols 18 to form intermediates 16 incorporating the hexahydroisobenzofuran core and five stereocenters of these marine diterpenes (Scheme 1).
Co-reporter:Alan Steven Dr.;Larry E. Overman Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 29) pp:
Publication Date(Web):25 JUN 2007
DOI:10.1002/anie.200700612
Our ability to access the more complex members of the cyclotryptamine family of alkaloids, and to exploit their disparate biological activities, is limited by the synthetic challenge posed by their oligomeric, polyindoline structures. A recurring structural theme within these molecules is the presence of multiple quaternary stereocenters in close proximity to one another. Over the last decade, we have developed a set of transformations that allow rapid access to polyindolines, a number of which exploit the ability of catalytic levels of palladium to orchestrate carbon–carbon bond formation with impressive levels of regio- and stereocontrol. This review tells the story behind the development of this toolbox of synthetic methods, and their validation through the total synthesis of a number of structurally complex cyclotryptamine alkaloids. It also highlights an aspect of asymmetric catalysis that has received little attention, the ability of catalytic asymmetric reactions to selectively elaborate complex, polyfunctional molecules.
Co-reporter:Alan Steven Dr.;Larry E. Overman Dr.
Angewandte Chemie 2007 Volume 119(Issue 29) pp:
Publication Date(Web):25 JUN 2007
DOI:10.1002/ange.200700612
Die Cyclotryptamine sind eine Familie von Alkaloiden mit sehr unterschiedlichen biologischen Aktivitäten. Die komplexeren Mitglieder dieser Naturstoffklasse sind durch benachbarte quartäre Kohlenstoffzentren charakterisiert und wegen der schwierig aufzubauenden, oligomeren Polyindolinstruktur schwer zugänglich. In den letzten Jahren konnten wir eine Reihe von Methoden entwickeln, die einen einfachen Zugang zu Polyindolinen ermöglichen; einige dieser Methoden nutzen Palladiumkatalysatoren zum Aufbau von Kohlenstoff-Kohlenstoff-Bindungen mit beeindruckenden Regio- und Stereoselektivitäten. In diesem Aufsatz schildern wir die Entwicklung eines Repertoires an Synthesemethoden und deren Anwendung in den Totalsynthesen einiger strukturell komplexer Cyclotryptamine. Insbesondere zeigen wir auf, wie es gelingen kann, mithilfe asymmetrischer Katalysen komplexe, hochfunktionalisierte Moleküle aufzubauen.
Co-reporter:Neil K. Garg Dr.;Sheldon Hiebert Dr.
Angewandte Chemie 2006 Volume 118(Issue 18) pp:
Publication Date(Web):27 MAR 2006
DOI:10.1002/ange.200600417
Zum krönenden Abschluss der ersten Totalsynthese des Naturstoffs Sarain A trug eine intramolekulare Stille-Kupplung entscheidend bei: Auf diese Weise wurden der ungesättigte Makrocyclus geschlossen und die unterbrochene Trien-Funktionalität eingeführt.
Co-reporter:Neil K. Garg Dr.;Sheldon Hiebert Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 18) pp:
Publication Date(Web):27 MAR 2006
DOI:10.1002/anie.200600417
The final synthetic challenges associated with sarain A have been overcome, thus leading to its first total synthesis. Critical to success was a late-stage intramolecular Stille coupling to construct the unsaturated macrocyclic ring and introduce the skipped triene functionality of the natural product.
Co-reporter:Christopher J. Douglas
PNAS 2004 Volume 101 (Issue 15 ) pp:5363-5367
Publication Date(Web):2004-04-13
DOI:10.1073/pnas.0307113101
Only a few catalytic asymmetric C—C bond-forming reactions have been shown to be useful for constructing all-carbon quaternary
stereocenters. This Perspective examines the current state of such methods.
Co-reporter:Emily A. Peterson
PNAS 2004 Volume 101 (Issue 33 ) pp:11943-11948
Publication Date(Web):2004-08-17
DOI:10.1073/pnas.0402416101
One element of structure that invariably increases the difficulty of a chemical synthesis is the presence in the target molecule
of contiguous all-carbon quaternary stereocenters. This Perspective will examine the most useful transformations and strategies devised recently for directly assembling this structural unit.
Co-reporter:Jeremy J. Kodanko Dr.
Angewandte Chemie 2003 Volume 115(Issue 22) pp:
Publication Date(Web):5 JUN 2003
DOI:10.1002/ange.200351261
Die Totalsynthese von Hodgkinsin und Hodgkinsin B ist mit nur zehn Stufen ausgesprochen kurz. Das auffälligste Merkmal ist die späte Stereodifferenzierung der meso-3a,3a′-Bispyrrolidinoindolin-Einheit in einer katalytischen asymmetrischen Reaktion. Durch enantioselektive Totalsynthese von Hodgkinsin B wurde die relative und absolute Konfiguration dieses Alkaloids bestätigt.
Co-reporter:Larry E. Overman Dr.;Emily A. Peterson
Angewandte Chemie 2003 Volume 115(Issue 22) pp:
Publication Date(Web):5 JUN 2003
DOI:10.1002/ange.200351260
Die größte Herausforderung bei der Synthese von Trispyrrolidinoindolin-Alkaloiden wie Idiospermulin bilden die stereogenen quartären Kohlenstoffzentren (siehe Bild). Durch Kombination des Lithium-Enolats von geschütztem Dihydroisoindigo mit einem Tartrat-Elektrophil werden die beiden benachbarten quartären Stereozentren dieser Verbindung in einem Reaktionsschritt eingeführt. Das dritte quartäre Stereozentrum wird durch eine katalytische asymmetrische Heck-Reaktion erhalten.
Co-reporter:Larry E. Overman Dr.;Emily A. Peterson
Angewandte Chemie International Edition 2003 Volume 42(Issue 22) pp:
Publication Date(Web):5 JUN 2003
DOI:10.1002/anie.200351260
Stereogenic quaternary carbons present the primary challenge in the syntheses of trispyrrolidinoindoline alkaloids such as idiospermuline. In its synthesis the two contiguous quaternary centers are expediently addressed by combining the lithium dienolate of a differentially protected dihydroisoindigo with a tartrate-derived electrophile. The third quaternary stereocenter is introduced by a catalytic asymmetric Heck reaction.
Co-reporter:Jeremy J. Kodanko Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 22) pp:
Publication Date(Web):5 JUN 2003
