Co-reporter:Junwei Wang, Qiao Song, Anhua Xu, Yu Bao, Yungen Xu, Qihua Zhu
European Journal of Medicinal Chemistry 2017 Volume 130(Volume 130) pp:
Publication Date(Web):21 April 2017
DOI:10.1016/j.ejmech.2017.02.029
Opioid receptors play an important role in both behavioral and mood functions. Based on the structural modification of LY2456302, a series of aminobenzyloxyarylamide derivatives were designed and synthesized as κ opioid receptor antagonists. The κ opioid receptor binding ability of these compounds were evaluated with opioid receptors binding assays. Compounds 1a-d showed high affinity for κ opioid receptor. Especially for compound 1c, exhibited a significant Ki value of 15.7 nM for κ opioid receptor binding and a higher selectivity over μ and δ opioid receptors compared to (±)LY2456302. In addition, compound 1c also showed potent κ antagonist activity with κ IC50 = 9.32 nM in [35S]GTP-γ-S functional assay. The potential use of the representative compounds as antidepressants was also investigated. The most potent compound 1c not only exhibited potent antidepressant activity in the mice forced swimming test, but also displayed the effect of anti-anxiety in the elevated plus-maze test.Download high-res image (188KB)Download full-size image
Co-reporter:Fan-Wei Peng, Ji Xuan, Ting-Ting Wu, Jia-Yu Xue, Zi-Wei Ren, Da-Ke Liu, Xiu-Qi Wang, Xin-Hang Chen, Jia-Wei Zhang, Yun-Gen Xu, Lei Shi
European Journal of Medicinal Chemistry 2016 Volume 109() pp:1-12
Publication Date(Web):15 February 2016
DOI:10.1016/j.ejmech.2015.12.033
•24 Novel hybrids were synthesized as dual VEGFR-2/HADC inhibitors.•The in vitro enzymatic and cellular activities were evaluated.•The primary structure–activity relationships were discussed.•Compound 6fd exhibited the most potent biological activities.A single agent that simultaneously inhibits multiple targets may offer greater therapeutic benefits in cancer than single-acting agents through interference with multiple pathways and potential synergistic action. In this work, a series of hybrids bearing N-phenylquinazolin-4-amine and hydroxamic acid moieties were designed and identified as dual VEGFR-2/HDAC inhibitors. Compound 6fd exhibited the most potent inhibitory activity against HDAC with IC50 of 2.2 nM and strong inhibitory effect against VEGFR-2 with IC50 of 74 nM. It also showed the most potent inhibitory activity against a human breast cancer cell line MCF-7 with IC50 of 0.85 μM. Docking simulation supported the initial pharmacophoric hypothesis and suggested a common mode of interaction at the active binding sites of VEGFR-2 and HDLP ((Histone Deacetylase-Like Protein), which demonstrates that compound 6fd is a potential agent for cancer therapy deserving further researching.
Co-reporter:Chen Zhou;Di Kang;Yungen Xu;Luyong Zhang;Xiaoming Zha
Chemical Biology & Drug Design 2015 Volume 85( Issue 6) pp:659-671
Publication Date(Web):
DOI:10.1111/cbdd.12461
Lysine-specific demethylase 1 (LSD1) plays an important role in regulating the lysine methylation at residues K4 and K9 on histone H3. High levels of LSD1 expression have been observed in several malignant tumors. In this study, we presented a pharmacophore-based virtual screening of a moderate database of 171 143 small molecules. A pharmacophore of LSD1 inhibitors was constructed for the first time and then used to screen the compound library combined with validated molecular docking tools followed by biochemical assays, led to the identification of 9 novel LSD1 inhibitors, showing their IC50 values in a range of 2.41–101 μm. Furthermore, compound XZ09 exhibited less inhibition against the homologous monoamine oxidase A (MAO-A) and B (MAO-B) displaying its moderate selectivity. Our study provides an effective virtual screening method to identify new LSD1 inhibitors and XZ09 represents a potent and selective lead compound to deserve further optimization for the treatment of LSD1 overexpressing cancers.
Co-reporter:Qihua Zhu, Junwei Wang, Xueguo Bian, Lingzhi Zhang, Ping Wei, and Yungen Xu
Organic Process Research & Development 2015 Volume 19(Issue 9) pp:1263-1267
Publication Date(Web):August 14, 2015
DOI:10.1021/acs.oprd.5b00144
A novel synthesis of antiobesity drug lorcaserin hydrochloride was accomplished in six steps. N-protection of 2-(4-chlorophenyl)ethanamine with di-tert-butyl dicarbonate, N-alkylation with allyl bromide, deprotection, intramolecular Friedel–Crafts alkylation, chiral resolution with l-(+)-tartaric acid, and the final salification led to the target molecule lorcaserin hydrochloride in 23.1% overall yield with 99.9% purity and excellent enantioselectivity (>99.8% ee). This convenient and economical procedure is remarkably applicable for scale-up production.
Co-reporter:Haiping Zhou;Qihua Zhu;Zongjie Gan;Guangping Dong
Medicinal Chemistry Research 2015 Volume 24( Issue 11) pp:3920-3931
Publication Date(Web):2015 November
DOI:10.1007/s00044-015-1431-8
DP antagonists are claimed to be useful in the treatment of allergic disorders. Laropiprant is a potent and selective DP antagonist to reduce the allergic disorders, especially niacin-induced flushing. In our study, a series of novel laropiprant derivatives (I-1–I-18) were synthesized and characterized by IR, 1H-NMR, 13C-NMR and HRMS spectrum. The potency of these compounds was evaluated in a murine model of niacin-induced flushing. The results indicated that most compounds exhibited faster-acting effect of suppressing vasodilation than laropiprant. Among them, I-1, I-2, I-3, I-9, I-13, I-15 and I-16 exhibited substantial flushing inhibitory effect. Especially, I-1 and I-2 showed higher potency than laropiprant and would be valuable for further investigation.
Co-reporter:Qiao Song, Dongmei Zhang, Qihua Zhu, and Yungen Xu
Organic Letters 2014 Volume 16(Issue 20) pp:5272-5274
Publication Date(Web):September 29, 2014
DOI:10.1021/ol502370r
p-Toluenesulfonohydrazide (PTSH) was shown to promote the highly efficient direct arylation of unactivated arenes with aryl iodides, bromides, or chlorides in the presence of potassium tert-butoxide without the assistance of any transition metals. The reaction proceeds through base-promoted homolytic aromatic substitution (BHAS) involving aryl radicals and arylradical anions as intermediates.
Co-reporter:Lei Shi, Ting-Ting Wu, Zhi Wang, Jia-Yu Xue, Yun-Gen Xu
European Journal of Medicinal Chemistry 2014 Volume 84() pp:698-707
Publication Date(Web):12 September 2014
DOI:10.1016/j.ejmech.2014.07.071
•21 Novel quinolin-4-amines containing benzimidazole moiety were synthesized.•The anticancer activities and inhibitory activities of VEGFR-2 were evaluated.•The primary structure–activity relationships were discussed.•Compound 7s exhibited the most potent inhibitory activities against VEGFR-2.•Compound 7s also showed potent anticancer activities against MCF-7 and Hep-G2.Inhibition of the VEGF signaling pathway has become a valuable approach in the treatment of cancers. In this work, a series of N-(2-phenyl-1H-benzo[d]imidazol-5-yl)quinolin-4-amine derivatives were designed and identified as potent inhibitors of VEGFR-2 (KDR) kinase. These compounds with quinoline scaffold and benzimidazole moiety were synthesized and their biological activities against VEGFR-2 and two human cancer cell lines were evaluated. Among them, compound 7s exhibited the most potent inhibitory activity against VEGFR-2 with IC50 of 0.03 μM and it also showed the highest anticancer activity against the tested cancer cell lines with IC50 of 1.2 μM against MCF-7 and 13.3 μM against Hep-G2. Docking simulation supported the initial pharmacophoric hypothesis and suggested a common mode of interaction at the ATP-binding site of VEGFR-2, which demonstrates that compound 7s is a potential agent for cancer therapy deserving further researching.A series of quinolin-4-amine derivatives containing benzimidazole moiety were discovered as potent inhibitors of VEGFR-2. Compound 7s exhibited the most potent inhibitory activity with IC50 of 0.03 μM against VEGFR-2.
Co-reporter:Lei Shi, Ting-Ting Wu, Zhi Wang, Jia-Yu Xue, Yun-Gen Xu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 17) pp:4735-4744
Publication Date(Web):1 September 2014
DOI:10.1016/j.bmc.2014.07.008
Both c-Met and VEGFR-2 are important targets for the treatment of cancers. In this study, a series of N-(2-phenyl-1H-benzo[d]imidazol-5-yl)quinazolin-4-amine derivatives were designed and identified as dual c-Met and VEGFR-2 inhibitors. Among these compounds bearing quinazoline and benzimidazole fragments, compound 7j exhibited the most potent inhibitory activity against c-Met and VEGFR-2 with IC50 of 0.05 μM and 0.02 μM, respectively. It also showed the highest anticancer activity against the tested cancer cell lines with IC50 of 1.5 μM against MCF-7 and 8.7 μM against Hep-G2. Docking simulation supported the initial pharmacophoric hypothesis and suggested a common mode of interaction at the ATP-binding site of c-Met and VEGFR-2, which demonstrates that compound 7j is a potential agent for cancer therapy deserving further researching.A series of quinazolin-4-amine derivatives containing benzimidazole moiety were discovered as potent dual inhibitors of c-Met and VEGFR-2. Compound 7j exhibited the most potent inhibitory activities with IC50 of 0.05 μM and 0.02 μM against c-Met and VEGFR-2, respectively.
Co-reporter:Kun Liu, Shuowei Yao, Yinghan Lou, Yungen Xu
Carbohydrate Research 2013 Volume 381() pp:83-92
Publication Date(Web):15 November 2013
DOI:10.1016/j.carres.2013.08.022
•Nineteen N-(3-aryl acryloyl)aminosaccharide derivatives were synthesized.•Their anti-angiogenetic activity was evaluated by MTT assay and CAM assay.•Compound 10s and topotecan had comparable effects on anti-angiogenesis.•Electron-withdrawing group and disaccharide structure are beneficial.In order to find novel potent inhibitors for signal pathways of FGF/FGFR, nineteen N-(3-aryl acryloyl)aminosaccharide derivatives were designed and synthesized based on the binding sites of FGF and oligosaccharides of heparin. Their structures were confirmed by IR, 1H NMR, 13C NMR, MS and elemental analysis. The nineteen target compounds were evaluated for biological activity against HUVEC cell. In vitro assays showed that compound 10s (IC50 = 5.3 μM) exhibited comparable inhibitory effects on endothelial cell growth with topotecan (IC50 = 2.7 μM). Compound 10s (10 μg/egg) also showed obvious anti-angiogenetic activity in the in vivo chicken chorio allantoic membrane (CAM) assay, and the potency was similar to topotecan (10 μg/egg).
Co-reporter:Haiping Zhou;Huiting Su;Zongjie Gan;Dechuan Wang
Research on Chemical Intermediates 2013 Volume 39( Issue 9) pp:4091-4098
Publication Date(Web):2013 November
DOI:10.1007/s11164-012-0925-y
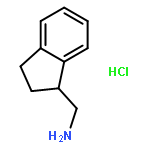
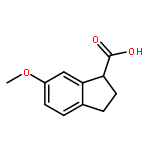
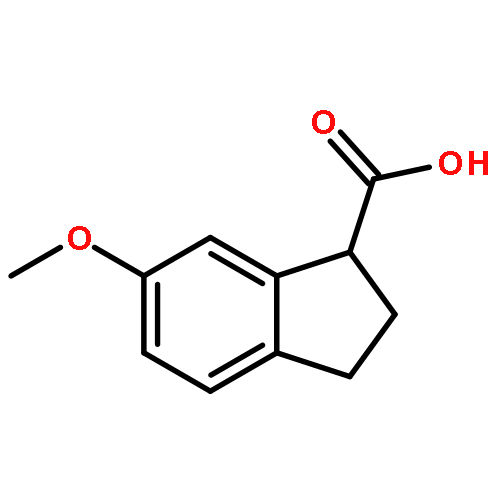
An efficient synthetic method for 2, 3-dihydro-1H-indene-1-methanamine and its derivatives is described. Six compounds of 2, 3-dihydro-1H-indene-1-methanamines were synthesized from corresponding 4-nitro-3-phenylbutanoic acid in satisfactory yields (50.9–57.9 % in 4 steps), and three compounds (1c, 1e, 1f) were newly reported. This procedure has the advantages of mild conditions, less pollution, and simple manipulation.


