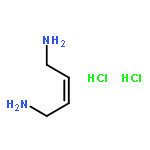
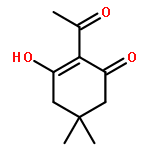
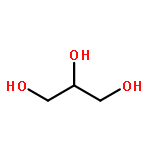
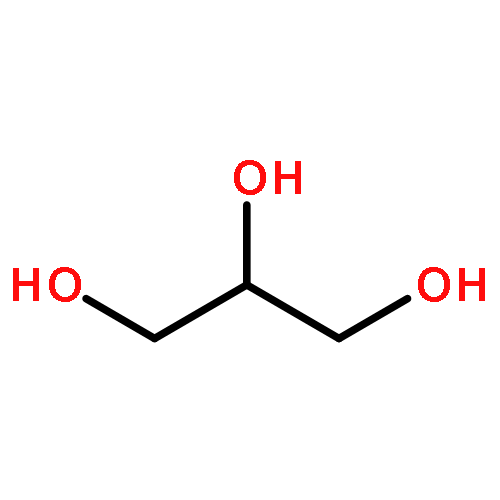
Co-reporter:Kai Tao, Bin Xue, Samuel Frere, Inna Slutsky, Yi Cao, Wei Wang, and Ehud Gazit
Chemistry of Materials May 23, 2017 Volume 29(Issue 10) pp:4454-4454
Publication Date(Web):May 3, 2017
DOI:10.1021/acs.chemmater.7b00966
Artificial photosynthesis shows a promising potential for sustainable supply of nutritional ingredients. While most studies focus on the assembly of the light-sensitive chromophores to 1-D architectures in an artificial photosynthesis system, other supramolecular morphologies, especially bioinspired ones, which may have more efficient light-harvesting properties, have been far less studied. Here, MCpP-FF, a bioinspired building block fabricated by conjugating porphyrin and diphenylalanine, was designed to self-assemble into nanofibers-based multiporous microspheres. The highly organized aromatic moieties result in extensive excitation red-shifts and notable electron transfer, thus leading to a remarkable attenuated fluorescence decay and broad-spectrum light sensitivity of the microspheres. Moreover, the enhanced photoelectron production and transfer capability of the microspheres are demonstrated, making them ideal candidates for sunlight-sensitive antennas in artificial photosynthesis. These properties induce a high turnover frequency of NADH, which can be used to produce bioproducts in biocatalytic reactions. In addition, the direct electron transfer makes external mediators unnecessary, and the insolubility of the microspheres in water allows their easy retrieval for sustainable applications. Our findings demonstrate an alternative to design new platforms for artificial photosynthesis, as well as a new type of bioinspired, supramolecular multiporous materials.
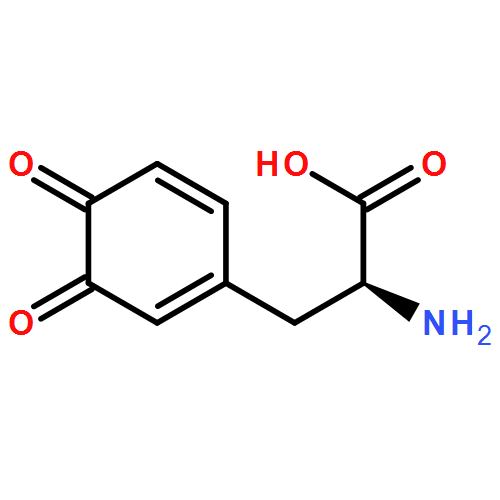
Co-reporter:Yiran Li, Jing Wen, Meng Qin, Yi Cao, Haibo Ma, and Wei Wang
ACS Biomaterials Science & Engineering June 12, 2017 Volume 3(Issue 6) pp:979-979
Publication Date(Web):May 11, 2017
DOI:10.1021/acsbiomaterials.7b00186
Metal coordination bonds are widely found in natural adhesives and load-bearing and protective materials, in which they are thought to be responsible for the high mechanical strength and toughness. However, it remains unknown how metal–ligand complexes could give rise to such superb mechanical properties. Here, we developed a single-chain nanoparticle based force spectroscopy to directly quantify the mechanical properties of individual catechol–Fe3+ complexes, the key elements accounting for the high toughness and extensibility of byssal threads of marine mussels. We found that catechol–Fe3+ complexes possess a unique combination of mechanical features, including high mechanical stability, fast reformation kinetics, and stoichiometry-dependent mechanics. Therefore, they can serve as sacrificial bonds to efficiently dissipate energy in the materials, quickly recover the mechanical properties when load is released, and respond to pH and Fe3+ concentrations. Especially, we revealed that the bis-catechol–Fe3+ complex is mechanically ∼90% stronger than the tris-catechol–Fe3+ complex. Quantum calculation study suggested that the distinction between mechanical strength and thermodynamic stability of catechol–Fe3+ complexes is due to their different mechanical rupture pathways. Our study provides the nanoscale mechanistic understanding of the coordination bond-mediated mechanical properties of biogenetic materials, and could guide future rational design and regulation of the mechanical properties of synthetic materials.Keywords: atomic force microscopy; dopa; load-bearing materials; mussel foot protein; surface adhesion;