DOI:10.1002/anie.200351261
A late-stage resolution of the meso-3a,3a′-bispyrrolidinoindoline unit of rac-7 in a catalytic asymmetric transformation earmarks the remarkably short, ten-step, total syntheses of hodgkinsine and hodgkinsine B. The enantioselective total synthesis of hodgkinsine B establishes the relative and absolute configuration of this trispyrrolidinoindoline alkaloid.
Co-reporter:Larry E. Overman Dr.;Carolyn E. Owen;G. Greg Zipp
Angewandte Chemie 2002 Volume 114(Issue 20) pp:
Publication Date(Web):18 OCT 2002
DOI:10.1002/1521-3757(20021018)114:20<4040::AID-ANGE4040>3.0.CO;2-G
Komplementäre Stereoselektion: Der Austausch eines Carbonylliganden in Tricarbonylchromkomplexen von 2-Phenyloxazolinen gegen einen Phosphanliganden ermöglicht die direkte ortho-Lithiierung am Phenylring. Je nach An- oder Abwesenheit von N,N,N′,N′-Tetramethylethylendiamin (TMEDA) werden bei der anschließenden Umsetzung mit Elektrophilen Komplexe mit entgegengesetzter Konfiguration erhalten, wobei die Diastereoselektivitäten zwischen 10:1 und 50:1 liegen.
Co-reporter:Martin Oestreich Dr.;Philip R. Dennison Dr.;Jeremy J. Kodanko Dr.
Angewandte Chemie 2001 Volume 113(Issue 8) pp:
Publication Date(Web):17 APR 2001
DOI:10.1002/1521-3757(20010417)113:8<1485::AID-ANGE1485>3.0.CO;2-F
Co-reporter:Larry E. Overman ;Mark D. Rosen
Angewandte Chemie 2000 Volume 112(Issue 24) pp:
Publication Date(Web):15 DEC 2000
DOI:10.1002/1521-3757(20001215)112:24<4768::AID-ANGE4768>3.0.CO;2-7
Eine Heck-Cyclisierung ist der Schlüsselschritt für eine neue stereokontrollierte Synthese von Spirotryprostatin-Alkaloiden. Die hierbei entstehende η3-Allylpalladiumzwischenstufe wird intramolekular durch ein angeknüpftes Diketopiperazin abgefangen. Die Vielseitigkeit dieses Konzeptes wird am Beispiel der enantioselektiven Totalsynthese des Pilzinhaltsstoffes (−)-Spirotryprostatin B 1 und dreier Isomere illustriert.
Co-reporter:Larry E. Overman ;Jay F. Larrow Dr.;Brian A. Stearns;Jennifer M. Vance
Angewandte Chemie 2000 Volume 112(Issue 1) pp:
Publication Date(Web):12 JAN 2000
DOI:10.1002/(SICI)1521-3757(20000103)112:1<219::AID-ANGE219>3.0.CO;2-V
Alle drei Stereoisomere der hexacyclischen 3a,3a′-Bispyrrolidino[2,3-b]indolin-Einheit, die in einigen Indolalkaloiden vorkommt, können hergestellt werden, wie anhand der Totalsynthesen von meso-Chimonanthin 1 und (+)-Chimonanthin 2 gezeigt wurde. Kern der asymmetrischen Synthese des C2-symmetrischen Stereoisomers ist eine hoch diastereoselektive Alkylierung, die auf die Kombination eines prostereogenen Enolats mit einem chiralen Elektrophil zurückzuführen ist, das ein sp3-Kohlenstoffzentrum enthält.
Co-reporter:Larry E. Overman ;Jay F. Larrow Dr.;Brian A. Stearns;Jennifer M. Vance
Angewandte Chemie International Edition 2000 Volume 39(Issue 1) pp:
Publication Date(Web):12 JAN 2000
DOI:10.1002/(SICI)1521-3773(20000103)39:1<213::AID-ANIE213>3.0.CO;2-Z
All three stereoisomers of the hexacyclic 3a,3a′-bispyrrolidino[2,3-b]indoline moiety found in complex indole alkaloids can be prepared, as illustrated by total syntheses of meso-chimonanthine (1) and (+)-chimonanthine (2). A rare example of high diastereoselectivity arising from the combination of a prostereogenic enolate with a chiral electrophile containing a sp3 carbon atom is the key feature of the asymmetric synthesis of the C2 stereoisomer.
Co-reporter:Andrew Madin;Christopher J. O'Donnell;Taeboem Oh;David W. Old;Matthew J. Sharp
Angewandte Chemie International Edition 1999 Volume 38(Issue 19) pp:
Publication Date(Web):24 SEP 1999
DOI:10.1002/(SICI)1521-3773(19991004)38:19<2934::AID-ANIE2934>3.0.CO;2-L
Acomplexmolecularreorganization (12), a sequential anionic aza-Cope rearrangement and Mannich cyclization, and an unprecedented intramolecular Heck reaction of the tetrasubstituted double bond of a vinylogous carbamate are key steps in a new total synthesis of (±)-gelsemine (3). MOM=methoxymethyl, DBU=1,8-diazabicyclo[5.4.0]undec-7-ene.
Co-reporter:Andrew Madin;Christopher J. O'Donnell;Taeboem Oh;David W. Old;Matthew J. Sharp
Angewandte Chemie 1999 Volume 111(Issue 19) pp:
Publication Date(Web):24 SEP 1999
DOI:10.1002/(SICI)1521-3757(19991004)111:19<3110::AID-ANGE3110>3.0.CO;2-F
Eine komplexemolekulareReorganisation (12), eine Sequenz aus Aza-Cope-Umlagerung und Mannich-Cyclisierung und eine neuartige intramolekulare Heck-Reaktion der tetrasubstituierten Doppelbindung eines vinylogen Carbamats sind die Schlüsselschritte einer neuen Totalsynthese von (±)-Gelsemin 3. MOM=Methoxymethyl, DBU=1,8-Diazabicyclo[5.4.0]undec-7-en.
Co-reporter:Marcus Baumann, André P. Dieskau, Brad M. Loertscher, Mary C. Walton, Sangkil Nam, Jun Xie, David Horne and Larry E. Overman
Chemical Science (2010-Present) 2015 - vol. 6(Issue 8) pp:NaN4457-4457
Publication Date(Web):2015/05/26
DOI:10.1039/C5SC01536G
Epipolythiodioxopiperazine (ETP) alkaloids are structurally elaborate alkaloids that show potent antitumor activity. However, their high toxicity and demonstrated interactions with various biological receptors compromises their therapeutic potential. In an effort to mitigate these disadvantages, a short stereocontrolled construction of tricyclic analogues of epidithiodioxopiperazine alkaloids was developed. Evaluation of a small library of such structures against two invasive cancer cell lines defined initial structure–activity relationships (SAR), which identified 1,4-dioxohexahydro-6H-3,8a-epidithiopyrrolo[1,2-a]pyrazine 3c and related structures as particularly promising antitumor agents. ETP alkaloid analogue 3c exhibits low nanomolar activity against both solid and blood tumors in vitro. In addition, 3c significantly suppresses tumor growth in mouse xenograft models of melanoma and lung cancer, without obvious signs of toxicity, following either intraperitoneal (IP) or oral administration. The short synthesis of molecules in this series will enable future mechanistic and translational studies of these structurally novel and highly promising clinical antitumor candidates.
.jpg)