![2-[[(3,5,6-TRIMETHYL-2-PYRAZINYL)METHYL]AMINO]BENZOIC ACID ETHYL ESTER](http://img.cochemist.com/ccimg/947300/947243-98-3.png)
![2-[[(3,5,6-TRIMETHYL-2-PYRAZINYL)METHYL]AMINO]BENZOIC ACID ETHYL ESTER](http://img.cochemist.com/ccimg/947300/947243-98-3_b.png)
![3-NITRO-4-[[(3,5,6-TRIMETHYL-2-PYRAZINYL)METHYL]AMINO]BENZOIC ACID ETHYL ESTER](http://img.cochemist.com/ccimg/947300/947243-97-2.png)
![3-NITRO-4-[[(3,5,6-TRIMETHYL-2-PYRAZINYL)METHYL]AMINO]BENZOIC ACID ETHYL ESTER](http://img.cochemist.com/ccimg/947300/947243-97-2_b.png)
![4-[[(3,5,6-TRIMETHYL-2-PYRAZINYL)METHYL]AMINO]BENZOIC ACID ETHYL ESTER](http://img.cochemist.com/ccimg/947300/947243-93-8.png)
![4-[[(3,5,6-TRIMETHYL-2-PYRAZINYL)METHYL]AMINO]BENZOIC ACID ETHYL ESTER](http://img.cochemist.com/ccimg/947300/947243-93-8_b.png)
![BENZOIC ACID, 2-[(3,5,6-TRIMETHYL-2-PYRAZINYL)METHOXY]-, ETHYL ESTER](http://img.cochemist.com/ccimg/947300/947243-90-5.png)
![BENZOIC ACID, 2-[(3,5,6-TRIMETHYL-2-PYRAZINYL)METHOXY]-, ETHYL ESTER](http://img.cochemist.com/ccimg/947300/947243-90-5_b.png)
![4-[(3,5,6-TRIMETHYL-2-PYRAZINYL)METHOXY]BENZOIC ACID ETHYL ESTER](http://img.cochemist.com/ccimg/947300/947243-80-3.png)
![4-[(3,5,6-TRIMETHYL-2-PYRAZINYL)METHOXY]BENZOIC ACID ETHYL ESTER](http://img.cochemist.com/ccimg/947300/947243-80-3_b.png)


![THIOUREA, N-[4-(1H-BENZIMIDAZOL-2-YLTHIO)PHENYL]-N'-ETHYL-](http://img.cochemist.com/ccimg/883500/883452-97-9.png)
![THIOUREA, N-[4-(1H-BENZIMIDAZOL-2-YLTHIO)PHENYL]-N'-ETHYL-](http://img.cochemist.com/ccimg/883500/883452-97-9_b.png)
![ACETAMIDE, N-[4-[[[(2-AMINOPHENYL)AMINO]THIOXOMETHYL]AMINO]PHENYL]-](http://img.cochemist.com/ccimg/865600/865526-59-6.png)
![ACETAMIDE, N-[4-[[[(2-AMINOPHENYL)AMINO]THIOXOMETHYL]AMINO]PHENYL]-](http://img.cochemist.com/ccimg/865600/865526-59-6_b.png)
![Acetamide, N-[4-(1H-benzimidazol-2-ylamino)phenyl]-](http://img.cochemist.com/ccimg/865600/865526-58-5.png)
![Acetamide, N-[4-(1H-benzimidazol-2-ylamino)phenyl]-](http://img.cochemist.com/ccimg/865600/865526-58-5_b.png)


![Thiourea, N-[4-(2-benzothiazolylthio)phenyl]-N'-propyl-](http://img.cochemist.com/ccimg/849500/849438-68-2.png)
![Thiourea, N-[4-(2-benzothiazolylthio)phenyl]-N'-propyl-](http://img.cochemist.com/ccimg/849500/849438-68-2_b.png)



![1H-Benzimidazole, 5-methoxy-2-[(4-nitrophenyl)thio]-](http://img.cochemist.com/ccimg/697900/697800-29-6.png)
![1H-Benzimidazole, 5-methoxy-2-[(4-nitrophenyl)thio]-](http://img.cochemist.com/ccimg/697900/697800-29-6_b.png)
![Benzenamine, 4-[(5-chloro-1H-benzimidazol-2-yl)thio]-](http://img.cochemist.com/ccimg/697900/697800-28-5.png)
![Benzenamine, 4-[(5-chloro-1H-benzimidazol-2-yl)thio]-](http://img.cochemist.com/ccimg/697900/697800-28-5_b.png)
![Benzenamine, 4-[(5-methoxy-1H-benzimidazol-2-yl)thio]-](http://img.cochemist.com/ccimg/697900/697800-27-4.png)
![Benzenamine, 4-[(5-methoxy-1H-benzimidazol-2-yl)thio]-](http://img.cochemist.com/ccimg/697900/697800-27-4_b.png)
![Thiourea, N-[4-[(5-chloro-1H-benzimidazol-2-yl)thio]phenyl]-N'-propyl-](http://img.cochemist.com/ccimg/697900/697800-26-3.png)
![Thiourea, N-[4-[(5-chloro-1H-benzimidazol-2-yl)thio]phenyl]-N'-propyl-](http://img.cochemist.com/ccimg/697900/697800-26-3_b.png)
![Thiourea, N-[4-[(5-chloro-1H-benzimidazol-2-yl)thio]phenyl]-N'-methyl-](http://img.cochemist.com/ccimg/697900/697800-25-2.png)
![Thiourea, N-[4-[(5-chloro-1H-benzimidazol-2-yl)thio]phenyl]-N'-methyl-](http://img.cochemist.com/ccimg/697900/697800-25-2_b.png)
![Thiourea,N-[4-[(5-methoxy-1H-benzimidazol-2-yl)thio]phenyl]-N'-phenyl-](http://img.cochemist.com/ccimg/697900/697800-24-1.png)
![Thiourea,N-[4-[(5-methoxy-1H-benzimidazol-2-yl)thio]phenyl]-N'-phenyl-](http://img.cochemist.com/ccimg/697900/697800-24-1_b.png)
![Thiourea, N-[4-[(5-methoxy-1H-benzimidazol-2-yl)thio]phenyl]-N'-propyl-](http://img.cochemist.com/ccimg/697900/697800-23-0.png)
![Thiourea, N-[4-[(5-methoxy-1H-benzimidazol-2-yl)thio]phenyl]-N'-propyl-](http://img.cochemist.com/ccimg/697900/697800-23-0_b.png)
![Thiourea,N-[4-[(5-methoxy-1H-benzimidazol-2-yl)thio]phenyl]-N'-methyl-](http://img.cochemist.com/ccimg/697900/697800-22-9.png)
![Thiourea,N-[4-[(5-methoxy-1H-benzimidazol-2-yl)thio]phenyl]-N'-methyl-](http://img.cochemist.com/ccimg/697900/697800-22-9_b.png)
![THIOUREA, N-[2-(1,3-BENZODIOXOL-5-YL)ETHYL]-N'-METHYL-](http://img.cochemist.com/ccimg/696600/696587-76-5.png)
![THIOUREA, N-[2-(1,3-BENZODIOXOL-5-YL)ETHYL]-N'-METHYL-](http://img.cochemist.com/ccimg/696600/696587-76-5_b.png)
![Thiourea, [2-(1,3-benzodioxol-5-yl)ethyl]-](http://img.cochemist.com/ccimg/696600/696587-75-4.png)
![Thiourea, [2-(1,3-benzodioxol-5-yl)ethyl]-](http://img.cochemist.com/ccimg/696600/696587-75-4_b.png)
![Thiourea, N-[4-(1H-benzimidazol-2-ylthio)phenyl]-N'-phenyl-](http://img.cochemist.com/ccimg/683800/683755-37-5.png)
![Thiourea, N-[4-(1H-benzimidazol-2-ylthio)phenyl]-N'-phenyl-](http://img.cochemist.com/ccimg/683800/683755-37-5_b.png)
![Thiourea, N-[4-(1H-benzimidazol-2-ylthio)phenyl]-N'-propyl-](http://img.cochemist.com/ccimg/683800/683755-36-4.png)
![Thiourea, N-[4-(1H-benzimidazol-2-ylthio)phenyl]-N'-propyl-](http://img.cochemist.com/ccimg/683800/683755-36-4_b.png)
![Thiourea, N-[4-(1H-benzimidazol-2-ylthio)phenyl]-N'-methyl-](http://img.cochemist.com/ccimg/683800/683755-35-3.png)
![Thiourea, N-[4-(1H-benzimidazol-2-ylthio)phenyl]-N'-methyl-](http://img.cochemist.com/ccimg/683800/683755-35-3_b.png)
![Thiourea, [4-(1H-benzimidazol-2-ylthio)phenyl]-](http://img.cochemist.com/ccimg/683800/683755-24-0.png)
![Thiourea, [4-(1H-benzimidazol-2-ylthio)phenyl]-](http://img.cochemist.com/ccimg/683800/683755-24-0_b.png)
![Benzamide,N-[[[4-(1H-benzimidazol-2-ylthio)phenyl]amino]thioxomethyl]-](http://img.cochemist.com/ccimg/683800/683755-23-9.png)
![Benzamide,N-[[[4-(1H-benzimidazol-2-ylthio)phenyl]amino]thioxomethyl]-](http://img.cochemist.com/ccimg/683800/683755-23-9_b.png)






![Benzenesulfonamide, 4-methoxy-N-[(4-methoxyphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/185200/185117-44-6.png)
![Benzenesulfonamide, 4-methoxy-N-[(4-methoxyphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/185200/185117-44-6_b.png)
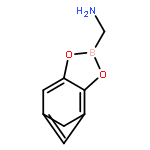
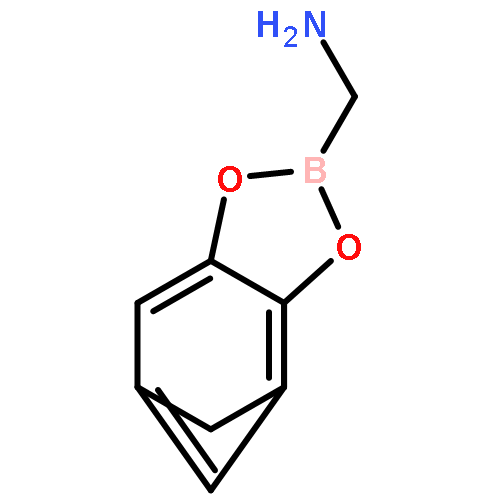