Co-reporter:Wenmao HuangMeng Qin, Ying Li, Yi Cao, Wei Wang
Langmuir 2017 Volume 33(Issue 6) pp:
Publication Date(Web):January 22, 2017
DOI:10.1021/acs.langmuir.6b04396
Cell-adhesion molecules (CAMs) often exist as homodimers under physiological conditions. However, owing to steric hindrance, simultaneous binding of two ligands to the homodimers at the same location can hardly be satisfied, and the molecular mechanism underlying this natural design is still unknown. Here, we present a theoretical model to understand the rupture behavior of cell-adhesion bonds formed by multiple binding ligands with a single receptor. We found that the dissociation forces for the cell-adhesion bond could be greatly enhanced in comparison with the monomer case through a ligand rebinding and exchange mechanism. We also confirmed this prediction by measuring dimeric cRGD (cyclic Arg-Gly-Asp) unbinding from integrin (αvβ3) using atomic force microscopy-based single-molecule force spectroscopy. Our finding addresses the mechanism of increasing the binding strength of cell-adhesion bonds through dimerization at the single-molecule level, representing a key step toward the understanding of complicated cell-adhesion behaviors. Moreover, our results also highlight a wealth of opportunities to design mechanically stronger bioconjunctions for drug delivery, biolabeling, and surface modification.
Co-reporter:Wenmao Huang, Zhenshu Zhu, Jing Wen, Xin Wang, Meng Qin, Yi CaoHaibo Ma, Wei Wang
ACS Nano 2017 Volume 11(Issue 1) pp:
Publication Date(Web):November 14, 2016
DOI:10.1021/acsnano.6b07119
Carbon–carbon double bonds (C═C) are ubiquitous in natural and synthetic polymers. In bulk studies, due to limited ways to control applied force, they are thought to be mechanically inert and not to contribute to the extensibility of polymers. Here, we report a single molecule force spectroscopy study on a polymer containing C═C bonds using atomic force microscope. Surprisingly, we found that it is possible to directly observe the cis-to-trans isomerization of C═C bonds at the time scale of ∼1 ms at room temperature by applying a tensile force ∼1.7 nN. The reaction proceeds through a diradical intermediate state, as confirmed by both a free radical quenching experiment and quantum chemical modeling. The force-free activation length to convert the cis C═C bonds to the transition state is ∼0.5 Å, indicating that the reaction rate is accelerated by ∼109 times at the transition force. On the basis of the density functional theory optimized structure, we propose that because the pulling direction is not parallel to C═C double bonds in the polymer, stretching the polymer not only provides tension to lower the transition barrier but also provides torsion to facilitate the rotation of cis C═C bonds. This explains the apparently low transition force for such thermally “forbidden” reactions and offers an additional explanation of the “lever-arm effect” of polymer backbones on the activation force for many mechanophores. This work demonstrates the importance of precisely controlling the force direction at the nanoscale to the force-activated reactions and may have many implications on the design of stress-responsive materials.Keywords: atomic force microscope; carbon−carbon double bonds; cis-to-trans isomerization; force-induced rotation; mechanochemistry;