![Benzenemethanamine, 4-methoxy-N-[4-(trimethylsilyl)-3-butynyl]-](http://img.cochemist.com/ccimg/112100/112069-96-2.png)
![Benzenemethanamine, 4-methoxy-N-[4-(trimethylsilyl)-3-butynyl]-](http://img.cochemist.com/ccimg/112100/112069-96-2_b.png)
![Pyrrolidine, 3-(1-iodoethylidene)-1-[(4-methoxyphenyl)methyl]-, (E)-](http://img.cochemist.com/ccimg/112100/112069-94-0.png)
![Pyrrolidine, 3-(1-iodoethylidene)-1-[(4-methoxyphenyl)methyl]-, (E)-](http://img.cochemist.com/ccimg/112100/112069-94-0_b.png)
![3-(1-IODOETHYLIDENE)-1-[(4-METHOXYPHENYL)METHYL]PIPERIDINE](/data/chemimg/361300/112069-89-3.png)
![3-(1-IODOETHYLIDENE)-1-[(4-METHOXYPHENYL)METHYL]PIPERIDINE](/data/chemimg/361300/112069-89-3_b.png)
![3-(1-BROMOETHYLIDENE)-1-[(4-METHOXYPHENYL)METHYL]PIPERIDINE](http://img.cochemist.com/ccimg/112100/112069-88-2.png)
![3-(1-BROMOETHYLIDENE)-1-[(4-METHOXYPHENYL)METHYL]PIPERIDINE](http://img.cochemist.com/ccimg/112100/112069-88-2_b.png)