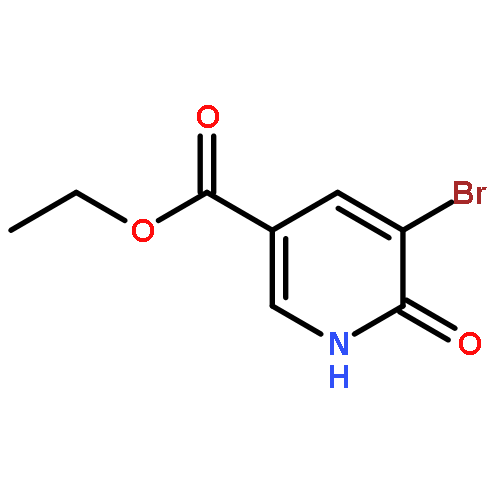
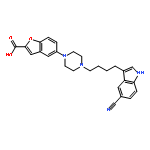
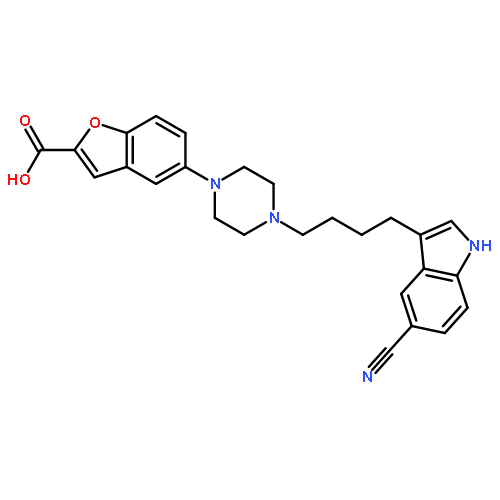
-](http://img.cochemist.com/ccimg/160700/160603-61-2.png)
-](http://img.cochemist.com/ccimg/160700/160603-61-2_b.png)
-](http://img.cochemist.com/ccimg/160700/160603-60-1.png)
-](http://img.cochemist.com/ccimg/160700/160603-60-1_b.png)
-](http://img.cochemist.com/ccimg/160700/160603-59-8.png)
-](http://img.cochemist.com/ccimg/160700/160603-59-8_b.png)
-](http://img.cochemist.com/ccimg/160700/160603-58-7.png)
-](http://img.cochemist.com/ccimg/160700/160603-58-7_b.png)
![Methanone,[3-[[bis(2-methylpropyl)amino]methyl]-4-hydroxy-5-methylphenyl](2-methyl-1H-indol-3-yl)-](http://img.cochemist.com/ccimg/160700/160603-57-6.png)
![Methanone,[3-[[bis(2-methylpropyl)amino]methyl]-4-hydroxy-5-methylphenyl](2-methyl-1H-indol-3-yl)-](http://img.cochemist.com/ccimg/160700/160603-57-6_b.png)
![Methanone,[3,5-bis[(diethylamino)methyl]-4-hydroxyphenyl](2-methyl-1H-indol-3-yl)-](http://img.cochemist.com/ccimg/160700/160603-55-4.png)
![Methanone,[3,5-bis[(diethylamino)methyl]-4-hydroxyphenyl](2-methyl-1H-indol-3-yl)-](http://img.cochemist.com/ccimg/160700/160603-55-4_b.png)
-](http://img.cochemist.com/ccimg/160700/160603-53-2.png)
-](http://img.cochemist.com/ccimg/160700/160603-53-2_b.png)
-](http://img.cochemist.com/ccimg/160700/160603-52-1.png)
-](http://img.cochemist.com/ccimg/160700/160603-52-1_b.png)
![Methanone,[3,5-bis[[bis(2-methylpropyl)amino]methyl]-4-hydroxyphenyl]-1H-indol-3-yl-](http://img.cochemist.com/ccimg/160700/160603-51-0.png)
![Methanone,[3,5-bis[[bis(2-methylpropyl)amino]methyl]-4-hydroxyphenyl]-1H-indol-3-yl-](http://img.cochemist.com/ccimg/160700/160603-51-0_b.png)
![Methanone,[4-hydroxy-3,5-bis(1-piperidinylmethyl)phenyl]-1H-indol-3-yl-](http://img.cochemist.com/ccimg/160700/160603-48-5.png)
![Methanone,[4-hydroxy-3,5-bis(1-piperidinylmethyl)phenyl]-1H-indol-3-yl-](http://img.cochemist.com/ccimg/160700/160603-48-5_b.png)
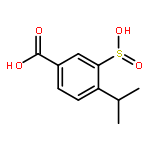



![Methanone, [3,5-diiodo-4-[2-(1-piperidinyl)ethoxy]phenyl]-1H-indol-3-yl-](http://img.cochemist.com/ccimg/138300/138222-07-8.png)
![Methanone, [3,5-diiodo-4-[2-(1-piperidinyl)ethoxy]phenyl]-1H-indol-3-yl-](http://img.cochemist.com/ccimg/138300/138222-07-8_b.png)
![METHANONE, [4-[2-(DIETHYLAMINO)ETHOXY]-3,5-DIIODOPHENYL]-1H-INDOL-3-YL-](http://img.cochemist.com/ccimg/138300/138222-06-7.png)
![METHANONE, [4-[2-(DIETHYLAMINO)ETHOXY]-3,5-DIIODOPHENYL]-1H-INDOL-3-YL-](http://img.cochemist.com/ccimg/138300/138222-06-7_b.png)
![[3,5-DIIODO-4-(2-PYRROLIDIN-1-YLETHOXY)PHENYL]-(1H-INDOL-3-YL)METHANONE](http://img.cochemist.com/ccimg/138300/138222-03-4.png)
![[3,5-DIIODO-4-(2-PYRROLIDIN-1-YLETHOXY)PHENYL]-(1H-INDOL-3-YL)METHANONE](http://img.cochemist.com/ccimg/138300/138222-03-4_b.png)
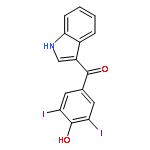



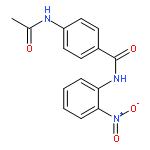

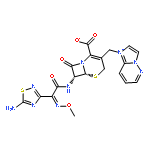
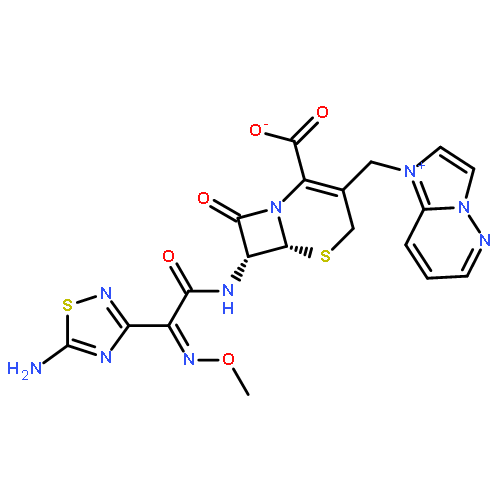




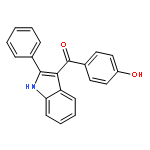
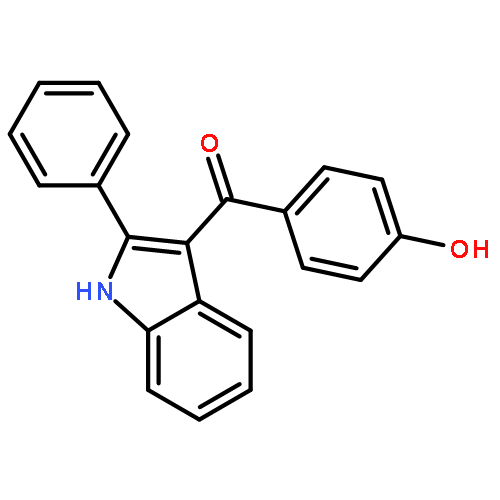


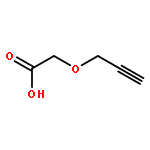
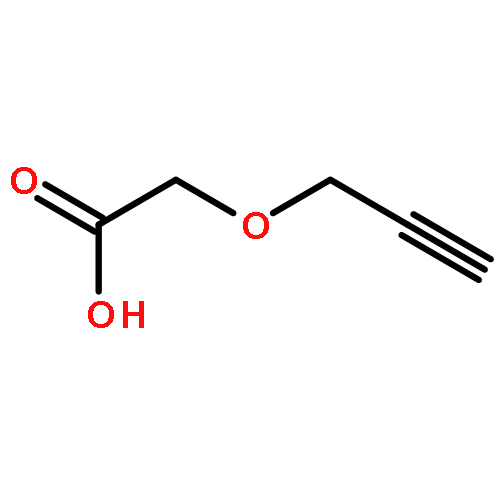
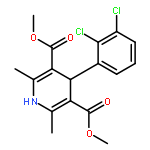

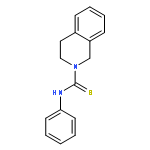

![2-Propenoic acid, 3-[3,4-bis(acetyloxy)phenyl]-, (E)-](http://img.cochemist.com/ccimg/88700/88623-81-8.png)
![2-Propenoic acid, 3-[3,4-bis(acetyloxy)phenyl]-, (E)-](http://img.cochemist.com/ccimg/88700/88623-81-8_b.png)

