Co-reporter:Tingting Li;Qing Yang;Hairong Ding;Jinlong Li;Honglai Liu
Industrial & Engineering Chemistry Research December 9, 2015 Volume 54(Issue 48) pp:12143-12149
Publication Date(Web):Publication Date (Web): November 11, 2015
DOI:10.1021/acs.iecr.5b02828
The effects of 18 different amino acid based ionic liquids (AAILs) on the phase behavior of an acetonitrile + water mixture were predicted through the COSMO-RS method, indicating that all investigated AAILs could enhance the relative volatility of acetonitrile and break the azeotrope, in which 1-ethyl-3-methylimidazolium proline ([EMIM][Pro]) was the most effective at a given condition. Furthermore, the properties of the interaction energy, charge-density profiles, and excess enthalpy were obtained, showing that the stronger interactions between AAIL and water lead to improvement of the separation efficiency. To verify the feasibility, the isobaric vapor–liquid equilibrium data for two ternary mixtures of acetonitrile + water + [EMIM][Pro] and + 1-ethyl-3-methylimidazolium acetate ([EMIM][OAC]) at fixed IL mole fraction (0.05 and 0.10) were measured at atmospheric pressure. The experimental results show that the two ILs can effectively enhance the relative volatility of acetonitrile and break the azeotrope at the mole fraction 0.05 and the higher the IL content in the mixture, the better the separation effect observed.
Co-reporter:Yan He;Ting Xu;Jun Hu;Qiang Yang;Hualin Wang;Honglai Liu
RSC Advances (2011-Present) 2017 vol. 7(Issue 48) pp:30500-30505
Publication Date(Web):2017/06/08
DOI:10.1039/C7RA04649A
Organic dyes and solvents are emerging pollutants; the development of new materials for their efficient adsorption and removal is thus of great significance. In this work, we report the application of an amine functionalized triptycene-based 3D polymer (TPP-NH2) as a novel adsorbent for the fast removal of organic dyes in aqueous solution and organic solvents with a view to understanding its adsorption kinetics, adsorption isotherms, desorption and adsorbent regeneration. The adsorption of organic dyes (cationic methylene blue, MEB, and anionic methyl orange, MO) on TPP-NH2 was fast, and most of the dyes were adsorbed in 60 min. The adsorption of MEB and MO follows pseudo-second-order kinetics and fits the Langmuir model, where the maximum adsorption capacity increased from 204.9 mg g−1 to 560.6 mg g−1 and 213.2 mg g−1 to 803.2 mg g−1 for MEB and MO, respectively, as the temperature increased, suggesting that the adsorption of MEB and MO on TPP-NH2 is favorable at high temperatures via an endothermic process. The pH has no obvious effect on the adsorption of MEB and MO. The used TPP-NH2 could be regenerated effectively and recycled at least five times without a significant loss of adsorption capacity. In addition, the TPP-NH2 can adsorb up to 33 times its own weight in organic solvents while wiping off the water. The high surface area, hierarchical porosity and π–π stacking interactions between the aromatic rings of MEB and MO and the aromatic rings of 3D TPP-NH2 were responsible for the efficient adsorption. Therefore, the TPP-NH2 that was synthesized using the facile strategy possesses significant potential in the treatment of wastewater.