Co-reporter:Yiran Li;Tiankuo Wang;Lei Xia;Lei Wang;Meng Qin;Ying Li;Wei Wang
Journal of Materials Chemistry B 2017 vol. 5(Issue 23) pp:4416-4420
Publication Date(Web):2017/06/14
DOI:10.1039/C7TB00131B
Using AFM based single-molecule force spectroscopy, we studied the synergy between Dopa and lysine for wet adhesion on titania (TiO2) and mica surfaces. We found that the binding forces for lysine–Dopa dipeptides are significantly higher when the positive charge of lysine is unprotected on both surfaces. However, such a synergistic effect is absent when the sequence is reversed. We attribute such differential synergistic effects to their distinct structure for load distribution within the molecules, which may represent a general principle for synergistic strong adhesion.
Co-reporter:Ying Li;Chao Liang;Ling Gao;Shiyu Li;Yizhe Zhang;Jiang Zhang
Materials Chemistry Frontiers 2017 vol. 1(Issue 12) pp:2664-2668
Publication Date(Web):2017/11/22
DOI:10.1039/C7QM00402H
Dopa and lysine are widely found in mussel foot proteins and are suggested to play synergistic roles in wet adhesion; yet, the detailed molecular mechanism remains unclear. Here, using PEG conjugated dipeptides as the model system, we found that the neighboring lysine can significantly enhance surface binding of Dopa through three distinct mechanisms: (1) displacing surface water and ions to increase the effective binding sites; (2) being directly involved in cooperative surface binding in a sequence dependent manner; (3) enhancing cohesion by Michael addition to oxidized species or forming cation–π interactions. This study may be helpful for rational design of biomimetic strong adhesives for biomedical applications.
Co-reporter:Bin Xue, Meng Qin, Junhua Wu, Dongjun Luo, Qing Jiang, Ying Li, Yi Cao, and Wei Wang
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 24) pp:15120-15127
Publication Date(Web):May 31, 2016
DOI:10.1021/acsami.6b04338
Bacteria contamination in drinking water and medical products can cause severe health problems. However, currently available sterilization methods, mainly based on the size-exclusion mechanism, are typically slow and require the entire contaminated water to pass through the filter. Here, we present an electroresponsive hydrogel based approach for bacteria adsorption and removal. We successfully engineered a series of graphene oxide hydrogels using redox-active ruthenium complexes as noncovalent cross-linkers. The resulting hydrogels can reversibly switch their physical properties in response to the applied electric field along with the changes of oxidation states of the ruthenium ions. The hydrogels display strong bacteria adsorbing capability. A hydrogel of 1 cm3 can adsorb a maximum of 1 × 108 E. coli. The adsorbed bacteria in the hydrogels can then be inactivated by a high voltage electric pulse and removed from the hydrogels subsequently. Owing to the high bacteria removal rate, reusability, and low production cost, these hydrogels represent promising candidates for the emergent sterilization of medical products or large-scale purification of drinking water.
