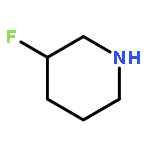
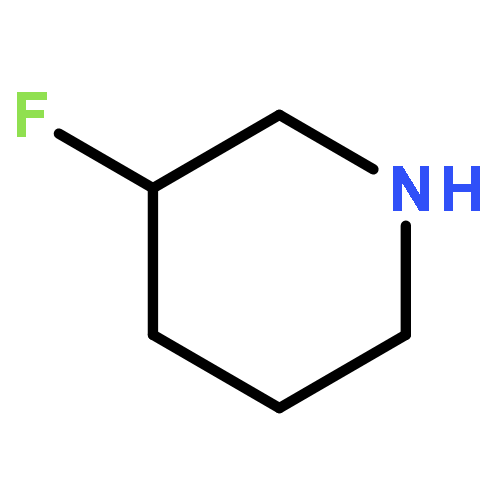
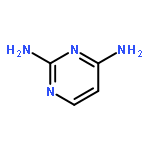
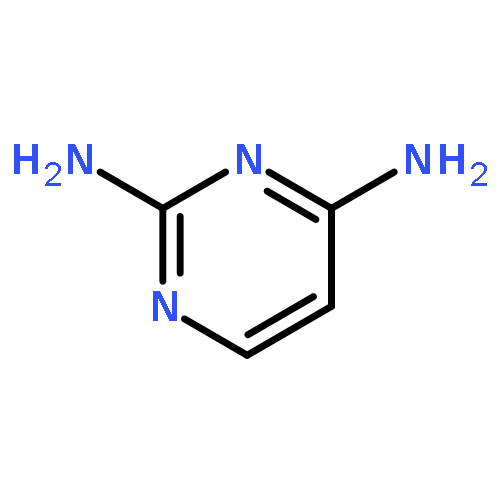
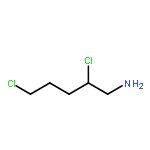
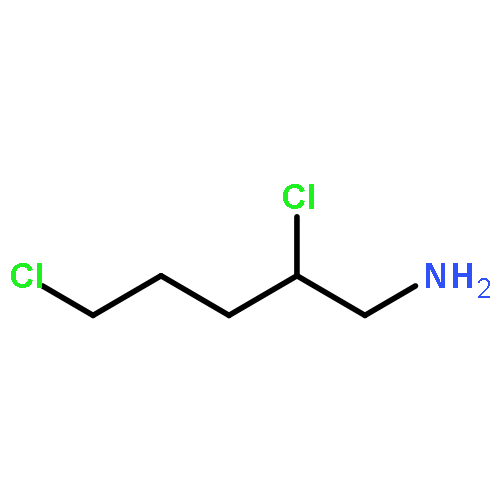
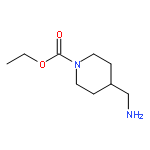
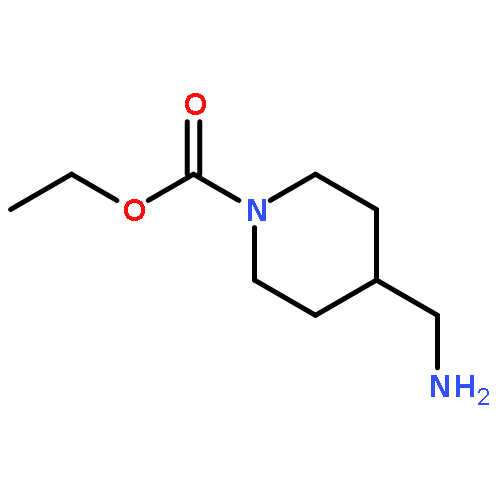
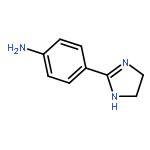
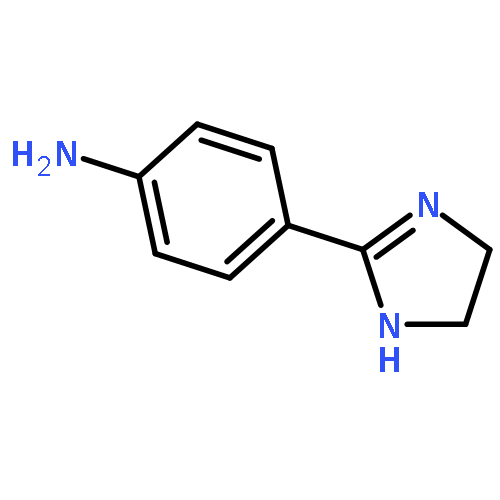
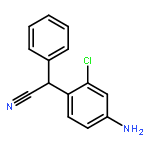
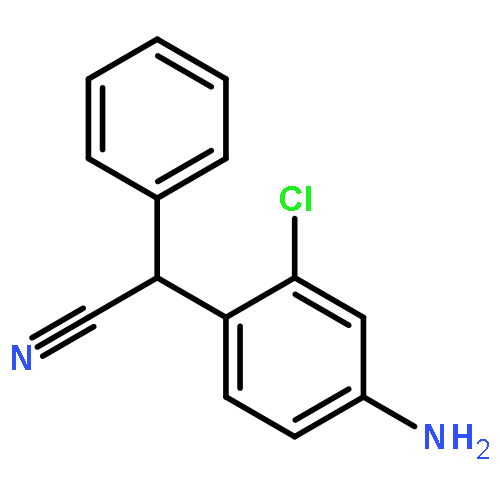
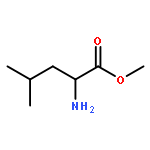
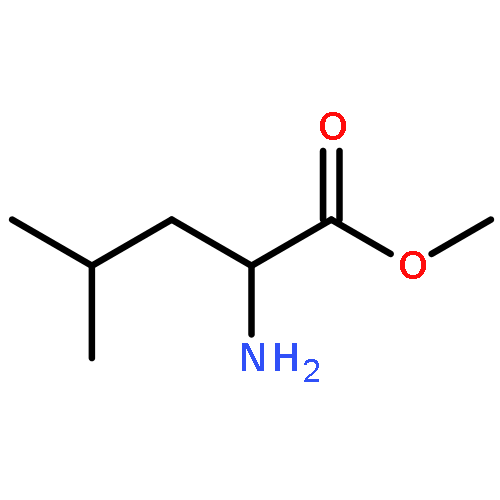
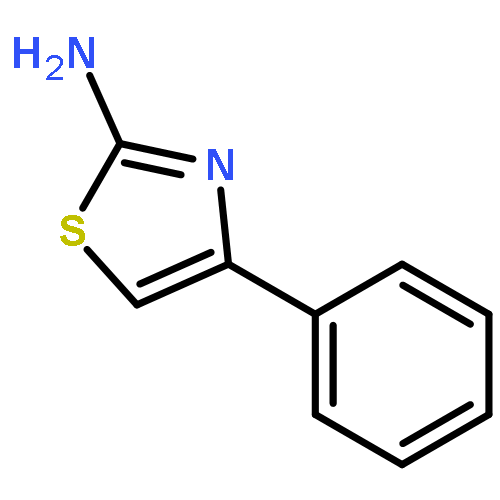
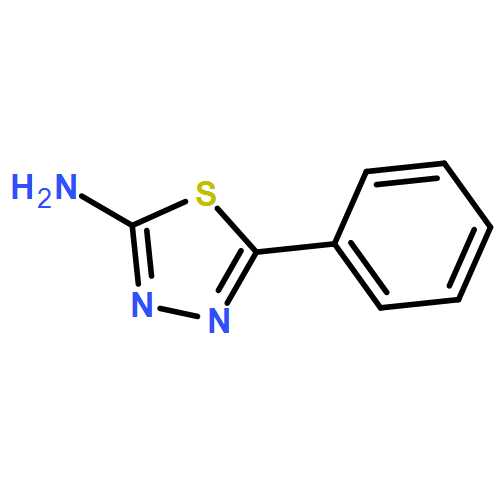
Co-reporter:Rongjun He, Li-Fan Zeng, Yantao He, Li Wu, Andrea Michelle Gunawan and Zhong-Yin Zhang
Chemical Communications 2013 vol. 49(Issue 20) pp:2064-2066
Publication Date(Web):24 Jan 2013
DOI:10.1039/C3CC38961H
Mycobacterium protein tyrosine phosphatase B (mPTPB) is essential for the survival and persistence of Mycobacterium in the host. Thus small molecule inhibitors of mPTPB are potential anti-TB agents. We developed an efficient organocatalytic multicomponent reaction (MCR) between pyrrole, formaldehyde and aniline, affording a potent and selective mPTPB inhibitor with an IC50 value of 1.5 μM and >50-fold specificity. Our studies provide a successful example of using organocatalysis as a discovery tool for the acquisition of PTP inhibitors.