Co-reporter:Yan He, Qinqin Liu, Jun Hu, Chengxi Zhao, Changjun Peng, Qiang Yang, Hualin Wang, Honglai Liu
Separation and Purification Technology 2017 Volume 180(Volume 180) pp:
Publication Date(Web):8 June 2017
DOI:10.1016/j.seppur.2017.01.026
•An amine functionalized polymer was obtained by post-synthetic modification method.•The maximum adsorption capacity of POP-NH2 for Pb(II) can reach 523.6 mg g−1.•FTIR and the theoretical calculation results illustrated the adsorption mechanism.•The used POP-NH2 can be regenerated effectively and recycled at least four times.Porous organic polymers (POPs), a new class of porous materials constructed by organic molecular building blocks, have attracted much attention. To design POPs with both good porosity and specific-task functionalization is still a critical challenge. In this work, using a simple Friedel-Crafts one-step reaction, one of the most common organic compound methylbenzene monomers were polymerized through the cross-linker formaldehyde dimethyl acetal (FDA) to produce a cost-effective non-functionalized porous polymer (POP-CH3). Moreover, amine groups were further incorporated into the network by the post-synthetic modification method. The resulted porous polymer POP-NH2 presented highly effective in removing Pb(II) from aqueous solution. The maximum Pb(II) adsorption capacity, qmax, evaluated from the Langmuir model was 523.6 mg g−1. The combination of FT-IR experimental results and the theoretical quantum calculations illustrated that the amine groups (NH2) can easily form coordination complexes with Pb(II), which were responsible for efficient adsorption. The generated POP-NH2 could be regenerated effectively and recycled at least four times without significant loss of adsorption efficiency. Hence, the outstanding Pb(II) adsorption capacity, excellent reusability as well as low cost of synthesis create potential polymer POP-NH2 to be an attractive adsorbent for removing toxic metal ions from aqueous solution.Download high-res image (116KB)Download full-size image
Co-reporter:Yan He, Qinqin Liu, Fei Liu, Chensheng Huang, Changjun Peng, Qiang Yang, Hualin Wang, Jun Hu, Honglai Liu
Microporous and Mesoporous Materials 2016 Volume 233() pp:10-15
Publication Date(Web):1 October 2016
DOI:10.1016/j.micromeso.2016.06.024
•A bifunctionalized TSP-NS was achieved through one-step Friedel-Crafts reaction.•The bifunctionalized TSP-NS was found to be highly effective in removing Cu (II).•The N and S heteroatoms on TSP-NS can easily form coordination complexes with Cu (II).•The used TSP-NS can be regenerated effectively and recycled at least five times.A triazine and thiophene bifunctionalized task-specific porous organic polymer with N and S atoms (TSP-NS) was synthesized from a simple one-step Friedel-Crafts reaction. The resulting novel porous polymer was determined to be highly effective in removing Cu (II) from aqueous solution. The maximum adsorption capacity, qmax, evaluated from the Langmuir model was 98.33 mg g−1. Moreover, the combination of experimental FTIR results and the theoretical quantum calculation demonstrated that the N and S heteroatoms of the bifunctionalized TSP-NS could easily form coordination complexes with Cu (II), which were responsible for efficient adsorption. The generated TSP-NS could be regenerated effectively and recycled at least five times without significant loss of adsorption capacity. Therefore, the convenience and low cost of synthesis, as well as the excellent adsorption capacity and reusability, made bifunctionalized polymer TSP-NS an attractive adsorbent for removing toxic metal ions from aqueous solution.
Co-reporter:Yan He, Xiang Zhu, Yankai Li, Changjun Peng, Jun Hu, Honglai Liu
Microporous and Mesoporous Materials 2015 Volume 214() pp:181-187
Publication Date(Web):15 September 2015
DOI:10.1016/j.micromeso.2015.05.013
•A series of TPPs were firstly achieved through one-step Friedel–Crafts reaction.•The TPPs have been designed with surface functionalized porosities.•The post-synthetic modification method was adopted to produce TPP-1-NH2.•TPP-1-NH2 showed excellent CO2 adsorption capacity and high CO2/N2 selectivity.Microporous organic polymers (MOPs) are an emerging class of materials constructed from organic molecular building blocks. However, to design MOPs with surface functionalized porosities and high CO2 adsorption capacity is still a critical challenge. In this work, starting from different chemical functionalized triptycene monomers, including amino (–NH2), formyl (–CHO), acetyl (–COCH3) and nitro (–NO2) functional groups, we achieved a series of three-dimensional rigid framework of pre-functionalized triptycene-based polymers (TPPs) through a simple one-step Friedel–Crafts reaction. Moreover, alkyl-substituted amino groups were further incorporated into the network by the post-synthetic modification of amine pre-functionalized polymer TPP-1. The resulted microporous organic polymer TPP-1-NH2 presented an excellent CO2 adsorption capacity (4.17 mmol g−1 at 273 K, 1 bar) and a high CO2/N2 selectivity (43.6 at 273 K), which was ascribed to the predominant micropores and high CO2 isosteric adsorption heat of 41 kJ mol−1. Therefore, it is believed that these MOPs with high physicochemical stability are promising candidates for selective CO2 capture by employing this functionalized modification method.