![1-Hexene, 6-[(2-methoxyethoxy)methoxy]-](http://img.cochemist.com/ccimg/102500/102436-85-1.png)
![1-Hexene, 6-[(2-methoxyethoxy)methoxy]-](http://img.cochemist.com/ccimg/102500/102436-85-1_b.png)


![PHENSERINE;(3AS,8AR)-1,2,3,3A,8,8A-HEXAHYDRO-1,3A,8-TRIMETHYLPYRROLO[2,3-B]INDOL-5-OL5-(N-PHENYLCARBAMATE)](http://img.cochemist.com/ccimg/101300/101246-66-6.png)
![PHENSERINE;(3AS,8AR)-1,2,3,3A,8,8A-HEXAHYDRO-1,3A,8-TRIMETHYLPYRROLO[2,3-B]INDOL-5-OL5-(N-PHENYLCARBAMATE)](http://img.cochemist.com/ccimg/101300/101246-66-6_b.png)
![(1r,2s,3s,4s,4ar,11br)-1,2,3,4,7-pentahydroxy-2,3,4,4a,5,11b-hexahydro-1h-[1,3]dioxolo[4,5-j]phenanthridin-6-one](http://img.cochemist.com/ccimg/96300/96203-70-2.png)
![(1r,2s,3s,4s,4ar,11br)-1,2,3,4,7-pentahydroxy-2,3,4,4a,5,11b-hexahydro-1h-[1,3]dioxolo[4,5-j]phenanthridin-6-one](http://img.cochemist.com/ccimg/96300/96203-70-2_b.png)
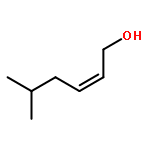
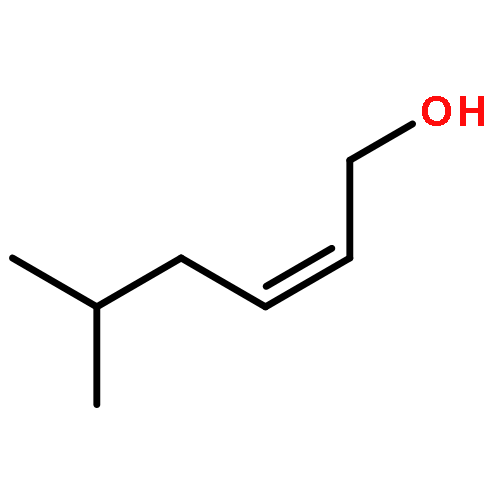
![Acetamide, 2,2,2-trichloro-N-[1-(1,1-dimethylethyl)-2-propenyl]-](http://img.cochemist.com/ccimg/93700/93667-63-1.png)
![Acetamide, 2,2,2-trichloro-N-[1-(1,1-dimethylethyl)-2-propenyl]-](http://img.cochemist.com/ccimg/93700/93667-63-1_b.png)
![2-Butyn-1-ol, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/92900/92808-80-5.png)
![2-Butyn-1-ol, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/92900/92808-80-5_b.png)
![1,4-Dioxaspiro[4.5]dec-7-ene-8-carboxylic acid](http://img.cochemist.com/ccimg/92600/92525-59-2.png)
![1,4-Dioxaspiro[4.5]dec-7-ene-8-carboxylic acid](http://img.cochemist.com/ccimg/92600/92525-59-2_b.png)
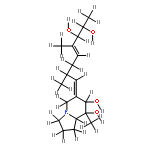

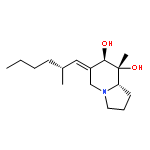


![(6E,7S,8R,8aS)-6-[(2R,4E,6R,7R)-6,7-dihydroxy-2,5-dimethyloct-4-en-1-ylidene]-8-methyloctahydroindolizine-7,8-diol](http://img.cochemist.com/ccimg/91600/91550-04-8.png)
![(6E,7S,8R,8aS)-6-[(2R,4E,6R,7R)-6,7-dihydroxy-2,5-dimethyloct-4-en-1-ylidene]-8-methyloctahydroindolizine-7,8-diol](http://img.cochemist.com/ccimg/91600/91550-04-8_b.png)

![Propanoic acid, 2-[bis(2,2,2-trifluoroethoxy)phosphinyl]-, methyl ester](http://img.cochemist.com/ccimg/88800/88738-84-5.png)
![Propanoic acid, 2-[bis(2,2,2-trifluoroethoxy)phosphinyl]-, methyl ester](http://img.cochemist.com/ccimg/88800/88738-84-5_b.png)

![BENZENAMINE, N-[(3Z)-4-(TRIMETHYLSILYL)-3-BUTENYL]-](http://img.cochemist.com/ccimg/87700/87682-59-5.png)
![BENZENAMINE, N-[(3Z)-4-(TRIMETHYLSILYL)-3-BUTENYL]-](http://img.cochemist.com/ccimg/87700/87682-59-5_b.png)
![2-AZASPIRO[4.5]DEC-1-ENE](http://img.cochemist.com/ccimg/85700/85674-93-7.png)
![2-AZASPIRO[4.5]DEC-1-ENE](http://img.cochemist.com/ccimg/85700/85674-93-7_b.png)



![Benzene, [(1-bromoethenyl)thio]-](http://img.cochemist.com/ccimg/80500/80485-53-6.png)
![Benzene, [(1-bromoethenyl)thio]-](http://img.cochemist.com/ccimg/80500/80485-53-6_b.png)




![1,4-DIOXASPIRO[4.5]DEC-7-EN-8-YLMETHANOL](http://img.cochemist.com/ccimg/72500/72445-21-7.png)
![1,4-DIOXASPIRO[4.5]DEC-7-EN-8-YLMETHANOL](http://img.cochemist.com/ccimg/72500/72445-21-7_b.png)