Co-reporter:Wei Wei; Yang Sun; Mingli Zhu; Xiangzhi Liu; Peiqing Sun; Feng Wang; Qiu Gui; Wuyi Meng; Yi Cao;Jing Zhao
Journal of the American Chemical Society 2015 Volume 137(Issue 49) pp:15358-15361
Publication Date(Web):November 13, 2015
DOI:10.1021/jacs.5b09895
The coordination bond between gold and sulfur (Au–S) has been widely studied and utilized in many fields. However, detailed investigations on the basic nature of this bond are still lacking. A gold-specific binding protein, GolB, was recently identified, providing a unique opportunity for the study of the Au–S bond at the molecular level. We probed the mechanical strength of the gold–sulfur bond in GolB using single-molecule force spectroscopy. We measured the rupture force of the Au–S bond to be 165 pN, much lower than Au–S bonds measured on different gold surfaces (∼1000 pN). We further solved the structures of apo-GolB and Au(I)–GolB complex using X-ray crystallography. These structures showed that the average Au–S bond length in GolB is much longer than the reported average value of Au–S bonds. Our results highlight the dramatic influence of the unique biological environment on the stability and strength of metal coordination bonds in proteins.
Co-reporter:Junhua Wu, Aiping Chen, Meng Qin, Rong Huang, Guang Zhang, Bin Xue, Jiwu Wei, Ying Li, Yi Cao and Wei Wang
Nanoscale 2015 vol. 7(Issue 5) pp:1655-1660
Publication Date(Web):28 Nov 2014
DOI:10.1039/C4NR05798H
The design of hydrogels with controllable drug-release properties remains challenging. Here we report a hydrogel made of hierarchical graphene oxide sheets and peptide assemblies for on-demand drug release. The hydrogel possesses high drug-sustainability and the release of drugs can be precisely and remotely controlled through external stimuli.
Co-reporter:Ying Li, Yang Sun, Meng Qin, Yi Cao and Wei Wang
Nanoscale 2015 vol. 7(Issue 13) pp:5638-5642
Publication Date(Web):26 Jan 2015
DOI:10.1039/C4NR07657E
The rigidity of peptide fibers is essential for their chemical and biological functions, despite that it remains largely unexplored. Here, we present the first direct measurement of the mechanics of individual fibers in peptide hydrogels by AFM imaging and statistical analysis and find that the intermolecular interactions play a considerable role.
Co-reporter:Bin Xue, Ying Li, Fan Yang, Chunfeng Zhang, Meng Qin, Yi Cao and Wei Wang
Nanoscale 2014 vol. 6(Issue 14) pp:7832-7837
Publication Date(Web):24 Feb 2014
DOI:10.1039/C4NR00295D
A peptide nanotube platform that integrates both light-harvesting and catalytic units was successfully engineered for artificial photosynthesis. Peptide nanotubes not only serve as a hub for physically combining both units, but also work as mediators that transfer the energy from photo-excited chromophores to catalytic centers. The direct conversion of NAD+ to NADH upon light illumination was demonstrated. This represents a promising step towards efficient and fully integrated artificial photosynthesis systems.
Co-reporter:Yiran Li, Meng Qin, Ying Li, Yi Cao, and Wei Wang
Langmuir 2014 Volume 30(Issue 15) pp:4358-4366
Publication Date(Web):2017-2-22
DOI:10.1021/la501189n
3,4-Dihydroxyphenylalanine (DOPA) is the noncanonical amino acid widely found in mussel holdfast proteins, which is proposed to be responsible for their strong wet adhesion. This feature has also inspired the successful development of a range of DOPA-containing synthetic polymers for wet adhesions and surface coating. Despite the increasing applications of DOPA in material science, the underlying mechanism of DOPA–wet surface interactions remains unclear. In this work, we studied DOPA–surface interactions one bond at a time using atomic force microscope (AFM) based single molecule force spectroscopy. With our recently developed “multiple fishhook” protocol, we were able to perform high-throughput quantification of the binding strength of DOPA to various types of surfaces for the first time. We found that the dissociation forces between DOPA and nine different types of organic and inorganic surfaces are all in the range of 60–90 pN at a pulling speed of 1000 nm s–1, suggesting the strong and versatile binding capability of DOPA to different types of surfaces. Moreover, by constructing the free energy landscape for the rupture events, we revealed several distinct binding modes between DOPA and different surfaces, which are directly related to the chemistry nature of the surfaces. These results explain the molecular origin of the versatile binding ability of DOPA. Moreover, we could quantitatively predict the relationship between DOPA contents and the binding strength based on the measured rupture kinetics. These serve as the bases for the quantitative prediction of the relationship between DOPA contents and adhesion strength to different wet surfaces, which is important for the design of novel DOPA based materials.