Co-reporter:Changchun He, Jinlong Li, Changjun Peng, Honglai Liu, and Ying Hu
Industrial & Engineering Chemistry Research 2012 Volume 51(Issue 7) pp:3137-3148
Publication Date(Web):January 20, 2012
DOI:10.1021/ie202237e
An equation of state for square-well chain fluids with variable well-width range (SWCF-VR EoS) [Li et al. Fluid Phase Equilib.2009, 276, 57] was applied to ionic liquid (IL) systems. ILs were treated as the square-well chain with hydrogen bonding. The corresponding association parameters were given according to our previous work [He et al. Fluid Phase Equilib.2011, 302, 139]. The nonassociation parameters were obtained by correlating the experimental liquid densities. Excellent agreements were observed between experimental and theoretical results for pure ILs, and the molecular parameters were linearly correlated with the molecular masses of the [Cnmim][NTf2] members (n = 2, 3, ..., 8, 10). It is found that the other thermodynamic properties such as the vapor pressure and the enthalpy of vaporization, etc., can be reasonably predicted by using the obtained molecular parameters. The phase behavior of the binary systems containing ILs was well-represented with a simple mixing rule. For the vapor–liquid equilibria (VLE) of a system of volatile fluid + IL at low pressures, a temperature-independent binary interaction parameter was adopted. Satisfactory results were achieved for both the self- and cross-associating systems. The influence of temperature on the binary interaction parameters was taken into account in the correlation for the gas–liquid equilibria (GLE) of CO2 + IL mixtures and liquid–liquid equilibria (LLE) of IL-containing systems. For CO2 + IL mixtures, the multipolar interactions between like and unlike molecules, and the cross-association between CO2 and IL molecules were neglected to reduce the computational complexity, and the correlated results agree well with the experimental ones over a wide range of temperatures and pressures. The LLE of alkanol + IL systems were acceptably reproduced with moderate deviations between the experimental and calculated mass fractions. In the water-rich phase of water + IL with LLE, the neglect of electrostatic interaction caused correlated results to deviate from experimental ones greatly.
Co-reporter:Jinlong Li, Xiaoqian Yang, Kexia Chen, Yeling Zheng, Changjun Peng, and Honglai Liu
Industrial & Engineering Chemistry Research 2012 Volume 51(Issue 27) pp:9376-9385
Publication Date(Web):June 1, 2012
DOI:10.1021/ie3000985
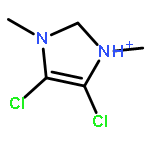
The phase behavior of the ternary acetonitrile + water + ionic liquid system was predicted using the COSMO-RS method. The effects of different ionic liquids on the separation of acetonitrile + water were discussed. It was found that the influence of anions [OAc]− and [Cl]− on the acetonitrile + water mixture is strong, while the cations have fewer effects on the mixture. The interaction energies and mixing enthalpies of binary acetonitrile + ionic liquid and water + ionic liquid systems were predicted indicating that the interaction energy between molecules is stronger in the water + ionic liquid mixture than in the acetonitrile + ionic liquid system. The excess enthalpies of binary mixtures mainly depend on the hydrogen bonds formed between water (or acetonitrile) and ionic liquids. Ionic liquids [EMIM][OAc] and [EMIM]Cl are expected to be favorable solvents and supposed to have practical applications in the separation of acetonitrile from aqueous solution.