Co-reporter:Yantao He ; Jie Xu ; Zhi-Hong Yu ; Andrea M. Gunawan ; Li Wu ; Lina Wang
Journal of Medicinal Chemistry 2013 Volume 56(Issue 3) pp:832-842
Publication Date(Web):January 10, 2013
DOI:10.1021/jm301781p
Mycobacterium tuberculosis (Mtb) protein tyrosine phosphatase B (mPTPB) is a virulence factor secreted by the pathogen and mediates mycobacterial survival in macrophages by targeting host cell immune responses. Consequently, mPTPB represents an exciting new target to combat tuberculosis (TB) infection. We describe a medicinal chemistry oriented approach that transforms a benzofuran salicylic acid scaffold into a highly potent (IC50 = 38 nM) and selective mPTPB inhibitor (>50 fold against a large panel of PTPs). Importantly, the inhibitor is capable of reversing the altered host immune responses induced by the bacterial phosphatase and restoring the macrophage’s full capacity to secrete IL-6 and undergo apoptosis in response to interferon-γ stimulation, validating the concept that chemical inhibition of mPTPB may be therapeutically useful for novel TB treatment. The study further demonstrates that bicyclic salicylic acid pharmacophores can be used to deliver PTP inhibitors with high potency, selectivity, and cellular efficacy.
Co-reporter:Dr. Li-Fan Zeng;Dr. Jie Xu;Dr. Yantao He;Dr. Rongjun He;Li Wu;Andrea M. Gunawan;Dr. Zhong-Yin Zhang
ChemMedChem 2013 Volume 8( Issue 6) pp:904-908
Publication Date(Web):
DOI:10.1002/cmdc.201300115
Co-reporter:Sheng Zhang ; Sijiu Liu ; Rongya Tao ; Dan Wei ; Lan Chen ; Weihua Shen ; Zhi-Hong Yu ; Lina Wang ; David R. Jones ; Xiaocheng C. Dong
Journal of the American Chemical Society 2012 Volume 134(Issue 43) pp:18116-18124
Publication Date(Web):October 17, 2012
DOI:10.1021/ja308212y
Protein tyrosine phosphatases (PTPs) constitute a large family of signaling enzymes that control the cellular levels of protein tyrosine phosphorylation. A detailed understanding of PTP functions in normal physiology and in pathogenic conditions has been hampered by the absence of PTP-specific, cell-permeable small-molecule agents. We present a stepwise focused library approach that transforms a weak and general non-hydrolyzable pTyr mimetic (F2Pmp, phosphonodifluoromethyl phenylalanine) into a highly potent and selective inhibitor of PTP-MEG2, an antagonist of hepatic insulin signaling. The crystal structures of the PTP-MEG2-inhibitor complexes provide direct evidence that potent and selective PTP inhibitors can be obtained by introducing molecular diversity into the F2Pmp scaffold to engage both the active site and unique nearby peripheral binding pockets. Importantly, the PTP-MEG2 inhibitor possesses highly efficacious cellular activity and is capable of augmenting insulin signaling and improving insulin sensitivity and glucose homeostasis in diet-induced obese mice. The results indicate that F2Pmp can be converted into highly potent and selective PTP inhibitory agents with excellent in vivo efficacy. Given the general nature of the approach, this strategy should be applicable to other members of the PTP superfamily.