![6H-Dibenzo[b,d]pyran-6-acetic acid, ethyl ester](http://img.cochemist.com/ccimg/83400/83359-04-0.png)
![6H-Dibenzo[b,d]pyran-6-acetic acid, ethyl ester](http://img.cochemist.com/ccimg/83400/83359-04-0_b.png)
![O,O-DIETHYL O-[4-(METHYLSULFANYL)PHENYL] PHOSPHOROTHIOATE](http://img.cochemist.com/ccimg/80000/79925-38-5.png)
![O,O-DIETHYL O-[4-(METHYLSULFANYL)PHENYL] PHOSPHOROTHIOATE](http://img.cochemist.com/ccimg/80000/79925-38-5_b.png)
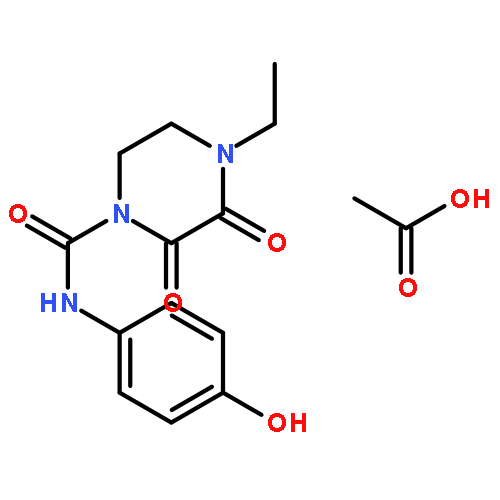
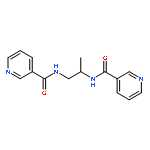
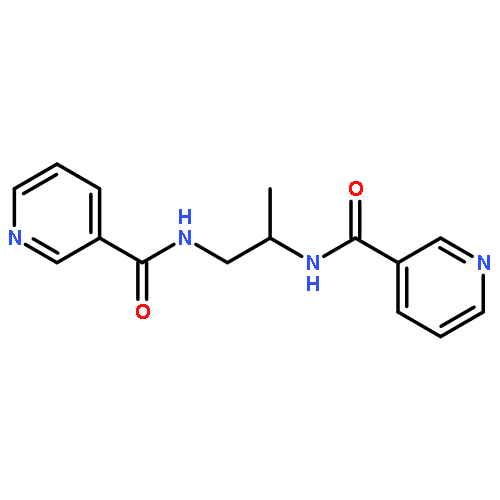
![1H-Benzimidazole, 5-chloro-2-[(4-nitrophenyl)thio]-](http://img.cochemist.com/ccimg/79400/79364-69-5.png)
![1H-Benzimidazole, 5-chloro-2-[(4-nitrophenyl)thio]-](http://img.cochemist.com/ccimg/79400/79364-69-5_b.png)
![4-BROMO-1,3-DIHYDRO-2H-PYRROLO[2,3-C]PYRIDIN-2-ONE](http://img.cochemist.com/ccimg/79100/79074-45-6.png)
![4-BROMO-1,3-DIHYDRO-2H-PYRROLO[2,3-C]PYRIDIN-2-ONE](http://img.cochemist.com/ccimg/79100/79074-45-6_b.png)
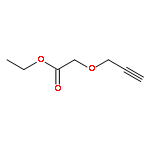
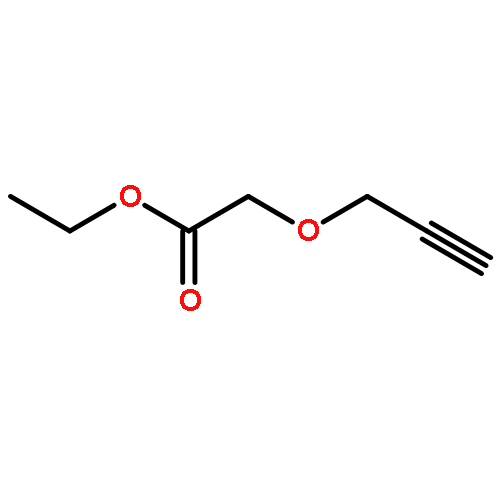
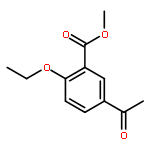
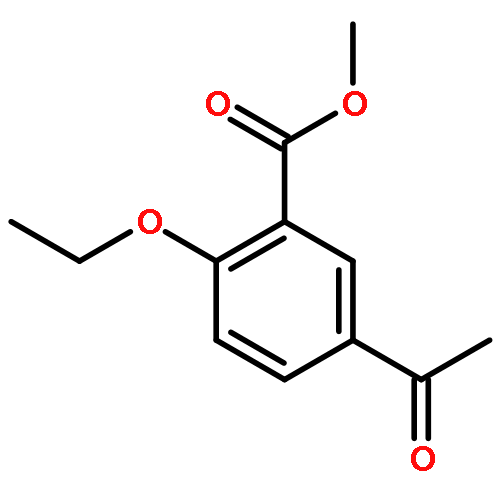
![2-[4-(DIETHOXYPHOSPHORYLMETHYL)PHENYL]BENZOTHIAZOLE](http://img.cochemist.com/ccimg/75900/75889-62-2.png)
![2-[4-(DIETHOXYPHOSPHORYLMETHYL)PHENYL]BENZOTHIAZOLE](http://img.cochemist.com/ccimg/75900/75889-62-2_b.png)
![4H-Pyrido[1,2-a]pyrimidin-4-one,9-methyl-3-(2H-tetrazol-5-yl)-](http://img.cochemist.com/ccimg/69400/69372-19-6.png)
![4H-Pyrido[1,2-a]pyrimidin-4-one,9-methyl-3-(2H-tetrazol-5-yl)-](http://img.cochemist.com/ccimg/69400/69372-19-6_b.png)
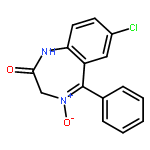
![1H,3H-[1,4]Oxazepino[4,3-a]benzimidazole(9CI)](http://img.cochemist.com/ccimg/69100/69056-79-7.png)
![1H,3H-[1,4]Oxazepino[4,3-a]benzimidazole(9CI)](http://img.cochemist.com/ccimg/69100/69056-79-7_b.png)
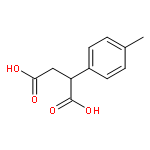
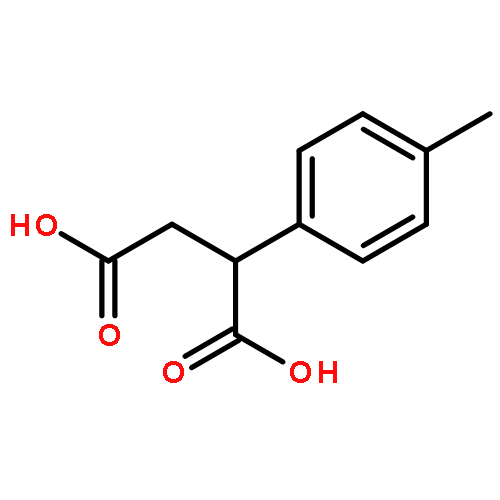
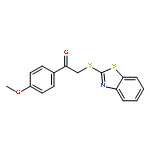
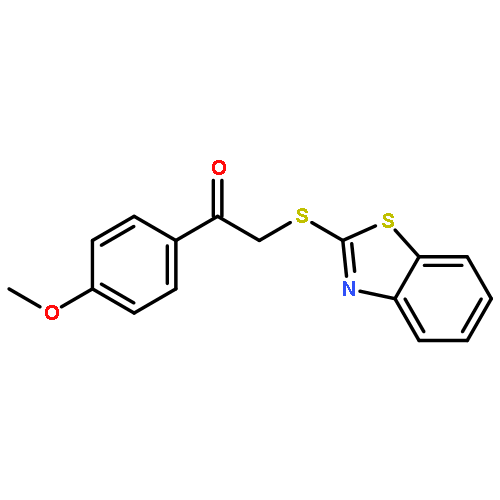
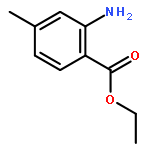
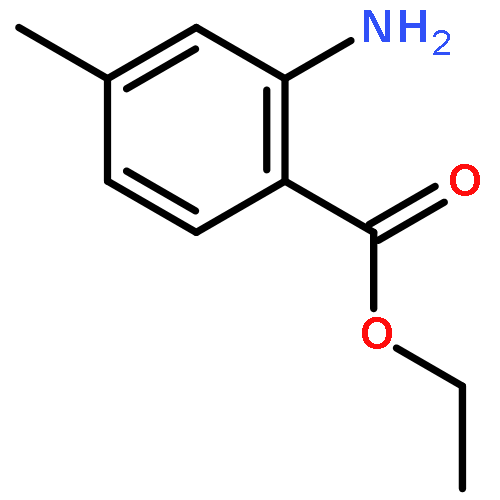
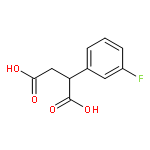
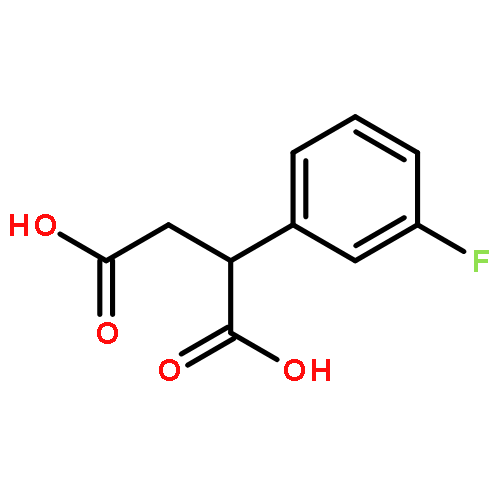
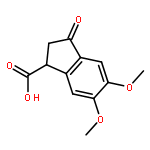


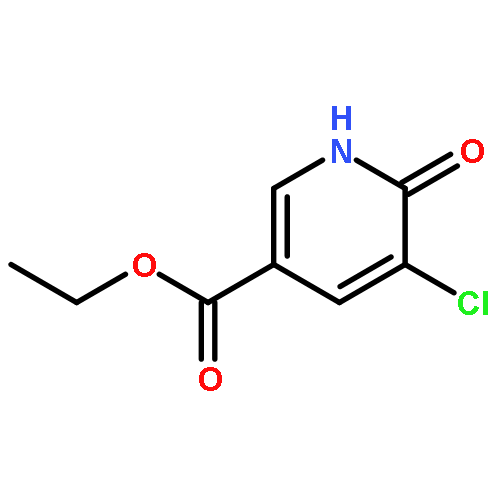


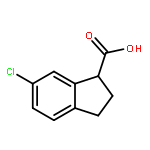
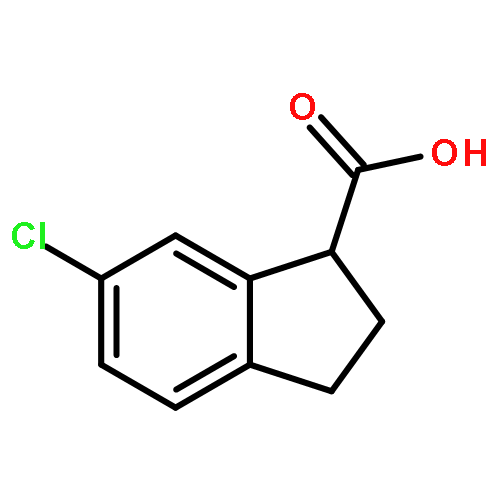
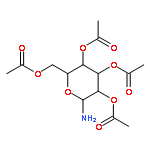
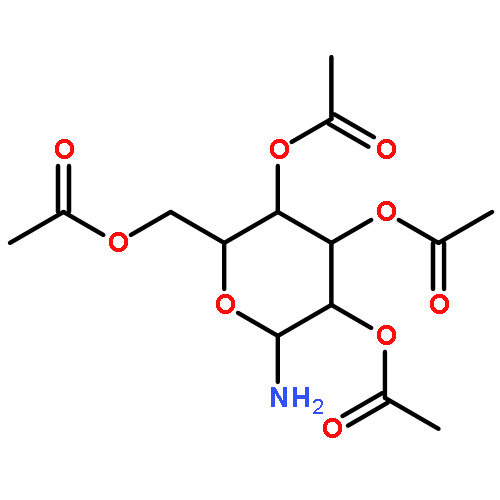