Co-reporter:Jinlong Li, Changchun He, Changjun Peng, Honglai Liu, Ying Hu, and Patrice Paricaud
Industrial & Engineering Chemistry Research 2011 Volume 50(Issue 11) pp:7027-7040
Publication Date(Web):April 28, 2011
DOI:10.1021/ie102156m
A new thermodynamic model based on an equation of state for square-well chain fluid with variable range (SWCF-VR) is proposed to describe the thermodynamic properties of aqueous solutions of ionic liquids. In this approach the electrostatic interactions are characterized via the mean spherical approximation and the ionic associations between the cations and anions of the ionic liquid are considered through the shield-sticky approach. The liquid densities, osmotic coefficients, and vapor pressures of aqueous solutions of nine ionic liquids (ILs) containing [Cxmim][Br] (x = 2, ..., 6), [C4mim][BF4] and [Cxmim][MSO4] (x = 1, 3, 4) have been modeled with the new model. Two ionic parameters for each anion and three for each cation of the ionic liquids were adjusted to experimental liquid densities and osmotic coefficients, and the corresponding average deviations are only 0.87% and 2.49%, respectively. Using the same ionic parameters, the vapor pressures of ionic liquid solutions are accurately predicted. The predicted equilibrium constants of the ionic association between the cations and anions of ILs in water were consistent with experimental observations.
Co-reporter:Yan Xiong, Jing Ding, Dahong Yu, Changjun Peng, Honglai Liu, and Ying Hu
Industrial & Engineering Chemistry Research 2011 Volume 50(Issue 24) pp:14155-14161
Publication Date(Web):October 31, 2011
DOI:10.1021/ie201784z
A new approach named volumetric connectivity index (VCI) based on the physical observable volumes of groups and the concept of molecular connectivity index is proposed for the prediction of the density of ionic liquids (ILs). The capability of VCI in the prediction by combination of group (formula units) volumes for a new and as yet not prepared modification can provide an estimate of the density of that compound. In this work, the densities at room temperature of 142 pure ILs including imidazolium, pyridinium, pyrrolidinium, piperidinium, quaternary ammonium, and quaternary phosphonium are estimated by this new model, and the results are compared with the experimental data collected from the most commonly used literatures. The average deviation for the prediction of all the 142 ILs is only 0.63%, and R2 and rmsd are 0.9971 and 0.01214 g·cm–3, respectively. Combined with a model called mass connectivity index (MCI), the new model can also be used to predict the densities of ILs at different temperatures accurately with the room temperature density obtained input in MCI.
Co-reporter:Changchun He, Jinlong Li, Jun Ma, Changjun Peng, Honglai Liu, Ying Hu
Fluid Phase Equilibria 2011 Volume 302(1–2) pp:139-152
Publication Date(Web):15 March 2011
DOI:10.1016/j.fluid.2010.10.023
The equation of state developed in our previous work for square-well chain molecules with variable range (SWCF-VR EOS) was further extended to associating fluids. The association contribution is directly calculated by Liu et al.’s association equation which was proposed based on the sticky-shield method. For the real pure associating fluids, the SWCF-VR EOS has six temperature-independent model parameters obtained by fitting the saturated pressures and/or liquid molar volumes. The results are satisfactory with the overall average absolute deviations (AADs) of 0.76% and 0.75% for saturated vapor pressures and liquid molar volumes of 78 pure fluids in wide temperature ranges. The heat of vaporization for 17 associating fluids were well predicted using these molecular parameters. The good relations between the molecular parameters and the molecular weights were observed. By using a simple mixing rule with only one adjustable binary interaction parameter kij for the cross well depth ɛij, the SWCF-VR EOS can satisfactorily reproduce the vapor–liquid equilibria (VLE) of binary mixtures at reduced and elevated pressures containing self- and cross-associating interaction. Furthermore, the VLE for ternary mixtures can be successfully predicted by using the obtained binary interaction parameters.