![3-BUTEN-2-OL, 2-METHYL-1-[(PHENYLMETHYL)AMINO]-](http://img.cochemist.com/ccimg/70000/69917-78-8.png)
![3-BUTEN-2-OL, 2-METHYL-1-[(PHENYLMETHYL)AMINO]-](http://img.cochemist.com/ccimg/70000/69917-78-8_b.png)
![Silane, trimethyl[4-[(tetrahydro-2H-pyran-2-yl)oxy]-1-butynyl]-](http://img.cochemist.com/ccimg/69400/69361-40-6.png)
![Silane, trimethyl[4-[(tetrahydro-2H-pyran-2-yl)oxy]-1-butynyl]-](http://img.cochemist.com/ccimg/69400/69361-40-6_b.png)
![7-Azaspiro[4.5]dec-6-ene](http://img.cochemist.com/ccimg/67700/67625-80-3.png)
![7-Azaspiro[4.5]dec-6-ene](http://img.cochemist.com/ccimg/67700/67625-80-3_b.png)
![2-Azaspiro[5.5]undec-1-ene](http://img.cochemist.com/ccimg/67700/67625-76-7.png)
![2-Azaspiro[5.5]undec-1-ene](http://img.cochemist.com/ccimg/67700/67625-76-7_b.png)










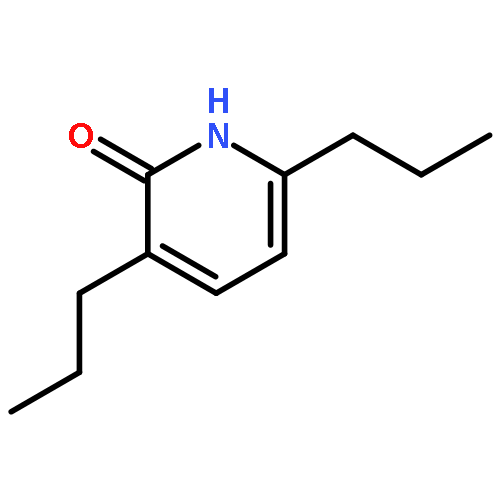
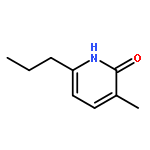
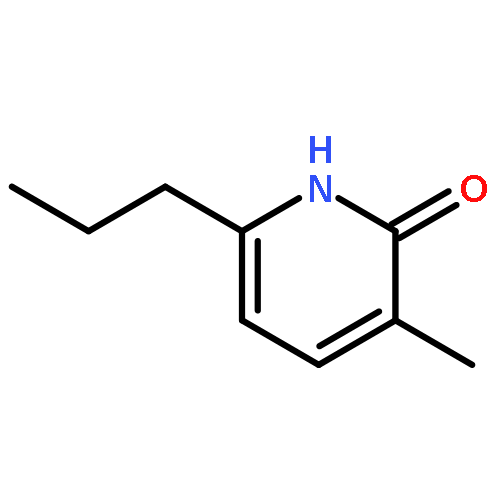
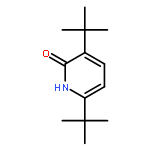













![2H-Cyclopenta[b]furan-2-one, 3,3a,4,6a-tetrahydro-, (3aS,6aR)-](http://img.cochemist.com/ccimg/56500/56487-70-8.png)
![2H-Cyclopenta[b]furan-2-one, 3,3a,4,6a-tetrahydro-, (3aS,6aR)-](http://img.cochemist.com/ccimg/56500/56487-70-8_b.png)












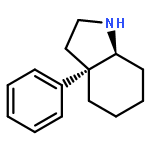
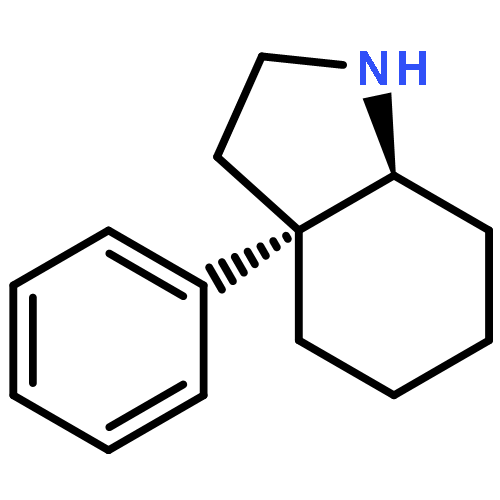
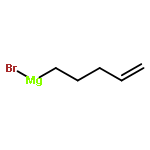
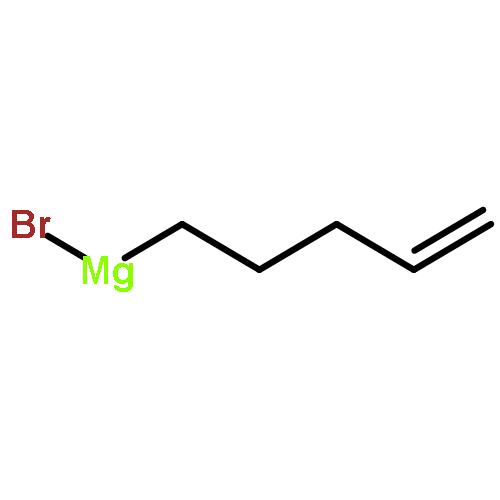




















![Silane, [1-cyclopentene-1,2-diylbis(oxy)]bis[trimethyl-](http://img.cochemist.com/ccimg/6900/6838-66-0.png)
![Silane, [1-cyclopentene-1,2-diylbis(oxy)]bis[trimethyl-](http://img.cochemist.com/ccimg/6900/6838-66-0_b.png)


![3,5-Ethano-3H-pyrrolo[2,3-d]carbazole-6-carboxylicacid, 4-ethyl-1,2,3a,4,5,7-hexahydro-, methyl ester, (3S,3aR,4S,5S,11bS)-](http://img.cochemist.com/ccimg/6800/6711-69-9.png)
![3,5-Ethano-3H-pyrrolo[2,3-d]carbazole-6-carboxylicacid, 4-ethyl-1,2,3a,4,5,7-hexahydro-, methyl ester, (3S,3aR,4S,5S,11bS)-](http://img.cochemist.com/ccimg/6800/6711-69-9_b.png)