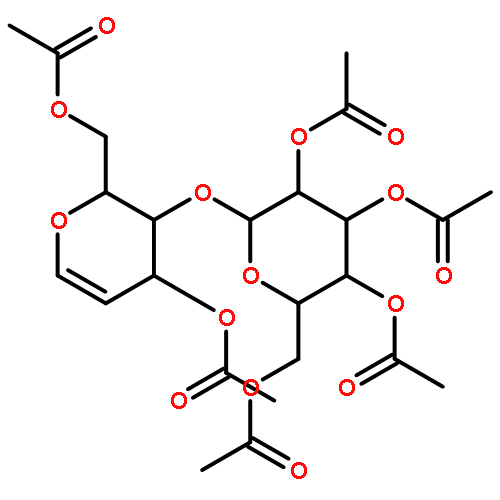

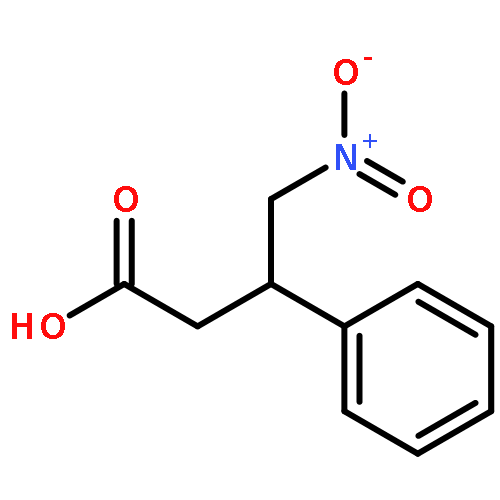
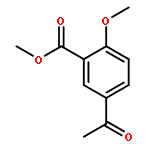
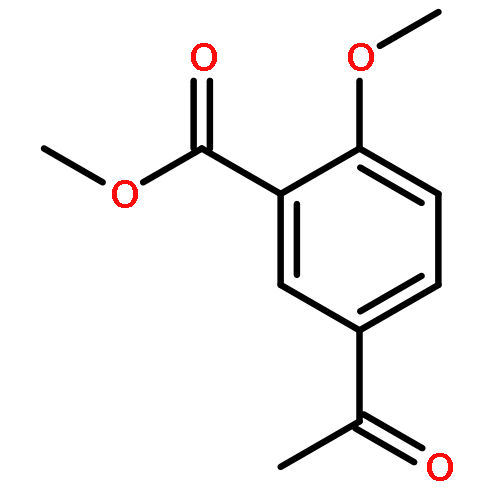
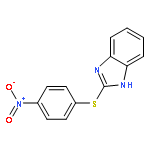
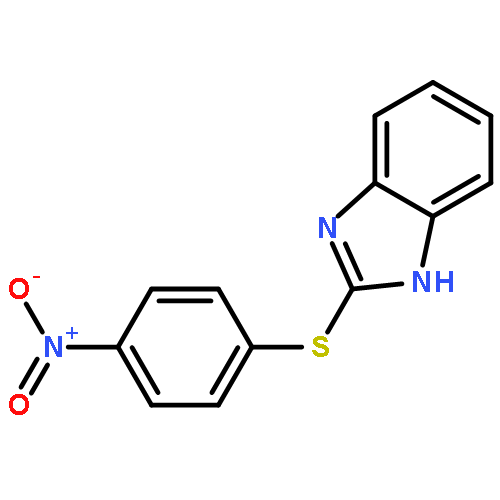


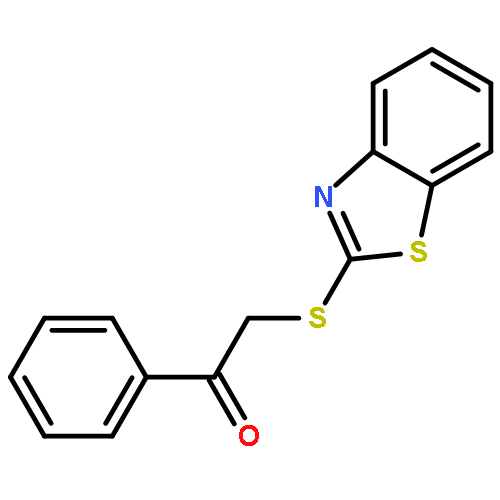
![Ethanone, 2-[(5-methoxy-1H-benzimidazol-2-yl)thio]-1-phenyl-](http://img.cochemist.com/ccimg/37100/37052-83-8.png)
![Ethanone, 2-[(5-methoxy-1H-benzimidazol-2-yl)thio]-1-phenyl-](http://img.cochemist.com/ccimg/37100/37052-83-8_b.png)

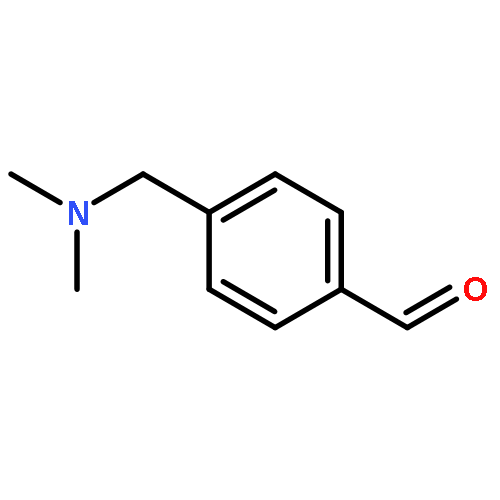
![2'-{(1R)-1-[(2R)-3-{[1-(4-CHLORO-3-FLUOROPHENYL)-2-METHYL-2-PROPA<WBR />NYL]AMINO}-2-HYDROXYPROPOXY]ETHYL}-3-METHYL-4-BIPHENYLCARBOXYLIC <WBR />ACID](http://img.cochemist.com/ccimg/35100/35008-62-9.png)
![2'-{(1R)-1-[(2R)-3-{[1-(4-CHLORO-3-FLUOROPHENYL)-2-METHYL-2-PROPA<WBR />NYL]AMINO}-2-HYDROXYPROPOXY]ETHYL}-3-METHYL-4-BIPHENYLCARBOXYLIC <WBR />ACID](http://img.cochemist.com/ccimg/35100/35008-62-9_b.png)
![2-Propenoic acid, 3-[4-(acetyloxy)-3-methoxyphenyl]-, (2E)-](/data/chemimg/1442400/34749-55-8.png)
![2-Propenoic acid, 3-[4-(acetyloxy)-3-methoxyphenyl]-, (2E)-](/data/chemimg/1442400/34749-55-8_b.png)

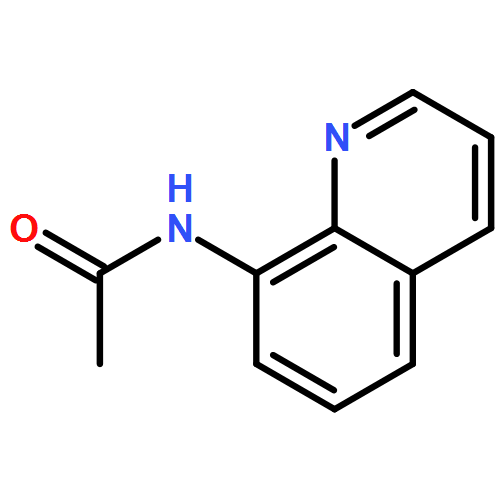
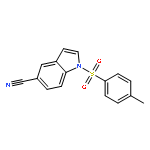
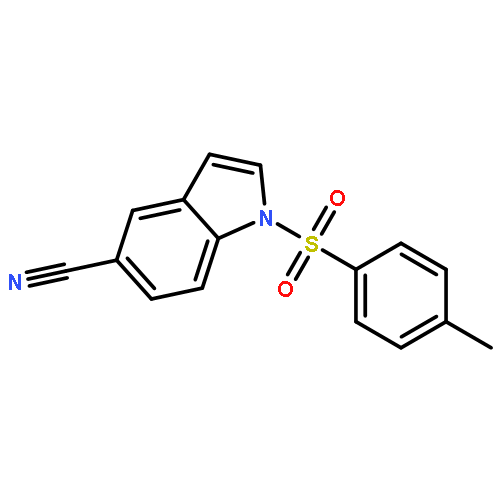
![THIOUREA, N-METHYL-N'-[2-(4-NITROPHENYL)ETHYL]-](http://img.cochemist.com/ccimg/25000/24954-56-1.png)
![THIOUREA, N-METHYL-N'-[2-(4-NITROPHENYL)ETHYL]-](http://img.cochemist.com/ccimg/25000/24954-56-1_b.png)
![Benzothiazole, 2-[4-(bromomethyl)phenyl]-](http://img.cochemist.com/ccimg/24300/24239-18-7.png)
![Benzothiazole, 2-[4-(bromomethyl)phenyl]-](http://img.cochemist.com/ccimg/24300/24239-18-7_b.png)
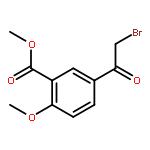
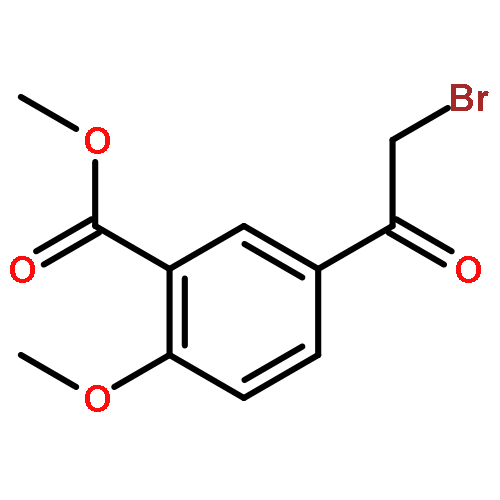


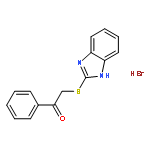

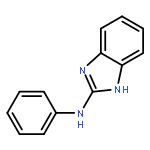
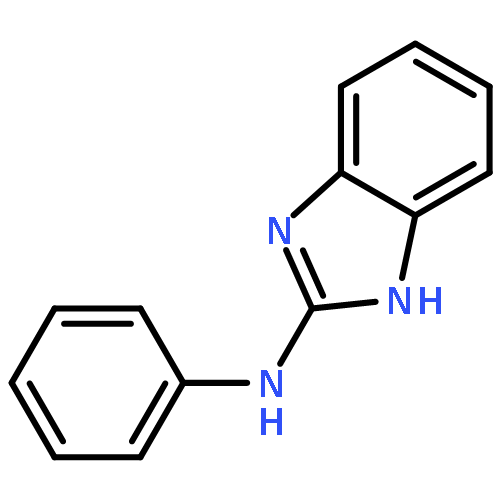





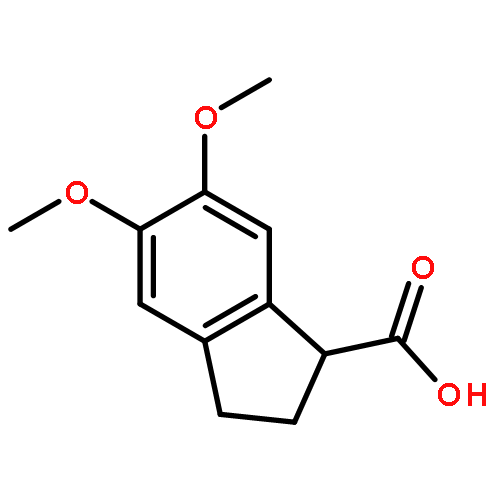
![Propanedioic acid,2-[[(4-methylphenyl)amino]methylene]-, 1,3-diethyl ester](http://img.cochemist.com/ccimg/19100/19056-84-9.png)
![Propanedioic acid,2-[[(4-methylphenyl)amino]methylene]-, 1,3-diethyl ester](http://img.cochemist.com/ccimg/19100/19056-84-9_b.png)