Co-reporter:Yantao He, Li-Fan Zeng, Zhi-Hong Yu, Rongjun He, Sijiu Liu, Zhong-Yin Zhang
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 6) pp:1940-1946
Publication Date(Web):15 March 2012
DOI:10.1016/j.bmc.2011.11.004
Protein tyrosine phosphatases (PTPs) constitute a large and structurally diverse family of signaling enzymes that control the cellular levels of protein tyrosine phosphorylation. Malfunction of PTP activity has significant implications in many human diseases, and the PTP protein family provides an exciting array of validated diabetes/obesity (PTP1B), oncology (SHP2), autoimmunity (Lyp), and infectious disease (mPTPB) targets. However, despite the fact that PTPs have been garnering attention as novel therapeutic targets, they remain largely an untapped resource. The main challenges facing drug developers by the PTPs are inhibitor specificity and bioavailability. Work over the last ten years has demonstrated that it is feasible to develop potent and selective inhibitors for individual members of the PTP family by tethering together small ligands that can simultaneously occupy both the active site and unique nearby peripheral binding sites. Recent results with the bicyclic salicylic acid pharmacophores indicate that the new chemistry platform may provide a potential solution to overcome the bioavailability issue that has plagued the PTP drug discovery field for many years. Structural analysis of PTP-inhibitor complexes reveals molecular determinants important for the development of more potent and selective PTP inhibitors, thus offering hope in the medicinal chemistry of a largely unexploited protein class with a wealth of attractive drug targets.
Co-reporter:Sijiu Liu, Zhihong Yu, Xiao Yu, Sheng-Xiong Huang, Yinggang Luo, Li Wu, Weihua Shen, Zhenyun Yang, Lina Wang, Andrea M. Gunawan, Rebecca J. Chan, Ben Shen, Zhong-Yin Zhang
Chemistry & Biology 2011 Volume 18(Issue 1) pp:101-110
Publication Date(Web):28 January 2011
DOI:10.1016/j.chembiol.2010.10.015
SHP2 phosphatase is a positive transducer of growth factor and cytokine signaling. SHP2 is also a bona fide oncogene; gain-of-function SHP2 mutations leading to increased phosphatase activity cause Noonan syndrome, as well as multiple forms of leukemia and solid tumors. We report that tautomycetin (TTN), an immunosuppressor in organ transplantation, and its engineered analog TTN D-1 are potent SHP2 inhibitors. TTN and TTN D-1 block T cell receptor–mediated tyrosine phosphorylation and ERK activation and gain-of-function mutant SHP2-induced hematopoietic progenitor hyperproliferation and monocytic differentiation. Crystal structure of the SHP2⋅TTN D-1 complex reveals that TTN D-1 occupies the SHP2 active site in a manner similar to that of a peptide substrate. Collectively, the data support the notion that SHP2 is a cellular target for TTN and provide a potential mechanism for the immunosuppressive activity of TTN. Moreover, the structure furnishes molecular insights upon which therapeutics targeting SHP2 can be developed on the basis of the TTN scaffold.Highlights► Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2) is the cellular target for an immunosuppressant tautomycetin (TTN) ► TTN and its engineered analog TTN D-1 block SHP2-mediated signaling in T cell ► TTN and TTN D-1 activate SHP2-induced hematopoietic progenitor hyperproliferation and monocytic differentiation ► Structure of SHP2 in complex with TTN D-1 provides molecular blueprint for novel therapeutics development
Co-reporter:Leandro Piovan, Li Wu, Zhong-Yin Zhang and Leandro H. Andrade
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 5) pp:1347-1351
Publication Date(Web):14 Jan 2011
DOI:10.1039/C0OB01050B
A series of organochalcogenanes was synthesized and evaluated as proteintyrosine phosphatases (PTPs) inhibitors. The results indicate that organochalcogenanes inactivate the PTPs in a time- and concentration-dependent fashion, most likely through covalent modification of the active site sulfur-moiety by the chalcogen atom. Consequently, organochalcogenanes represent a new class of mechanism-based probes to modulate the PTP-mediated cellular processes.