![(3aR,3a'R,8aR,8a'R)-1,1'-dimethyl-2,2',3,3',8,8',8a,8a'-octahydro-1H,1'H-3a,3a'-bipyrrolo[2,3-b]indole](http://img.cochemist.com/ccimg/5600/5545-89-1.png)
![(3aR,3a'R,8aR,8a'R)-1,1'-dimethyl-2,2',3,3',8,8',8a,8a'-octahydro-1H,1'H-3a,3a'-bipyrrolo[2,3-b]indole](http://img.cochemist.com/ccimg/5600/5545-89-1_b.png)














![2-Cyclohexen-1-one, 2-[[2-[[tris(1-methylethyl)silyl]oxy]phenyl]amino]-](http://img.cochemist.com/ccimg/863400/863312-49-6.png)
![2-Cyclohexen-1-one, 2-[[2-[[tris(1-methylethyl)silyl]oxy]phenyl]amino]-](http://img.cochemist.com/ccimg/863400/863312-49-6_b.png)



![Acetamide, N-[(1S)-4-(acetyloxy)-1-ethenylbutyl]-2,2,2-trichloro-](http://img.cochemist.com/ccimg/611200/611182-49-1.png)
![Acetamide, N-[(1S)-4-(acetyloxy)-1-ethenylbutyl]-2,2,2-trichloro-](http://img.cochemist.com/ccimg/611200/611182-49-1_b.png)
![Acetamide, 2,2,2-trichloro-N-[(1S)-1-ethenyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/611200/611182-44-6.png)
![Acetamide, 2,2,2-trichloro-N-[(1S)-1-ethenyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/611200/611182-44-6_b.png)

![Acetamide, N-[(1R)-1-ethenylbutyl]-2,2,2-trifluoro-N-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/562900/562870-84-2.png)
![Acetamide, N-[(1R)-1-ethenylbutyl]-2,2,2-trifluoro-N-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/562900/562870-84-2_b.png)
![Benzamide, N-[(1S)-1-ethenylbutyl]-N-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/217700/217653-92-4.png)
![Benzamide, N-[(1S)-1-ethenylbutyl]-N-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/217700/217653-92-4_b.png)
![9-[1-amino-4-({4-[(diaminomethylidene)amino]butoxy}carbonyl)-3,5,6,7-tetrahydropyrrolo[1,2-c]pyrimidin-3-yl]nonyl (7S)-7-heptyl-4-methyl-2,2a,5,7,8,8a-hexahydro-1H-5,6,8b-triazaacenaphthylene-3-carboxylate](http://img.cochemist.com/ccimg/161600/161503-23-7.png)
![9-[1-amino-4-({4-[(diaminomethylidene)amino]butoxy}carbonyl)-3,5,6,7-tetrahydropyrrolo[1,2-c]pyrimidin-3-yl]nonyl (7S)-7-heptyl-4-methyl-2,2a,5,7,8,8a-hexahydro-1H-5,6,8b-triazaacenaphthylene-3-carboxylate](http://img.cochemist.com/ccimg/161600/161503-23-7_b.png)
![Silane, trimethyl[1-[(tributylstannyl)methyl]ethenyl]-](http://img.cochemist.com/ccimg/151100/151073-99-3.png)
![Silane, trimethyl[1-[(tributylstannyl)methyl]ethenyl]-](http://img.cochemist.com/ccimg/151100/151073-99-3_b.png)


![5,10b:11,4b-Bis(iminoethano)dibenzo[c,h][2,6]naphthyridine,5,6,11,12-tetrahydro-13,18-dimethyl-1,7-bis[(3aR,8aR)-2,3,8,8a-tetrahydro-1-methylpyrrolo[2,3-b]indol-3a(1H)-yl]-,(4bR,5R,10bS,11S)-](http://img.cochemist.com/ccimg/144500/144424-79-3.png)
![5,10b:11,4b-Bis(iminoethano)dibenzo[c,h][2,6]naphthyridine,5,6,11,12-tetrahydro-13,18-dimethyl-1,7-bis[(3aR,8aR)-2,3,8,8a-tetrahydro-1-methylpyrrolo[2,3-b]indol-3a(1H)-yl]-,(4bR,5R,10bS,11S)-](http://img.cochemist.com/ccimg/144500/144424-79-3_b.png)
![1,4-DIOXASPIRO[4.5]DEC-7-ENE, 8-[(2-IODOPHENOXY)METHYL]-](http://img.cochemist.com/ccimg/142700/142613-82-9.png)
![1,4-DIOXASPIRO[4.5]DEC-7-ENE, 8-[(2-IODOPHENOXY)METHYL]-](http://img.cochemist.com/ccimg/142700/142613-82-9_b.png)