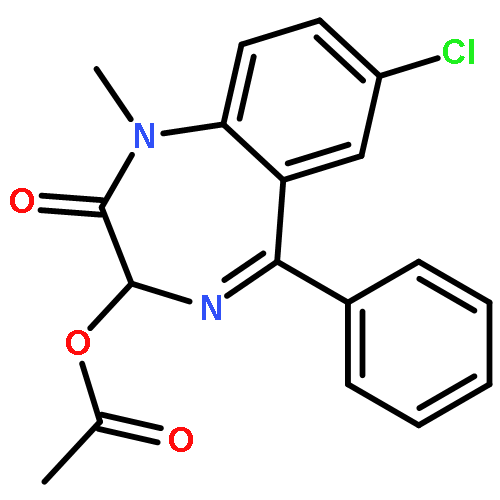


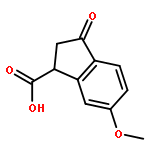

![2-(p-Tolyl)benzo[d]thiazole](/data/chemimg/517000/16112-21-3.png)
![2-(p-Tolyl)benzo[d]thiazole](/data/chemimg/517000/16112-21-3_b.png)
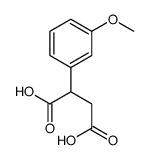
![Zinc, bis[2-(amino-kN)benzenethiolato-kS]-, (T-4)-](http://img.cochemist.com/ccimg/14700/14650-81-8.png)
![Zinc, bis[2-(amino-kN)benzenethiolato-kS]-, (T-4)-](http://img.cochemist.com/ccimg/14700/14650-81-8_b.png)




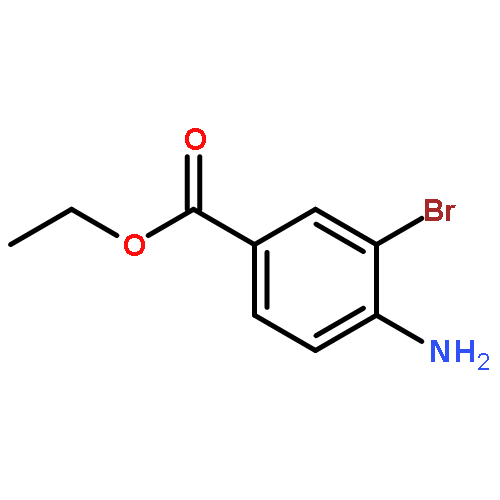
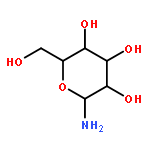
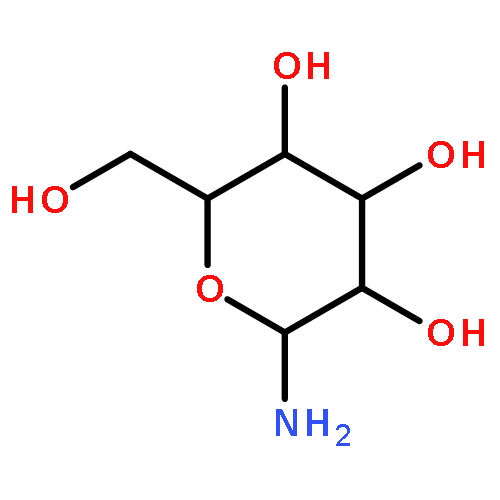

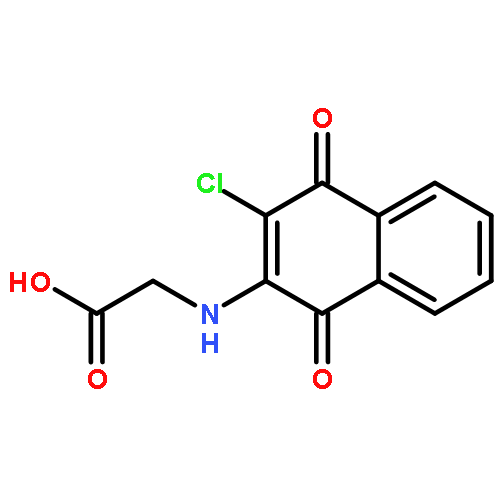


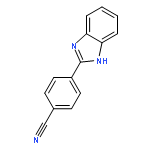
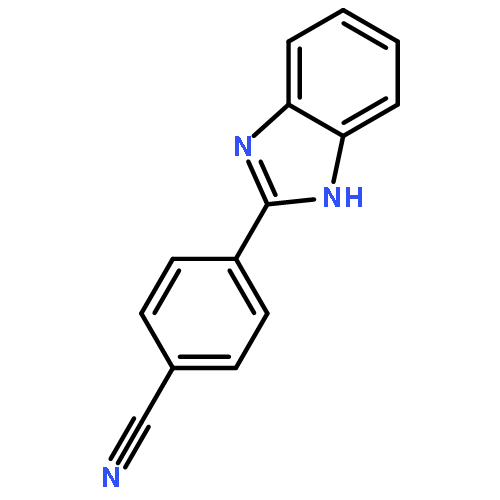
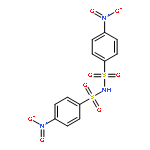

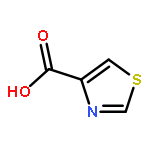
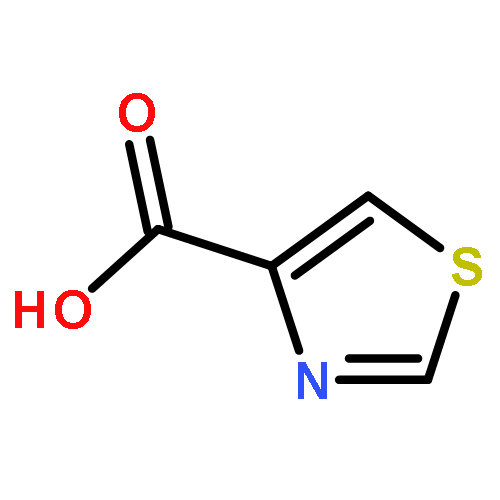
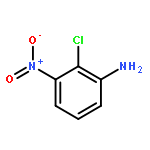

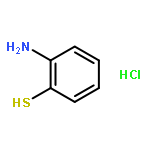
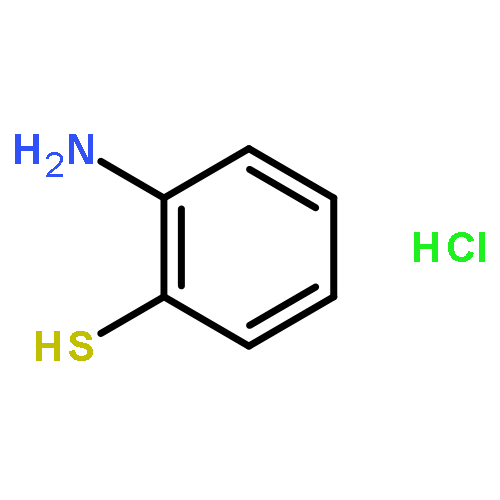
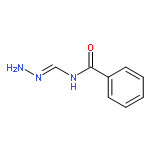
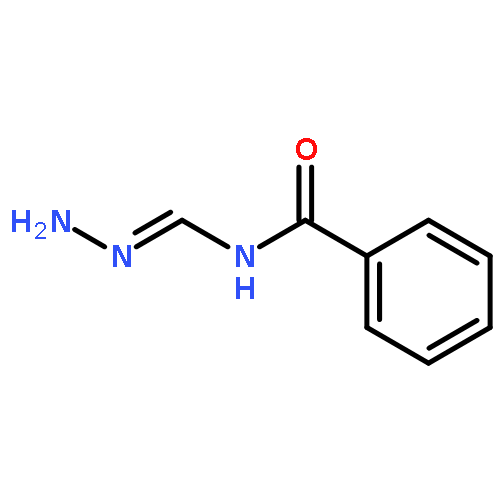
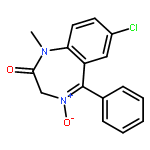
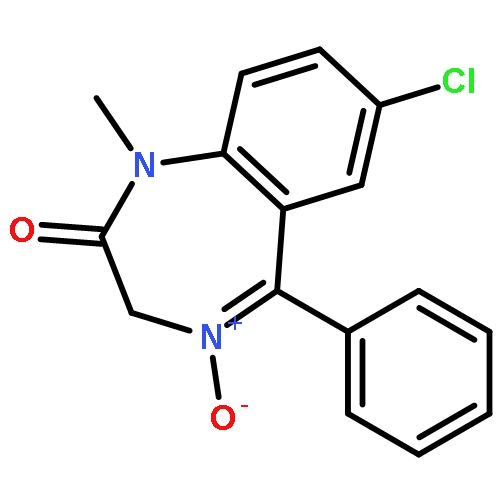


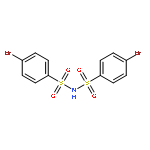
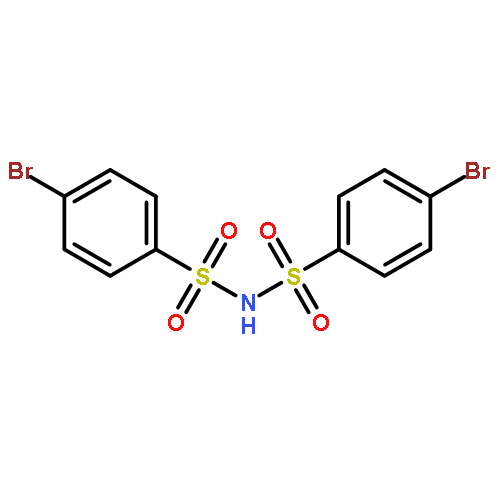
![Benzenesulfonamide,4-chloro-N-[(4-chlorophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/2800/2725-55-5.png)
![Benzenesulfonamide,4-chloro-N-[(4-chlorophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/2800/2725-55-5_b.png)



![Benzenesulfonamide, 4-fluoro-N-[(4-fluorophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/1500/1495-58-5.png)
![Benzenesulfonamide, 4-fluoro-N-[(4-fluorophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/1500/1495-58-5_b.png)














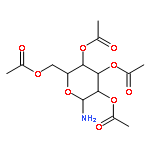
![Thiourea, N-[4-(2-benzothiazolylthio)phenyl]-N'-methyl-](http://img.cochemist.com/ccimg/849500/849438-67-1.png)
![Thiourea, N-[4-(2-benzothiazolylthio)phenyl]-N'-methyl-](http://img.cochemist.com/ccimg/849500/849438-67-1_b.png)
![Thiourea, [4-(2-benzothiazolylthio)phenyl]-](http://img.cochemist.com/ccimg/849500/849438-66-0.png)
![Thiourea, [4-(2-benzothiazolylthio)phenyl]-](http://img.cochemist.com/ccimg/849500/849438-66-0_b.png)
![Benzamide, N-[[[4-(2-benzothiazolylthio)phenyl]amino]thioxomethyl]-](http://img.cochemist.com/ccimg/849500/849438-65-9.png)
![Benzamide, N-[[[4-(2-benzothiazolylthio)phenyl]amino]thioxomethyl]-](http://img.cochemist.com/ccimg/849500/849438-65-9_b.png)


![2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenol](http://img.cochemist.com/ccimg/117100/117048-59-6.png)
![2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenol](http://img.cochemist.com/ccimg/117100/117048-59-6_b.png)



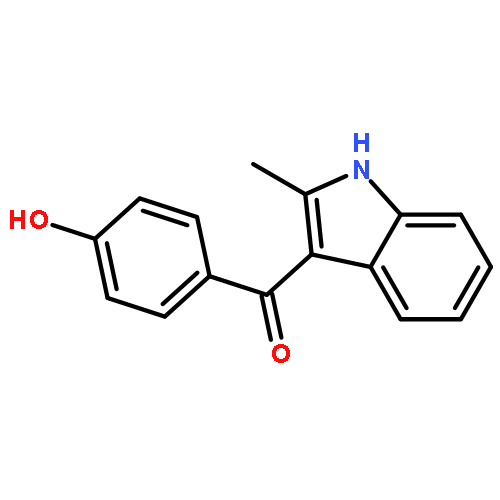
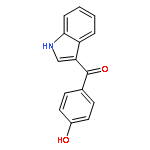