Co-reporter:Zhi-Hong Yu, Lan Chen, Li Wu, Sijiu Liu, Lina Wang, Zhong-Yin Zhang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 14) pp:4238-4242
Publication Date(Web):15 July 2011
DOI:10.1016/j.bmcl.2011.05.078
SHP2, encoded by PTPN11, is a non-receptor protein tyrosine phosphatase (PTP) containing two tandem Src homology-2 (SH2) domains. It is expressed ubiquitously and plays critical roles in growth factor mediated processes, primarily by promoting the activation of the RAS/ERK signaling pathway. Genetic and biochemical studies have identified SHP2 as the first bona fide oncoprotein in the PTP superfamily, and a promising target for anti-cancer and anti-leukemia therapy. Here, we report a structure-based approach to identify SHP2 inhibitors with a novel scaffold. Through sequential virtual screenings and in vitro inhibition assays, a reversible competitive SHP2 inhibitor (C21) was identified. C21 is structurally distinct from all known SHP2 inhibitors. Combining molecular dynamics simulation and binding free energy calculation, a most likely binding mode of C21 with SHP2 is proposed, and further validated by site-directed mutagenesis and structure–activity relationship studies. This binding mode is consistent with the observed potency and specificity of C21, and reveals the molecular determinants for further optimization based on the new scaffold.Graphical abstract
Co-reporter:Xian Zhang ; Yantao He ; Sijiu Liu ; Zhihong Yu ; Zhong-Xing Jiang ; Zhenyun Yang ; Yuanshu Dong ; Sarah C. Nabinger ; Li Wu ; Andrea M. Gunawan ; Lina Wang ; Rebecca J. Chan
Journal of Medicinal Chemistry 2010 Volume 53(Issue 6) pp:2482-2493
Publication Date(Web):February 19, 2010
DOI:10.1021/jm901645u
The Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2) plays a pivotal role in growth factor and cytokine signaling. Gain-of-function SHP2 mutations are associated with Noonan syndrome, various kinds of leukemias, and solid tumors. Thus, there is considerable interest in SHP2 as a potential target for anticancer and antileukemia therapy. We report a salicylic acid based combinatorial library approach aimed at binding both active site and unique nearby subpockets for enhanced affinity and selectivity. Screening of the library led to the identification of a SHP2 inhibitor II-B08 (compound 9) with highly efficacious cellular activity. Compound 9 blocks growth factor stimulated ERK1/2 activation and hematopoietic progenitor proliferation, providing supporting evidence that chemical inhibition of SHP2 may be therapeutically useful for anticancer and antileukemia treatment. X-ray crystallographic analysis of the structure of SHP2 in complex with 9 reveals molecular determinants that can be exploited for the acquisition of more potent and selective SHP2 inhibitors.
Co-reporter:Lan Chen, Bo Zhou, Sheng Zhang, Li Wu, Yuehong Wang, Scott G. Franzblau, and Zhong-Yin Zhang
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 7) pp:355
Publication Date(Web):July 7, 2010
DOI:10.1021/ml1001135
Mycobacterium protein tyrosine phosphatase B (mPTPB) is an essential virulence factor required for Mycobacterium tuberculosis (Mtb) survival in host macrophages. Consequently, mPTPB represents an exciting new target with a completely novel mechanism of action. We screened a library of 7500 compounds against mPTPB and identified several 2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamide and piperazinyl-thiophenyl-ethyl-oxalamide derivatives as two distinct classes of mPTPB inhibitors. We showed that both classes of inhibitors are capable of blocking the mPTPB-mediated ERK1/2 inactivation. We further demonstrated that both classes of mPTPB inhibitors are effective in inhibiting the growth of Mtb in macrophages. Thus, improvement of the lead compounds may produce a novel class of anti-TB agents.Keywords (keywords): anti-TB agents; high throughput screening; mPTPB inhibitors; Mycobacterium tuberculosis; protein tyrosine phosphatase
Co-reporter:Bo Zhou;Jie Xu;Yantao He;Xian Zhang;Yong Luo;Yuehong Wang;Zhenyun Yang;Yan Liu;Scott G. Franzblau;Rebecca J. Chan;Jianyu Zheng
PNAS 2010 Volume 107 (Issue 10 ) pp:4573-4578
Publication Date(Web):2010-03-09
DOI:10.1073/pnas.0909133107
Protein tyrosine phosphatases are often exploited and subverted by pathogenic bacteria to cause human diseases. The tyrosine
phosphatase mPTPB from Mycobacterium tuberculosis is an essential virulence factor that is secreted by the bacterium into the cytoplasm of macrophages, where it mediates mycobacterial
survival in the host. Consequently, there is considerable interest in understanding the mechanism by which mPTPB evades the
host immune responses, and in developing potent and selective mPTPB inhibitors as unique antituberculosis (antiTB) agents.
We uncovered that mPTPB subverts the innate immune responses by blocking the ERK1/2 and p38 mediated IL-6 production and promoting
host cell survival by activating the Akt pathway. We identified a potent and selective mPTPB inhibitor I-A09 with highly efficacious
cellular activity, from a combinatorial library of bidentate benzofuran salicylic acid derivatives assembled by click chemistry.