![12H-2,7a-Ethenoindolizino[8a,1-b]indole-14-methanol,3-ethylidene-1,2,3,4,6,7-hexahydro-, (2S,3E,7aR,12aS)-](http://img.cochemist.com/ccimg/125400/125362-84-7.png)
![12H-2,7a-Ethenoindolizino[8a,1-b]indole-14-methanol,3-ethylidene-1,2,3,4,6,7-hexahydro-, (2S,3E,7aR,12aS)-](http://img.cochemist.com/ccimg/125400/125362-84-7_b.png)
![Silane, [(3-methyl-1,3-cyclohexadien-1-yl)oxy]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/120100/120016-39-9.png)
![Silane, [(3-methyl-1,3-cyclohexadien-1-yl)oxy]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/120100/120016-39-9_b.png)
![2-Butanone, 4-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/113300/113249-47-1.png)
![2-Butanone, 4-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/113300/113249-47-1_b.png)
![3a(1H),7':3'a,3''a(1'H,1''H):7'',3'''a(1'''H)-Quaterpyrrolo[2,3-b]indole,2,2',2'',2''',3,3',3'',3''',8,8',8'',8''',8a,8'a,8''a,8'''a-hexadecahydro-1,1',1'',1'''-tetramethyl-,stereoisomer (9CI)](http://img.cochemist.com/ccimg/112300/112295-93-9.png)
![3a(1H),7':3'a,3''a(1'H,1''H):7'',3'''a(1'''H)-Quaterpyrrolo[2,3-b]indole,2,2',2'',2''',3,3',3'',3''',8,8',8'',8''',8a,8'a,8''a,8'''a-hexadecahydro-1,1',1'',1'''-tetramethyl-,stereoisomer (9CI)](http://img.cochemist.com/ccimg/112300/112295-93-9_b.png)
![Silane, (1,1-dimethylethyl)[(1R)-3-iodo-1-methylpropoxy]dimethyl-](http://img.cochemist.com/ccimg/109700/109613-36-7.png)
![Silane, (1,1-dimethylethyl)[(1R)-3-iodo-1-methylpropoxy]dimethyl-](http://img.cochemist.com/ccimg/109700/109613-36-7_b.png)




![Propanoic acid, 2-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, ethyl ester, (2S)-](http://img.cochemist.com/ccimg/102800/102732-44-5.png)
![Propanoic acid, 2-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, ethyl ester, (2S)-](http://img.cochemist.com/ccimg/102800/102732-44-5_b.png)





![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/87200/87184-81-4.png)
![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/87200/87184-81-4_b.png)
![Acetonitrile, [methyl(2-oxocyclohexyl)amino]-](http://img.cochemist.com/ccimg/83200/83196-13-8.png)
![Acetonitrile, [methyl(2-oxocyclohexyl)amino]-](http://img.cochemist.com/ccimg/83200/83196-13-8_b.png)
![BENZENECARBOXIMIDOYL CHLORIDE, N-[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/80900/80819-62-1.png)
![BENZENECARBOXIMIDOYL CHLORIDE, N-[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/80900/80819-62-1_b.png)
![(6E,7R,8R,8aS)-8-methyl-6-[(2R)-2-methylhexylidene]octahydroindolizine-7,8-diol](http://img.cochemist.com/ccimg/73400/73376-38-2.png)
![(6E,7R,8R,8aS)-8-methyl-6-[(2R)-2-methylhexylidene]octahydroindolizine-7,8-diol](http://img.cochemist.com/ccimg/73400/73376-38-2_b.png)
![1,4-Dioxaspiro[4.5]dec-7-ene-8-carboxaldehyde](http://img.cochemist.com/ccimg/72500/72445-22-8.png)
![1,4-Dioxaspiro[4.5]dec-7-ene-8-carboxaldehyde](http://img.cochemist.com/ccimg/72500/72445-22-8_b.png)

![(6Z,8S)-6-[(4E,6S)-6-hydroxy-2,5-dimethyloct-4-en-1-ylidene]-8-methyloctahydroindolizin-8-ol](http://img.cochemist.com/ccimg/67100/67054-00-6.png)
![(6Z,8S)-6-[(4E,6S)-6-hydroxy-2,5-dimethyloct-4-en-1-ylidene]-8-methyloctahydroindolizin-8-ol](http://img.cochemist.com/ccimg/67100/67054-00-6_b.png)
![(3aS,8aR)-5-methoxy-1,3a,8-trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole](http://img.cochemist.com/ccimg/65200/65166-97-4.png)
![(3aS,8aR)-5-methoxy-1,3a,8-trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole](http://img.cochemist.com/ccimg/65200/65166-97-4_b.png)

![1-[(1e)-buta-1,3-dien-1-yl]prolinamide](http://img.cochemist.com/ccimg/61800/61759-62-4.png)
![1-[(1e)-buta-1,3-dien-1-yl]prolinamide](http://img.cochemist.com/ccimg/61800/61759-62-4_b.png)

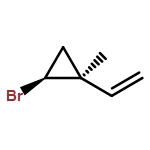
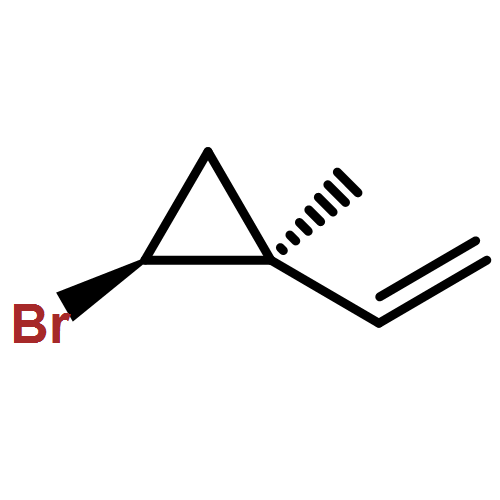