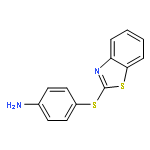

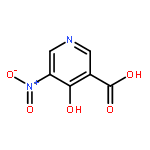
![Benzothiazole,2-[(4-nitrophenyl)thio]-](http://img.cochemist.com/ccimg/19400/19387-54-3.png)
![Benzothiazole,2-[(4-nitrophenyl)thio]-](http://img.cochemist.com/ccimg/19400/19387-54-3_b.png)











![(2S,4R)-4-[(3R,5S,6R,7R,8R,9S,10S,12S,13R,14S,17R)-6-ethyl-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid](http://img.cochemist.com/ccimg/1199800/1199796-29-6.png)
![(2S,4R)-4-[(3R,5S,6R,7R,8R,9S,10S,12S,13R,14S,17R)-6-ethyl-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid](http://img.cochemist.com/ccimg/1199800/1199796-29-6_b.png)
![BENZYL (4-CHLORO-1H-PYRROLO[2,3-B]PYRIDIN-1-YL)ACETATE](http://img.cochemist.com/ccimg/1197400/1197300-24-5.png)
![BENZYL (4-CHLORO-1H-PYRROLO[2,3-B]PYRIDIN-1-YL)ACETATE](http://img.cochemist.com/ccimg/1197400/1197300-24-5_b.png)
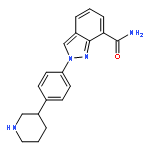



![2-[(2R)-2-Methylpyrrolidin-2-yl]-1H-benimidazole-4-carboxamide](http://img.cochemist.com/ccimg/912500/912444-00-9.png)
![2-[(2R)-2-Methylpyrrolidin-2-yl]-1H-benimidazole-4-carboxamide](http://img.cochemist.com/ccimg/912500/912444-00-9_b.png)





![(R)-8-Chloro-1-methyl-2,3,4,5-tetrahydro-1H-benzo[d]azepine (2R,3R)-2,3-dihydroxysuccinate](http://img.cochemist.com/ccimg/824500/824430-78-6.png)
![(R)-8-Chloro-1-methyl-2,3,4,5-tetrahydro-1H-benzo[d]azepine (2R,3R)-2,3-dihydroxysuccinate](http://img.cochemist.com/ccimg/824500/824430-78-6_b.png)







![2-[4-(benzyloxy)phenyl]-2-oxoethyl [(phenylsulfonyl)amino]acetate](http://img.cochemist.com/ccimg/335400/335390-66-4.png)
![2-[4-(benzyloxy)phenyl]-2-oxoethyl [(phenylsulfonyl)amino]acetate](http://img.cochemist.com/ccimg/335400/335390-66-4_b.png)
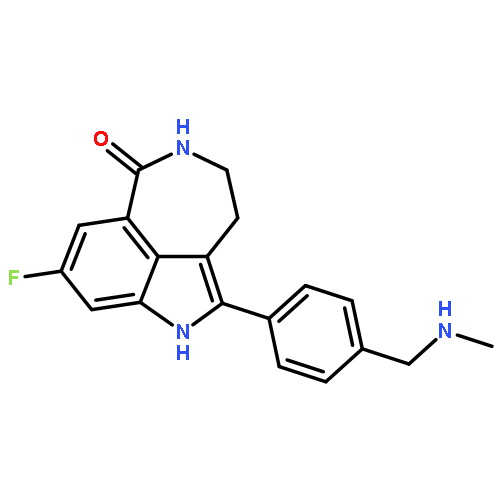


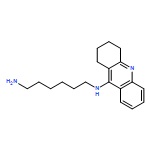


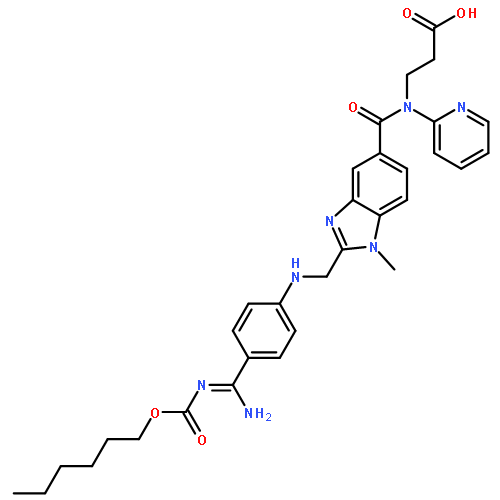
![3-[[2-[[(4-carbamimidoylphenyl)amino]methyl]-1-methyl-benzoimidazole-5-carbonyl]-pyridin-2-yl-amino]propanoic acid](http://img.cochemist.com/ccimg/212000/211914-51-1.png)
![3-[[2-[[(4-carbamimidoylphenyl)amino]methyl]-1-methyl-benzoimidazole-5-carbonyl]-pyridin-2-yl-amino]propanoic acid](http://img.cochemist.com/ccimg/212000/211914-51-1_b.png)
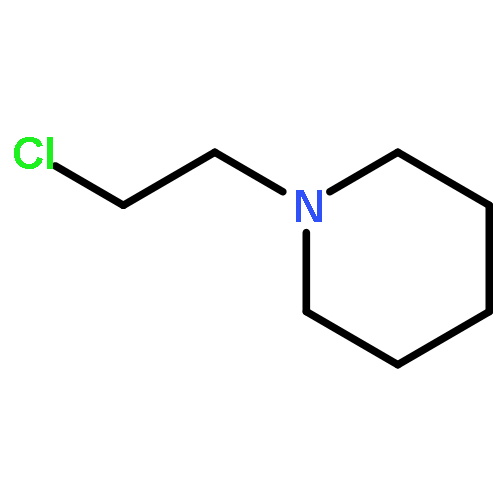
![tert-butyl N-[2-(4-chlorophenyl)ethyl]carbamate](http://img.cochemist.com/ccimg/167900/167886-56-8.png)
![tert-butyl N-[2-(4-chlorophenyl)ethyl]carbamate](http://img.cochemist.com/ccimg/167900/167886-56-8_b.png)
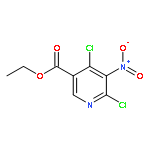
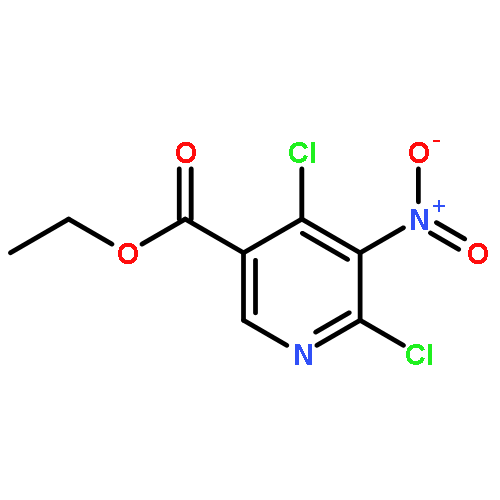
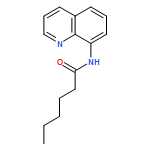
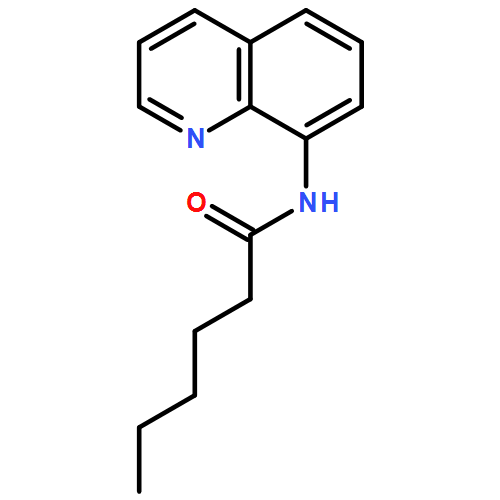
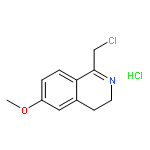
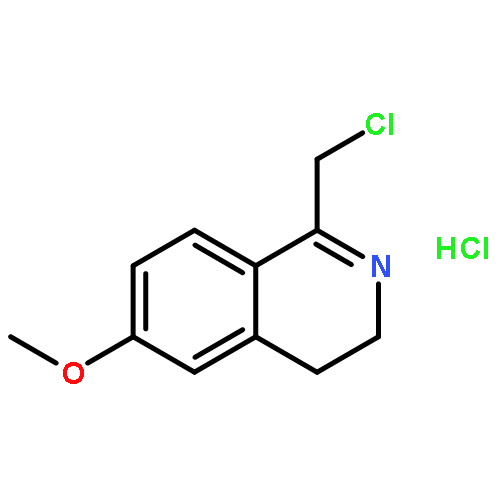
![4-FLUORO-N-{(1S)-2,2,2-TRIFLUORO-1-[4'-(METHYLSULFONYL)-4-BIPHENYLYL]ETHYL}-L-LEUCINE](http://img.cochemist.com/ccimg/127700/127606-02-4.png)
![4-FLUORO-N-{(1S)-2,2,2-TRIFLUORO-1-[4'-(METHYLSULFONYL)-4-BIPHENYLYL]ETHYL}-L-LEUCINE](http://img.cochemist.com/ccimg/127700/127606-02-4_b.png)
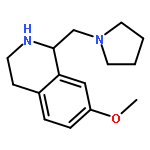
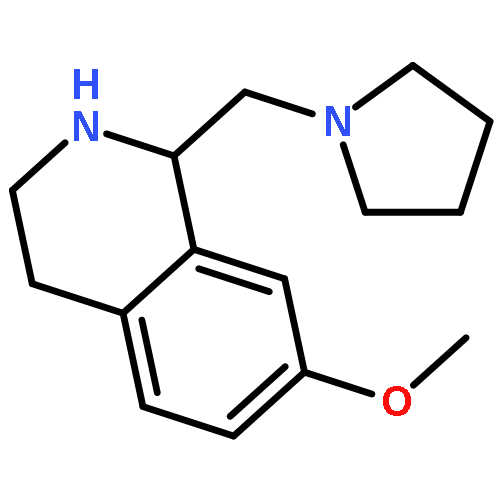
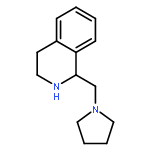







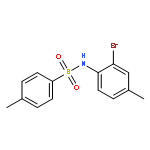

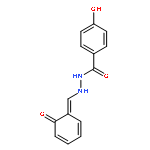



![2-(3,4-dichlorophenyl)-N-methyl-N-[2-(pyrrolidin-1-yl)cyclohexyl]acetamide](http://img.cochemist.com/ccimg/67200/67198-13-4.png)
![2-(3,4-dichlorophenyl)-N-methyl-N-[2-(pyrrolidin-1-yl)cyclohexyl]acetamide](http://img.cochemist.com/ccimg/67200/67198-13-4_b.png)