We demonstrated that inhibition of mPTPB with I-A09 in macrophages reverses the altered host immune responses induced by the
bacterial phosphatase and prevents TB growth in host cells. The results provide the necessary proof-of-principle data to support
the notion that specific inhibitors of the mPTPB may serve as effective antiTB therapeutics.
Co-reporter:Dr. Rongjun He;Dr. Zhihong Yu;Dr. Yantao He;Dr. Li-Fan Zeng;Dr. Jie Xu;Li Wu;Andrea M. Gunawan;Lina Wang;Dr. Zhong-Xing Jiang;Dr. Zhong-Yin Zhang
ChemMedChem 2010 Volume 5( Issue 12) pp:2051-2056
Publication Date(Web):
DOI:10.1002/cmdc.201000348
Abstract
Tuberculosis (TB), which is caused by Mycobacterium tuberculosis (Mtb), is a major worldwide threat to public health. Mycobacterium protein tyrosine phosphatase B (mPTPB) is a virulent phosphatase secreted by Mtb, which is essential for the survival and persistence of the bacterium in the host. Consequently, small-molecule inhibitors of mPTPB are expected to serve as anti-TB agents with a novel mode of action. Herein, we report the discovery of highly potent and selective mPTPB inhibitors using a novel, double Click chemistry strategy. The most potent mPTPB inhibitor from this approach possesses a Ki value of 160 nM and a >25-fold selectivity for mPTPB over 19 other protein tyrosine phosphatases (PTBs). Molecular docking study of the enzyme–inhibitor complex provides a rationale for the high potency and selectivity of the lead compound and reveals an unusual binding mode, which may guide further optimization effort.
Co-reporter:Sheng Zhang ; Lan Chen ; Yong Luo ; Andrea Gunawan ; David S. Lawrence
Journal of the American Chemical Society 2009 Volume 131(Issue 36) pp:13072-13079
Publication Date(Web):August 19, 2009
DOI:10.1021/ja903733z
Protein tyrosine phosphatases (PTPs) regulate a broad range of cellular processes including proliferation, differentiation, migration, apoptosis, and immune responses. Dysfunction of PTP activity is associated with cancers, metabolic syndromes, and autoimmune disorders. Consequently, small molecule PTP inhibitors should serve not only as powerful tools to delineate the physiological roles of these enzymes in vivo but also as lead compounds for therapeutic development. We describe a novel stepwise fluorophore-tagged combinatorial library synthesis and competitive fluorescence polarization screening approach that transforms a weak and general PTP inhibitor into an extremely potent and selective TC-PTP inhibitor with highly efficacious cellular activity. The result serves as a proof-of-concept in PTP inhibitor development, as it demonstrates the feasibility of acquiring potent, yet highly selective, cell permeable PTP inhibitory agents. Given the general nature of the approach, this strategy should be applicable to other PTP targets.
Co-reporter:Yong Luo, Fubo Liang and Zhong-Yin Zhang
Biochemistry 2009 Volume 48(Issue 8) pp:
Publication Date(Web):February 6, 2009
DOI:10.1021/bi8020789
The PRL (phosphatase of regenerating liver) phosphatases represent a distinct class of protein tyrosine phosphatases, which are implicated in tumorigenesis and metastasis processes. Accumulating evidence indicates that alteration of PRL1 expression affects cell motility and tumor metastasis, although the biochemical pathways regulated by PRL1 remain less well defined. We find that elevated expression of PRL1 increases the levels of the matrix metalloproteinases MMP2 and MMP9. We have studied whether MMP2 and MMP9 are regulated by PRL1 and participate in PRL1-dependent cell migration and invasion. To this end, knockdown or inhibition of MMP2 and MMP9 by either siRNA or a specific small molecule inhibitor blocks PRL1-mediated cell migration and invasion. In addition, we report that upregulation of PRL1 activates the Src kinase through increased Tyr416 phosphorylation, which culminates in the phosphorylation of focal adhesion proteins FAK and p130Cas, as well as ERK1/2 activation. We provide evidence that both the Src and ERK1/2 pathways contribute to the increased motility of the PRL1 cells. We further demonstrate that Src and ERK1/2 activities are required for the PRL1-induced increase in the levels of MMP2 and MMP9, likely through activation of transcription factors AP1 and Sp1. Accordingly, increased PRL1 expression results in activation of Src and ERK1/2, which stimulates MMP2 and MMP9 production, leading to increased cell migration and invasion.