Co-reporter:Dongfu Fan, Jinlong Li, Jibin Shi, Changjun Peng, Honglai Liu, Ying Hu, and Patrice Paricaud
Journal of Chemical & Engineering Data 2011 Volume 56(Issue 4) pp:1323-1329
Publication Date(Web):February 2, 2011
DOI:10.1021/je101131x
A static total pressure method was employed to determine the vapor pressures of pure propyl ethanoate (propyl acetate), pure ethanoic acid, and the corresponding propyl acetate + ethanoic acid binary system at different feeding compositions over the temperature range (323.15 to 353.15) K. The isothermal vapor−liquid equilibrium (VLE) data of the binary mixture at various temperatures were obtained from classical thermodynamic relations and mass-balance equations. The nonrandom two-liquid (NRTL), Wilson, and universal quasi-chemical (UNIQUAC) models were used to represent the nonideality of the liquid phase, and a modified Peng−Robinson equation of state was used to compute the properties of the vapor phase. The overall average relative deviations between the experimental equilibrium pressures and the NRTL, Wilson, and UNIQUAC models were (0.95, 0.96, and 0.94) %, respectively. The three models gave about the same equilibrium pressures and vapor compositions. A new version of the conductor-like screening model−segment activity coefficient (COSMO−SAC) model was applied to predict the VLE of propyl acetate + ethanoic acid mixtures, and good agreement with the experimental data was obtained.
Co-reporter:Yingqian Ren, Yue Wang, Guangming Liu, Changjun Peng, Honglai Liu, and Ying Hu
Journal of Chemical & Engineering Data 2011 Volume 56(Issue 4) pp:1341-1347
Publication Date(Web):February 28, 2011
DOI:10.1021/je1010592
Solubilities of potassium chloride (KCl) and sodium chloride (NaCl) in aqueous systems containing the ionic liquid 1-butyl-3-methylimidazolium chloride ([Bmim]Cl) were determined by cloud-point measurements using the laser beam scattering method in a temperature range of (298.15 to 343.15) K at atmospheric pressure. The solubility data were correlated and calculated with the Pitzer model modified by Harvie and Weare. The interaction parameters of the mixed-electrolyte solution were obtained using experimental results. It was found that the addition of [Bmim]Cl leads to salting-out, triggering a marked decrease of salt solubility in the aqueous solutions. The results also show that [Bmim]Cl has different salting-out ratios on KCl and NaCl in the mass-fraction range of w[Bmim]Cl = (0.025 to 0.20). It is also shown that the modified Pitzer model gives a satisfactory prediction for the solubilities of KCl and NaCl in aqueous systems containing ionic liquid [Bmim]Cl.
Co-reporter:Yazhuo Shang, Tengfang Wang, Xia Han, Changjun Peng and Honglai Liu
Industrial & Engineering Chemistry Research 2010 Volume 49(Issue 18) pp:8852-8857
Publication Date(Web):August 5, 2010
DOI:10.1021/ie100896z
The effect of ionic liquids CnmimBr (n = 4, 6, 8) on the properties of cationic Gemini surfactant trimethylene-1,3-bis(dodecyl ammonium bromide) (Gemini 12-3-12) aqueous solution was investigated. The critical micelle concentration (cmc) of Gemini 12-3-12 solutions containing different amounts of ionic liquids were obtained by measurement of surface tension. The composition of mixed micelle and the interaction parameter between ionic liquids and Gemini 12-3-12 were calculated by Rubingh regular solution model; the effective area of Gemini 12-3-12 molecule at air/water interface was calculated by Gibbs adsorption isotherm. The results show that the addition of ionic liquid has important effect on the properties of surfactant aqueous solution. The cmc of surfactant solution and the effective area of surfactant molecules at the air/water interface vary parabolically with the addition of ionic liquids owing to the two extra opposite effects of ionic liquids in surfactant aqueous solutions besides counterion effect, participation of micelles formation, and the effect on the properties of solvent. The effect of ionic liquids on surfactant solution can be regarded as the comprehensive effects of inorganic salt, cosurfactant, and cosolvent on surfactant solution.