Co-reporter:Chunmei Lv, Dawei Zou, Meng Qin, Wei Meng, Yi Cao, and Wei Wang
Langmuir 2013 Volume 29(Issue 34) pp:10624-10629
Publication Date(Web):August 14, 2013
DOI:10.1021/la4023689
Many cellular processes, such as the diffusion of biomacromolecules, the movement of molecular motors, and the conformational dynamics of proteins, are subjected to hydrodynamic forces because of the high viscosities of cellular environments. However, it is still unknown how hydrodynamic forces are related to the physical properties of different viscogens. Here, using the atomic force microscope-based force spectroscopy technique, we directly measured the hydrodynamic forces acting on a moving cantilever in various viscogen solutions. We found that the hydrodynamic force is not only dependent on the viscosity but also related to the molecular weight of viscogens. Counterintuitively, at the same macroscopic viscosity, the hydrodynamic force rises with the increasing molecular weight of viscogens, although the local microscopic viscosity of the solution decreases. This finding provides insights into the origin of hydrodynamic forces in biomolecule solutions and could inspire many force-spectroscopy-based techniques to measure the molecular weight and conformational changes of biomacromolecules in biological settings directly.
Co-reporter:Dr. Junyi Liang;Yang Yang;Puguang Yin;Dr. Yin Ding; Yan Shen; Meng Qin; Jun Wang; Qiang Xu; Yi Cao; Wei Wang
ChemBioChem 2013 Volume 14( Issue 12) pp:1423-1426
Publication Date(Web):
DOI:10.1002/cbic.201300199
Co-reporter:Junyi Liang, Meng Qin, Rui Xu, Xiang Gao, Yan Shen, Qiang Xu, Yi Cao and Wei Wang
Chemical Communications 2012 vol. 48(Issue 32) pp:3890-3892
Publication Date(Web):24 Feb 2012
DOI:10.1039/C2CC30531C
We report a genetically encoded fluorescent sensor for in vivo copper(I) imaging based on engineered intramolecular mechanical strain and structural distortion of EGFP.
Co-reporter:Wei Meng, Xinlu Guo, Meng Qin, Hai Pan, Yi Cao, and Wei Wang
Langmuir 2012 Volume 28(Issue 46) pp:16133-16140
Publication Date(Web):October 30, 2012
DOI:10.1021/la303466w
Protein PEGylation (attaching PEG chains to proteins) has been widely used in pharmaceuticals and nanotechnology. Although it is widely known that PEGylation can increase the thermodynamic stability of proteins, the underlying mechanism remains elusive. In this Article, we studied the effect of PEGylation on the thermodynamic and kinetic stability of a protein, SH3. We show that the thermodynamic stability of SH3 is enhanced upon PEGylation, mainly due to the slowing of the unfolding rate. Moreover, PEGylation can decrease the solvent-accessible surface area of SH3, leading to an increase of the m-value (the change in free energy with respect to denaturant concentration, which is a measure of the transition cooperativity between corresponding states). Such an effect also causes an enhancement of the thermodynamic stability. We quantitatively measured how the physical properties of PEG, such as the molecular weight and the number of PEGylation sites, affect the stabilization effect. We found that the stabilization effect is largely dependent on the number of PEGylation sites but only has a weak correlation with the molecular weight of the attached PEG. These experimental findings inspire us to derive a physical model based on excluded volume effect, which can satisfactorily describe all experimental observations. This model allows quantitatively calculating the free energy change upon PEGylation based on the change of water excluded zone on the protein surface. Although it is still unknown whether such a mechanism can be extended to other proteins, our work represents a key step toward the understanding of the nature of protein stabilization upon PEGylation.