Co-reporter:Zhong-Xing Jiang
Cancer and Metastasis Reviews 2008 Volume 27( Issue 2) pp:263-272
Publication Date(Web):2008 June
DOI:10.1007/s10555-008-9113-3
Protein tyrosine phosphorylation plays a major role in cellular signaling. The level of tyrosine phosphorylation is controlled by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). Disturbance of the normal balance between PTK and PTP activity results in aberrant tyrosine phosphorylation, which has been linked to the etiology of several human diseases, including cancer. A number of PTPs have been implicated in oncogenesis and tumor progression and therefore are potential drug targets for cancer chemotherapy. These include PTP1B, which may augment signaling downstream of HER2/Neu; SHP2, which is the first oncogene in the PTP superfamily and is essential for growth factor-mediated signaling; the Cdc25 phosphatases, which are positive regulators of cell cycle progression; and the phosphatase of regenerating liver (PRL) phosphatases, which promote tumor metastases. As PTPs have emerged as drug targets for cancer, a number of strategies are currently been explored for the identification of various classes of PTP inhibitors. These efforts have resulted many potent, and in some cases selective, inhibitors for PTP1B, SHP2, Cdc25 and PRL phosphatases. Structural information derived from these compounds serves as a solid foundation upon which novel anti-cancer agents targeted to these PTPs can be developed.
Co-reporter:Fubo Liang, Sanjai Kumar and Zhong-Yin Zhang
Molecular BioSystems 2007 vol. 3(Issue 5) pp:308-316
Publication Date(Web):21 Mar 2007
DOI:10.1039/B700704N
Protein tyrosine phosphatases (PTPs) constitute a large family of enzymes that play key roles in cell signaling. Deregulation of PTP activity results in aberrant tyrosine phosphorylation, which has been linked to the etiology of several human diseases, including cancer. Since phosphate removal by the PTPs can both enhance and antagonize cellular signaling, it is essential to elucidate the physiological context in which PTPs operate. Two powerful proteomic approaches have been developed to rapidly establish the exact functional roles for every PTP, both in normal cellular physiology and in pathogenic conditions. In the first, an affinity-based substrate-trapping approach has been employed for PTP substrate identification. Identification and characterization of specific PTP–substrate interactions will associate functions with PTP as well as implicate PTP to specific signaling pathways. In the second, a number of activity-based PTP probes have been developed that can provide a direct readout of the functional state of the PTPs in complex proteomes. The ability to profile the entire PTP family on the basis of changes in their activity is expected to yield new functional insights into pathways regulated by the PTPs and contribute to the discovery of PTPs as novel therapeutic targets. Effective application of these proteomic techniques will accelerate the functional characterization of PTPs, thereby facilitating our understanding of PTPs in cell signaling and in diseases.
Co-reporter:Xiao Yu;Jin-Peng Sun;Sijiu Liu;Xiaoling Guo;Bo Zhou;Andy Hudmon;Yantao He
PNAS 2007 Volume 104 (Issue 50 ) pp:19767-19772
Publication Date(Web):2007-12-11
DOI:10.1073/pnas.0706233104
The lymphoid-specific tyrosine phosphatase (Lyp) has generated enormous interest because a single-nucleotide polymorphism
in the gene (PTPN22) encoding Lyp produces a gain-of-function mutant phosphatase that is associated with several autoimmune diseases, including
type I diabetes, rheumatoid arthritis, Graves disease, and systemic lupus erythematosus. Thus, Lyp represents a potential
target for a broad spectrum of autoimmune disorders. Unfortunately, no Lyp inhibitor has been reported. In addition, little
is known about the structure and biochemical mechanism that directly regulates Lyp function. Here, we report the identification
of a bidentate salicylic acid-based Lyp inhibitor I-C11 with excellent cellular efficacy. Structural and mutational analyses
indicate that the inhibitor binds both the active site and a nearby peripheral site unique to Lyp, thereby furnishing a solid
foundation upon which inhibitors with therapeutic potency and selectivity can be developed. Moreover, a comparison of the
apo- and inhibitor-bound Lyp structures reveals that the Lyp-specific region S35TKYKADK42, which harbors a PKC phosphorylation site, could adopt either a loop or helical conformation. We show that Lyp is phosphorylated
exclusively at Ser-35 by PKC both in vitro and in vivo. We provide evidence that the status of Ser-35 phosphorylation may dictate the conformational state of the insert region
and thus Lyp substrate recognition. We demonstrate that Ser-35 phosphorylation impairs Lyp's ability to inactivate the Src
family kinases and down-regulate T cell receptor signaling. Our data establish a mechanism by which PKC could attenuate the
cellular function of Lyp, thereby augmenting T cell activation.