Co-reporter:Li Yang, Stanley I. Sandler, Changjun Peng, Honglai Liu, and Ying Hu
Industrial & Engineering Chemistry Research 2010 Volume 49(Issue 24) pp:12596-12604
Publication Date(Web):November 9, 2010
DOI:10.1021/ie1013647
Here we examine the application of the COSMO-SAC model to phase equilibrium calculations of solutions containing ionic liquids (ILs). As there is uncertainty about the degree of ionization in these liquids, we consider three methods: treating the IL as a neutral molecule, as a separated anion and cation, and essentially as an ion pair. In the first case, the needed quantum mechanics calculation is done for the neutral molecule referred to as COSMO-SAC(molecule), and in the other two cases, the calculations are performed for the separated ions in which the IL is treated as two separate components, COSMO-SAC(ions) or COSMO-SAC(CA), in which the separate cosmo files are combined and the IL is treated as a single-component neutral ion pair. There is generally good agreement between the predicted results and the experimental data using COSMO-SAC(CA) and COSMO-SAC(ions), while for COSMO-SAC(molecule) the results are less satisfactory and the calculations are more difficult because of the problems with geometry optimization and the density functional calculations for the large intact IL molecules. The results show that the COSMO-SAC model can give reasonably good predictions for the vapor−liquid equilibria, liquid−liquid equilibria, and infinite dilution activity coefficients for solutions containing ionic liquids.
Co-reporter:Tengfang Wang, Changjun Peng, Honglai Liu, Ying Hu
Journal of Molecular Liquids 2009 Volume 146(Issue 3) pp:89-94
Publication Date(Web):30 June 2009
DOI:10.1016/j.molliq.2009.02.007
The phase behavior and microstructure of the system consisting of ionic liquid BmimPF6, copolymer F127, H2O and short-chain alcohols including ethanol, n-propanol or n-butanol were investigated at 30 °C. Large areas of single-phase microemulsion were observed in all the three systems. Specially, lyotropic liquid crystal appeared in the n-butanol system while it disappeared when the other two alcohols were used. The water-in-BmimPF6, bicontinuous, and BmimPF6-in-water regions of the microemulsions were identified by the usual electrical conductivity measurement. As an attempt, UV–vis spectroscopy was successfully used to distinguish different type of microemulsions using methyl orange as solvatochromic probe. Furthermore, dynamic light scattering (DLS) measurements were also skillfully introduced to vividly track the structural variation of different microemulsions. On the other hand, the hydrodynamic diameter of the BmimPF6-in-water microemulsion was accurately obtained. An unusual phenomenon was demonstrated that Z-average size of BmimPF6-in-water microemulsions decreases with increasing BmimPF6 (oil) concentration at fixed (F127 + n-propanol) concentration.
Co-reporter:Yanfang Geng, Siliu Chen, Tengfang Wang, Dahong Yu, Changjun Peng, Honglai Liu, Ying Hu
Journal of Molecular Liquids 2008 Volume 143(2–3) pp:100-108
Publication Date(Web):20 October 2008
DOI:10.1016/j.molliq.2008.06.014
Densities, viscosities and electrical conductivities of ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate ([C4mim][PF6]) in monoethanolamine (MEA) and N, N-dimethylethanolamine (DMEA) have been determined from (288.15 to 323.15) K. The results show that the densities of both binary mixtures linearly decrease with increasing temperature. The dependence of temperature on the viscosity has been fitted to the Arrhenius equation with high precision. A viscosity model based on the equation of state for chain-like fluids and a solute aggregation model were used to calculate the viscosity of binary mixture. The dependence of temperature on the electrical conductivity has also been fitted in the form of Arrhenius equation. The effect of concentration of ionic liquid on the electrical conductivity has been examined using the Walden rule. Excess molar volumes and viscosity deviations from a mole fraction average have been obtained and fitted to the Redlich–Kister equation.