Co-reporter:Xiaotian Han, Meng Qin, Hai Pan, Yi Cao, and Wei Wang
Langmuir 2012 Volume 28(Issue 26) pp:10020-10025
Publication Date(Web):June 12, 2012
DOI:10.1021/la301903z
Despite the powerfulness of atomic force microscopy (AFM)-based single-molecule force spectroscopy in the study of ligand–receptor interactions, complicated cantilever functionalization and data interpretation have often been a great hurdle for its widespread application. Here, we present a much simplified experimental scheme by using a “multiple fishhooks” approach. In this strategy, multiple ligands are labeled on a single polymer chain, which forms complexes with receptors anchored on the substrate surface. Therefore, multiple single-bond rupture events can be captured in the same force–extension curves, similar to the widely used polyprotein approach. This method also allows nonsingle-molecule events and nonspecific interactions between cantilever and surface to be readily excluded from real data pool and greatly increases the quality and quantity of single-molecule data. The biggest advantage of our approach over the previously reported one is the choice of a naturally occurring polysaccharide, hyaluronan, the conformation of which in solution can be fine-tuned by pH, as the polymer backbone of the “multiple fishhooks” handle. Furthermore, our approach greatly simplifies the chemical synthesis of the polymer handle, allowing bioactive molecules to be easily one-step labeled on the handles in aqueous solution. We validate this strategy using the widely studied streptavidin–biotin system, and our single-molecule AFM results are in good agreement with previously reported ones. We anticipate that this novel strategy can be used as a versatile tool to study other complex and challenging ligand–receptor interactions.
Co-reporter:Hai Pan, Meng Qin, Wei Meng, Yi Cao, and Wei Wang
Langmuir 2012 Volume 28(Issue 35) pp:12779-12787
Publication Date(Web):August 13, 2012
DOI:10.1021/la302258k
Owing to their many outstanding features, such as small size, large surface area, and cell penetration ability, nanoparticles have been increasingly used in medicine and biomaterials as drug carriers and diagnostic or therapeutic agents. However, our understanding of the interactions of biological entities, especially proteins, with nanoparticles is far behind the explosive development of nanotechnology. In typical protein–nanoparticle interactions, two processes (i.e., surface binding and conformational changes in proteins) are intermingled with each other and have not yet been quantitatively described. Here, by using a stopped-flow fast mixing technique, we were able to shed light on the kinetics of the adsorption-induced protein unfolding on nanoparticle surfaces in detail. We observed a biphasic denaturation behavior of protein GB1 on latex nanoparticle surfaces. Such kinetics can be adequately described by a fast equilibrium adsorption followed by a slow reversible unfolding of GB1. On the basis of this model, we quantitatively measured all rate constants that are involved in this process, from which the free-energy profile is constructed. This allows us to evaluate the effects of environmental factors, such as pH and ionic strength, on both the adsorption and the conformational change in GB1 on the latex nanoparticle surface. These studies provide a general physical picture of the adsorption-induced unfolding of proteins on nanoparticle surfaces and a quantitative description of the energetics of each transition. We anticipate that it will greatly advance our current understanding of protein–nanoparticle interactions and will be helpful for the rational control of such interactions in biomedical applications.