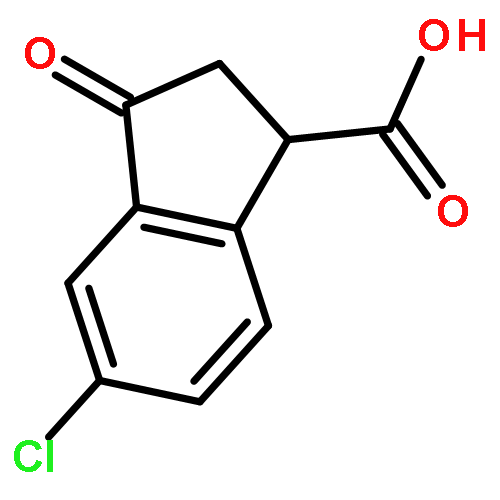
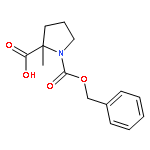
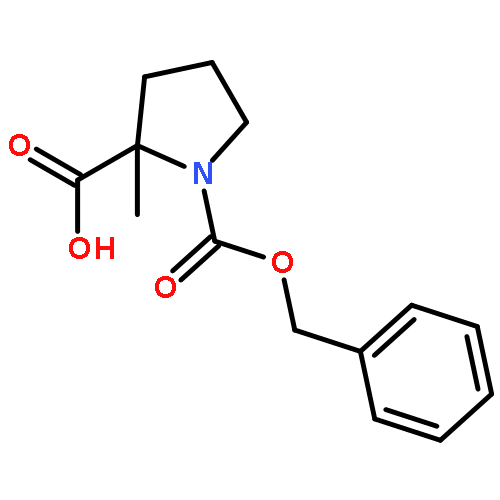
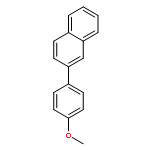



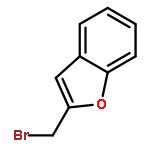
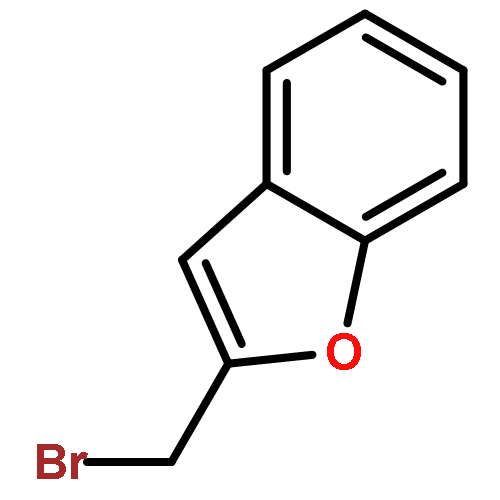
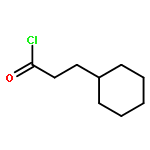
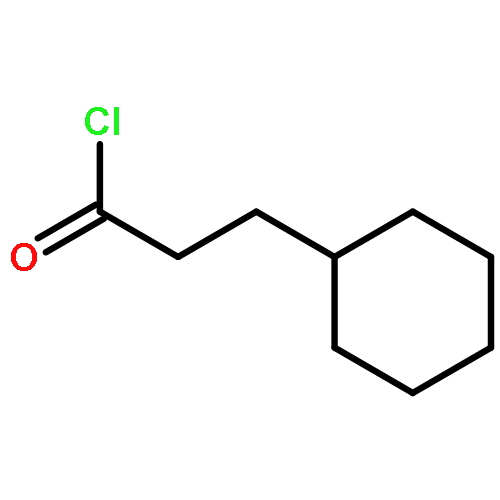

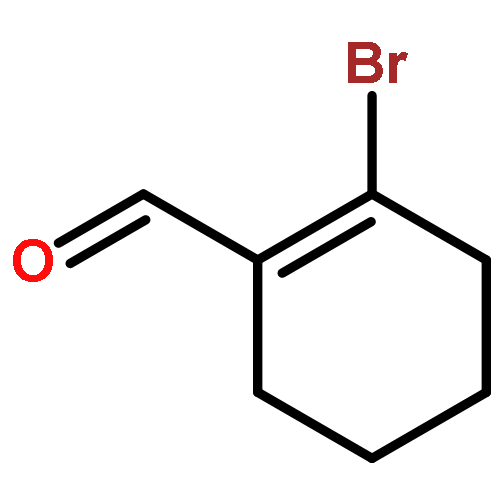
![2-CHLORO-N-[2-(3-METHOXYPHENYL)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/34200/34162-12-4.png)
![2-CHLORO-N-[2-(3-METHOXYPHENYL)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/34200/34162-12-4_b.png)
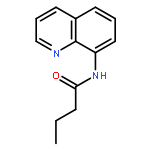
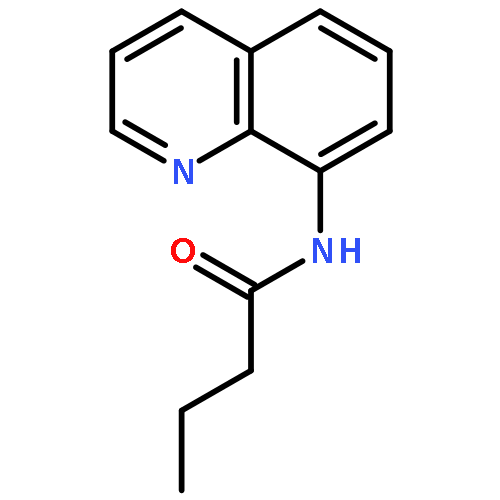

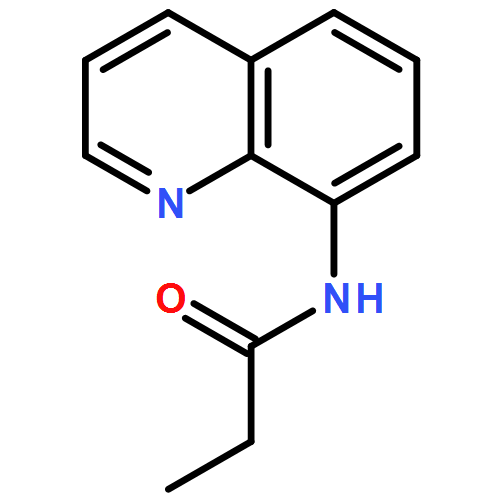
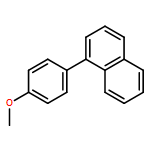
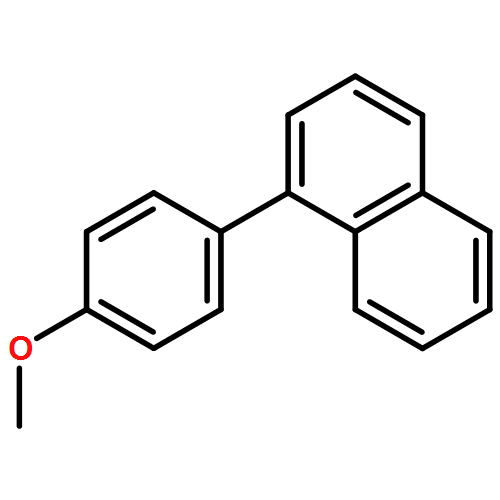

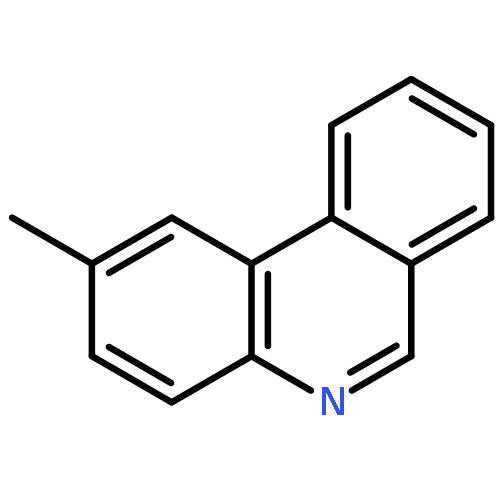
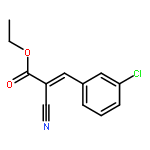
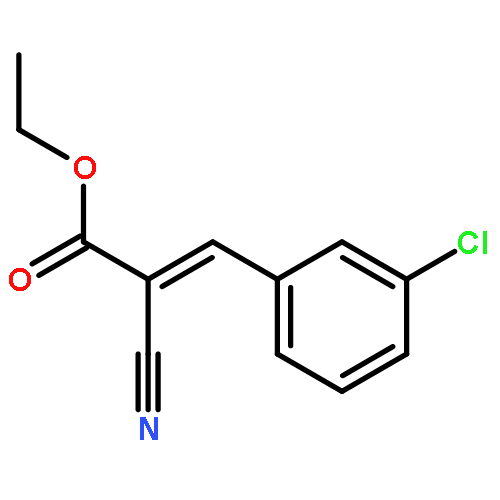


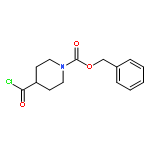


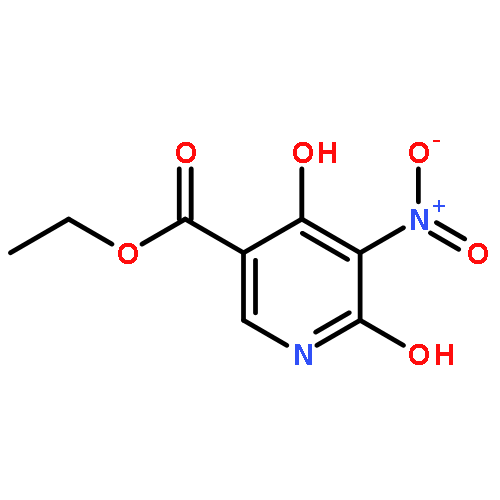
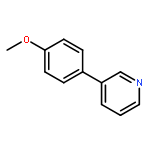
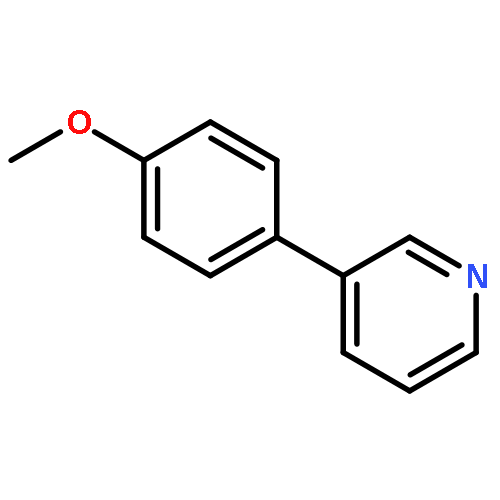
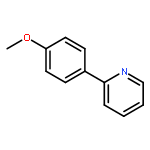
![11-chloro-7,8,9,10-tetrahydro-6H-cyclohepta[b]quinoline](http://img.cochemist.com/ccimg/5800/5778-71-2.png)
![11-chloro-7,8,9,10-tetrahydro-6H-cyclohepta[b]quinoline](http://img.cochemist.com/ccimg/5800/5778-71-2_b.png)


![4H,6H,8H-Pyrano[4',3':3,3a]isobenzofuro[5,4-f][2]benzopyran-4,11,13(7H,14H)-trione,9-(3-furanyl)-1,2a,3,6b,8a,9,12b,14a-octahydro-1,1,8a,12b-tetramethyl-,(2aS,6aR,6bR,8aR,9R,12bR,14aR)- (9CI)](http://img.cochemist.com/ccimg/1000/989-23-1.png)
![4H,6H,8H-Pyrano[4',3':3,3a]isobenzofuro[5,4-f][2]benzopyran-4,11,13(7H,14H)-trione,9-(3-furanyl)-1,2a,3,6b,8a,9,12b,14a-octahydro-1,1,8a,12b-tetramethyl-,(2aS,6aR,6bR,8aR,9R,12bR,14aR)- (9CI)](http://img.cochemist.com/ccimg/1000/989-23-1_b.png)