Co-reporter:Qin Xin, Changjun Peng, Honglai Liu and Ying Hu
Industrial & Engineering Chemistry Research 2008 Volume 47(Issue 23) pp:9678-9686
Publication Date(Web):October 31, 2008
DOI:10.1021/ie800924r
The molecular thermodynamic model of polymer solutions based on a close-packed lattice model presented in a previous work has been generally extended to multicomponent chainlike fluid mixtures. The Helmholtz function of mixing contains three terms, i.e., the contribution of athermal mixing of polymer chains, which is calculated by Guggenheim’s theory; the contribution of nearest-neighbor interactions between monomers, which is calculated by Yang et al.’s model of the Helmholtz function of mixing for a multicomponent Ising lattice; and the contribution of the formation of polymer chains from monomers, which is obtained according to the sticky-point theory of Cummings, Zhou, and Stell. The liquid−liquid phase equilibria of ternary chainlike mixtures predicted by this model are in good agreement with Monte Carlo simulation results and superior to the results calculated by Flory−Huggins (FH) theory and revised Freed theory (RFT) obviously. This model not only can describe types 1−3 phase separations of Treybal classification satisfactorily, but can also correlate well the coexistence curves of binary polymer blends systems with an upper critical solution temperature (UCST) or a lower critical solution temperature (LCST). Meanwhile, model parameters correlated from the binary system can be further extended to predict the corresponding liquid−liquid equilibrium of ternary mixtures, including systems of ionic liquids.
Co-reporter:Qin Xin, Changjun Peng, Honglai Liu, Ying Hu
Fluid Phase Equilibria 2008 Volume 267(Issue 2) pp:163-171
Publication Date(Web):25 May 2008
DOI:10.1016/j.fluid.2008.03.001
A new Helmholtz energy model of mixing for random copolymer solutions based on a close-packed lattice has been developed. The model contains three terms: the contribution of the athermal mixing of polymer chain and solvent, the Helmoltz energy of mixing in a multi-component Ising lattice where the interactions between segments is accounted for, and the contribution of the dissociation of the polymer and the association of monomers. The Guggenheim model, Yang et al.'s model and the sticky-point model of Cummings, Zhou and Stell are used respectively, for the above three contributions. Comparisons between Monte Carlo simulated coexistence curves with those predicted by various theories for random copolymer solutions with various chain lengths, chain compositions and inter-segment interaction parameters show that the agreement between simulations and the predictions of this work is nearly perfect. The model can be used satisfactorily to correlate the liquid–liquid equilibria of practical random copolymer solutions.
Co-reporter:Xiaochun Xu, Honglai Liu, Changjun Peng, Ying Hu
Fluid Phase Equilibria 2008 Volume 265(1–2) pp:112-121
Publication Date(Web):25 March 2008
DOI:10.1016/j.fluid.2008.01.011
In previous work, we have developed a close-packed lattice model for binary solutions of chain-like molecules. As a continuation, considering the effect of volume change, we present here a new molecular-thermodynamic model based on lattice fluid theory. The resulting equation of state shows good performance in describing thermodynamic properties such as pVT behavior, vapor pressure and liquid volume of pure normal fluids. Equation-of-state parameters are obtained by correlation of experimental data. The model is extended to calculate vapor–liquid equilibria of binary mixtures with only one binary interaction parameter. Comparison between the present model and other theories is also presented.
Co-reporter:Aiguo Xuan, Yuanxin Wu, Changjun Peng, Peisheng Ma
Fluid Phase Equilibria 2006 Volume 240(Issue 1) pp:15-21
Publication Date(Web):10 February 2006
DOI:10.1016/j.fluid.2005.11.027
A novel model has been presented for correlating the dynamic viscosity of Newtonian liquids at high pressures. The proposed model was started with the activation volume, which could simultaneously be influenced by temperature and pressure. The core of the model was based on Hu and Liu's work to calculate the compressibility factor. The final expression may contain two adjustable parameters, namely κ1 and κ2, which had been determined by fitting literature viscosity data. The results show that the agreement between experimental data of viscosity and the calculated ones with the proposed model was reasonably good for the selected systems. It was found that the logarithm of parameter κ1 was a linear function of the reciprocal of the temperature, and κ1 was approximately equal to viscosity of liquid at 0.1 MPa. Besides, for linear chain hydrocarbons, the logarithm of the parameter κ1 was completely a linear function of the number of the carbon atoms under certain temperature. A comparison between model with two-parameter and one with one-parameter had been given to show their applicability. The results calculated by not only two-parameter but also one-parameter were superior to corresponding ones by previous pressure equation for estimating viscosity.