Co-reporter:Xiang Gao, Meng Qin, Puguang Yin, Junyi Liang, Jun Wang, Yi Cao, Wei Wang
Biophysical Journal (2 May 2012) Volume 102(Issue 9) pp:
Publication Date(Web):2 May 2012
DOI:10.1016/j.bpj.2012.03.042
Per-ARNT-Sim (PAS) domains serve as versatile binding motifs in many signal-transduction proteins and are able to respond to a wide spectrum of chemical or physical signals. Despite their diverse functions, PAS domains share a conserved structure. It has been suggested that the structure of PAS domains is flexible and thus adaptable to many binding partners. However, direct measurement of the flexibility of PAS domains has not yet been provided. Here, we quantitatively measure the mechanical unfolding of a PAS domain, ARNT PAS-B, using single-molecule atomic force microscopy. Our force spectroscopy results indicate that the structure of ARNT PAS-B can be unraveled under mechanical forces as low as ∼30 pN due to its broad potential well for the mechanical unfolding transition of ∼2 nm. This allows the PAS-B domain to extend by up to 75% of its resting end-to-end distance without unfolding. Moreover, we found that the ARNT PAS-B domain unfolds in two distinct pathways via a kinetic partitioning mechanism. Sixty-seven percent of ARNT PAS-B unfolds through a simple two-state pathway, whereas the other 33% unfolds with a well-defined intermediate state in which the C-terminal β-hairpin is detached. We propose that the structural flexibility and force-induced partial unfolding of PAS-B domains may provide a unique mechanism for them to recruit diverse binding partners and lower the free-energy barrier for the formation of the binding interface.
Co-reporter:Chunmei Lv, Cheng Tan, Meng Qin, Dawei Zou, Yi Cao, Wei Wang
Biophysical Journal (18 April 2012) Volume 102(Issue 8) pp:
Publication Date(Web):18 April 2012
DOI:10.1016/j.bpj.2012.03.028
Some small proteins, such as HP35, fold at submicrosecond timescale with low folding cooperativity. Although these proteins have been extensively investigated, still relatively little is known about their folding mechanism. Here, using single-molecule force spectroscopy and steered molecule dynamics simulation, we study the unfolding of HP35 under external force. Our results show that HP35 unfolds at extremely low forces without a well-defined unfolding transition state. Subsequently, we probe the structure of unfolded HP35 using the persistence length obtained in the force spectroscopy. We found that the persistence length of unfolded HP35 is around 0.72 nm, >40% longer than typical unstructured proteins, suggesting that there are a significant amount of residual secondary structures in the unfolded HP35. Molecular dynamics simulation further confirmed this finding and revealed that many native contacts are preserved in HP35, even its two ends have been extended up to 8 nm. Our results therefore suggest that retaining a significant amount of secondary structures in the unfolded state of HP35 may be an efficient way to reduce the entropic cost for the formation of tertiary structure and increase the folding speed, although the folding cooperativity is compromised. Moreover, we anticipate that the methods we used in this work can be extended to the study of other proteins with complex folding behaviors and even intrinsically disordered ones.
Co-reporter:Yiran Li, Tiankuo Wang, Lei Xia, Lei Wang, Meng Qin, Ying Li, Wei Wang and Yi Cao
Journal of Materials Chemistry A 2017 - vol. 5(Issue 23) pp:NaN4420-4420
Publication Date(Web):2017/02/28
DOI:10.1039/C7TB00131B
Using AFM based single-molecule force spectroscopy, we studied the synergy between Dopa and lysine for wet adhesion on titania (TiO2) and mica surfaces. We found that the binding forces for lysine–Dopa dipeptides are significantly higher when the positive charge of lysine is unprotected on both surfaces. However, such a synergistic effect is absent when the sequence is reversed. We attribute such differential synergistic effects to their distinct structure for load distribution within the molecules, which may represent a general principle for synergistic strong adhesion.
Co-reporter:Junyi Liang, Meng Qin, Rui Xu, Xiang Gao, Yan Shen, Qiang Xu, Yi Cao and Wei Wang
Chemical Communications 2012 - vol. 48(Issue 32) pp:NaN3892-3892
Publication Date(Web):2012/02/24
DOI:10.1039/C2CC30531C
We report a genetically encoded fluorescent sensor for in vivo copper(I) imaging based on engineered intramolecular mechanical strain and structural distortion of EGFP.