![Benzene, [1-(chloromethyl)ethenyl]-](/data/chemimg/554200/3360-52-9.png)
![Benzene, [1-(chloromethyl)ethenyl]-](/data/chemimg/554200/3360-52-9_b.png)






![Acetamide, 2,2,2-trichloro-N-[(1S)-1-(2-phenylethyl)-2-propenyl]-](http://img.cochemist.com/ccimg/611200/611182-48-0.png)
![Acetamide, 2,2,2-trichloro-N-[(1S)-1-(2-phenylethyl)-2-propenyl]-](http://img.cochemist.com/ccimg/611200/611182-48-0_b.png)
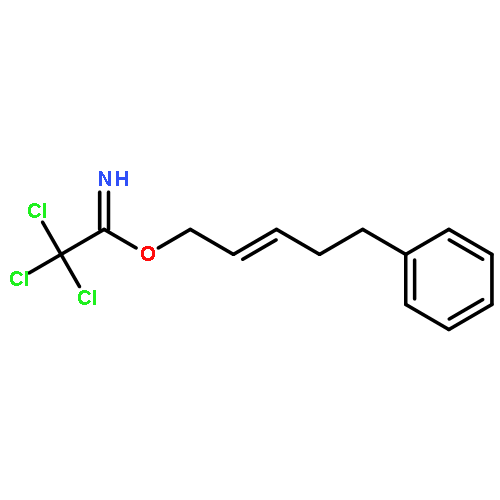
![Acetamide, 2,2,2-trichloro-N-[(1R)-1-ethenylbutyl]-](http://img.cochemist.com/ccimg/221700/221624-08-4.png)
![Acetamide, 2,2,2-trichloro-N-[(1R)-1-ethenylbutyl]-](http://img.cochemist.com/ccimg/221700/221624-08-4_b.png)
![Benzamide, N-[(1S)-1-ethenylbutyl]-N-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/201700/201612-24-0.png)
![Benzamide, N-[(1S)-1-ethenylbutyl]-N-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/201700/201612-24-0_b.png)
![Benzenamine, 2-[[tris(1-methylethyl)silyl]oxy]-](http://img.cochemist.com/ccimg/194900/194869-04-0.png)
![Benzenamine, 2-[[tris(1-methylethyl)silyl]oxy]-](http://img.cochemist.com/ccimg/194900/194869-04-0_b.png)
![Lithium, [1-(1,3-benzodioxol-5-yl)ethenyl]-](http://img.cochemist.com/ccimg/79100/79076-14-5.png)
![Lithium, [1-(1,3-benzodioxol-5-yl)ethenyl]-](http://img.cochemist.com/ccimg/79100/79076-14-5_b.png)










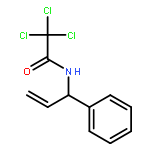
![Acetamide, 2,2,2-trichloro-N-[(1S)-1-ethenylbutyl]-](http://img.cochemist.com/ccimg/611200/611182-43-5.png)
![Acetamide, 2,2,2-trichloro-N-[(1S)-1-ethenylbutyl]-](http://img.cochemist.com/ccimg/611200/611182-43-5_b.png)
![7-[(2aS,4R,7R,8aS)-7-methyl-2,2a,3,4,6,7,8,8a-octahydro-1H-5,6,8b-triazaacenaphthylen-4-yl]heptan-2-yl (2aS,3S,4R,8aS)-4-methyl-7-nonyl-2,2a,3,4,6,7,8,8a-octahydro-1H-5,6,8b-triazaacenaphthylene-3-carboxylate](http://img.cochemist.com/ccimg/188200/188112-82-5.png)
![7-[(2aS,4R,7R,8aS)-7-methyl-2,2a,3,4,6,7,8,8a-octahydro-1H-5,6,8b-triazaacenaphthylen-4-yl]heptan-2-yl (2aS,3S,4R,8aS)-4-methyl-7-nonyl-2,2a,3,4,6,7,8,8a-octahydro-1H-5,6,8b-triazaacenaphthylene-3-carboxylate](http://img.cochemist.com/ccimg/188200/188112-82-5_b.png)
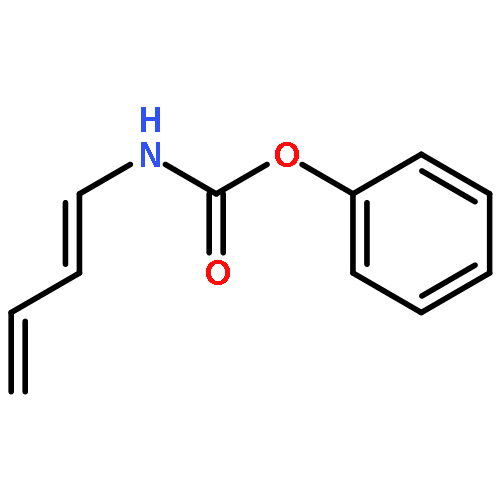
![N-[(1E)-buta-1,3-dien-1-yl]-2,2,2-trichloroacetamide](http://img.cochemist.com/ccimg/59500/59403-01-9.png)
![N-[(1E)-buta-1,3-dien-1-yl]-2,2,2-trichloroacetamide](http://img.cochemist.com/ccimg/59500/59403-01-9_b.png)



![(3aS,8aR)-1,3a,8-Trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl methylcarbamate](http://img.cochemist.com/ccimg/100/57-47-6.png)
![(3aS,8aR)-1,3a,8-Trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl methylcarbamate](http://img.cochemist.com/ccimg/100/57-47-6_b.png)