Co-reporter:Jun MA, Jinlong LI, Dongfu FAN, Changjun PENG, Honglai LIU, Ying HU
Chinese Journal of Chemical Engineering (December 2011) Volume 19(Issue 6) pp:1009-1016
Publication Date(Web):1 December 2011
DOI:10.1016/S1004-9541(11)60084-0
Combining Peng-Robinson (PR) equation of state (EoS) with an association model derived from shield-sticky method (SSM) by Liu et al., a new cubic-plus-association (CPA) EoS is proposed to describe the thermodynamic properties of pure ionic liquids (ILs) and their mixtures. The new molecular parameters for 25 ILs are obtained by fitting the experimental density data over a wide temperature and pressure range, and the overall average deviation is 0.22%. The model parameter b for homologous ILs shows a good linear relationship with their molecular mass, so the number of model parameters is reduced effectively. Using one temperature-independent binary adjustable parameter kij, satisfactory correlations of vapor-liquid equilibria (VLE) for binary mixtures of ILs + non-associating solvents and + associating solvents are obtained with the overall average deviation of vapor pressure 2.91% and 7.01%, respectively. In addition, VLE results for ILs + non-associating mixtures from CPA, lattice-fluid (LF) and square-well chain fluids with variable range (SWCF-VR) EoSs are compared.
Co-reporter:Jinlong LI, Qing HE, Changchun HE, Changjun PENG, Honglai LIU
Chinese Journal of Chemical Engineering (December 2009) Volume 17(Issue 6) pp:983-989
Publication Date(Web):1 December 2009
DOI:10.1016/S1004-9541(08)60306-7
An equation of state (EOS) for square-well chain fluids with variable range (SWCF-VR) developed based on statistical mechanics for chemical association was employed for the calculations of pressure-volume-temperature (pVT) and phase equilibrium of pure ionic liquids (ILs) and their mixtures. The new molecular parameters for 23 ILs were obtained by fitting their experimental density data over a wide temperature and pressure ranges. The molecular parameters of ILs composed of homologous organic cation and an identical anion such as [Cxmim][NTf2] are good linear with respect to their molecular weight, indicating that the molecular parameters of homologous substances, subsequently pVT and vapor-liquid equilibria vapor-liquid equilibria (VLE) can be predicted using the generalized parameter when no experimental data were available. The new set of parameters were satisfactorily used for calculations of the property of solvent and ILs mixture and the solubility of gas in various ILs at low pressure only using one temperature-independent binary interaction parameter.
Co-reporter:Jinlong LI, Changchun HE, Jun MA, Changjun PENG, Honglai LIU, Ying HU
Chinese Journal of Chemical Engineering (August 2011) Volume 19(Issue 4) pp:533-542
Publication Date(Web):1 August 2011
DOI:10.1016/S1004-9541(11)60018-9
The equation of state (EOS) for square-well chain fluid with variable range (SWCF-VR) developed in our previous work based on statistical mechanical theory for chemical association is employed for the correlations of surface tension and viscosity of common fluids and ionic liquids (ILs). A model of surface tension for multi-component mixtures is presented by combining the SWCF-VR EOS and the scaled particle theory and used to produce the surface tension of binary and ternary mixtures. The predicted surface tensions are in excellent agreement with the experimental data with an overall average absolute relative deviation (AAD) of 0.36%. A method for the calculation of dynamic viscosity of common fluids and ILs at high pressure is presented by combining Eyring's rate theory of viscosity and the SWCF-VR EOS. The calculated viscosities are in good agreement with the experimental data with the overall AAD of 1.44% for 14 fluids in 84 cases. The salient feature is that the molecular parameters used in these models are self-consistent and can be applied to calculate different thermodynamic properties such as pVT, vapor-liquid equilibrium, caloric properties, surface tension, and viscosity.