Co-reporter:Sijiu Liu;Jin-Peng Sun;Bo Zhou;
Proceedings of the National Academy of Sciences 2006 103(14) pp:5326-5331
Publication Date(Web):March 27, 2006
DOI:10.1073/pnas.0510506103
Mitogen-activated protein (MAP) kinases are central components of signal transduction pathways for cell proliferation, stress
responses, and differentiation. Signaling efficiency and specificity are modulated in large part by docking interactions between
individual MAP kinase and the kinase interaction motif (KIM), (R/K)2–3-X1–6-ΦA-X-ΦB, in its cognate kinases, phosphatases, scaffolding proteins, and substrates. We have determined the crystal structure of
extracellular signal-regulated protein kinase 2 bound to the KIM peptide from MAP kinase phosphatase 3, an extracellular signal-regulated
protein kinase 2-specific phosphatase. The structure reveals that the KIM docking site, situated in a noncatalytic region
opposite of the kinase catalytic pocket, is comprised of a highly acidic patch and a hydrophobic groove, which engage the
basic and ΦA-X-ΦB residues, respectively, in the KIM sequence. The specific docking interactions observed in the structure consolidate all
known biochemical data. In addition, structural comparison indicates that the KIM docking site is conserved in all MAP kinases.
The results establish a structural model for understanding how MAP kinases interact with their regulators and substrates and
provide new insights into how MAP kinase docking specificity can be achieved.
Co-reporter:Sheng Zhang, Zhong-Yin Zhang
Drug Discovery Today (May 2007) Volume 12(Issues 9–10) pp:373-381
Publication Date(Web):1 May 2007
DOI:10.1016/j.drudis.2007.03.011
Protein tyrosine phosphatase 1B (PTP1B) is an effective target for the treatment of both type 2 diabetes and obesity; however, targeting PTP1B for drug discovery is challenging because of the highly conserved and positively charged active-site pocket. Tremendous progress has been made in the development of potent and selective PTP1B inhibitors that engage both the active site and no catalytic sites. Several strategies are being pursued to improve the pharmacological properties of PTP1B inhibitors. These new developments suggest that it is feasible to acquire PTP1B-based, small-molecule therapeutics with the requisite potency and selectivity. Future efforts will probably transform the potent and selective PTP1B inhibitors into orally available drugs with desirable physicochemical properties and in vivo efficacies.
Co-reporter:Leandro Piovan, Li Wu, Zhong-Yin Zhang and Leandro H. Andrade
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 5) pp:NaN1351-1351
Publication Date(Web):2011/01/14
DOI:10.1039/C0OB01050B
A series of organochalcogenanes was synthesized and evaluated as proteintyrosine phosphatases (PTPs) inhibitors. The results indicate that organochalcogenanes inactivate the PTPs in a time- and concentration-dependent fashion, most likely through covalent modification of the active site sulfur-moiety by the chalcogen atom. Consequently, organochalcogenanes represent a new class of mechanism-based probes to modulate the PTP-mediated cellular processes.
Co-reporter:Rongjun He, Li-Fan Zeng, Yantao He, Li Wu, Andrea Michelle Gunawan and Zhong-Yin Zhang
Chemical Communications 2013 - vol. 49(Issue 20) pp:NaN2066-2066
Publication Date(Web):2013/01/24
DOI:10.1039/C3CC38961H
Mycobacterium protein tyrosine phosphatase B (mPTPB) is essential for the survival and persistence of Mycobacterium in the host. Thus small molecule inhibitors of mPTPB are potential anti-TB agents. We developed an efficient organocatalytic multicomponent reaction (MCR) between pyrrole, formaldehyde and aniline, affording a potent and selective mPTPB inhibitor with an IC50 value of 1.5 μM and >50-fold specificity. Our studies provide a successful example of using organocatalysis as a discovery tool for the acquisition of PTP inhibitors.