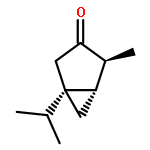
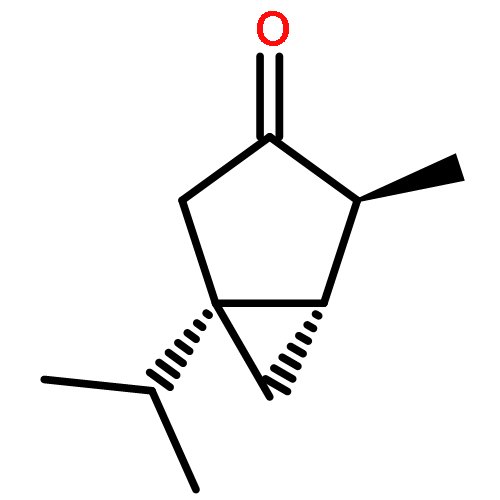
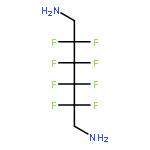
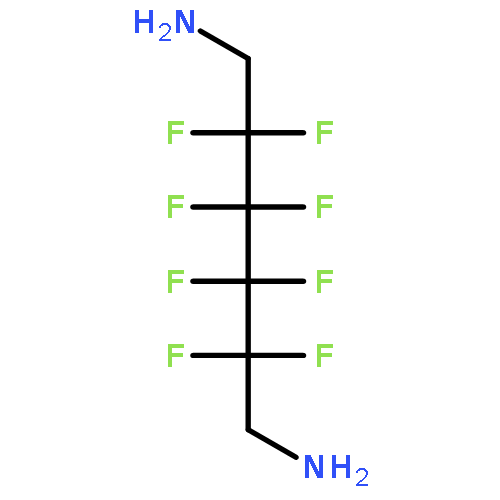
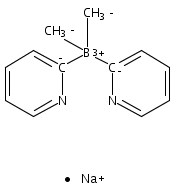
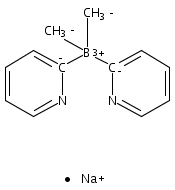
Co-reporter:Jeff Joseph A. Celaje, Xingyue Zhang, Forrest Zhang, Lisa Kam, Jessica R. Herron, and Travis J. Williams
ACS Catalysis February 3, 2017 Volume 7(Issue 2) pp:1136-1136
Publication Date(Web):December 30, 2016
DOI:10.1021/acscatal.6b03088
A (pyridyl)phosphine-ligated ruthenium(II) catalyst is reported for the chemoselective benzylic N-alkylation of amines, via a hydrogen-borrowing mechanism. The catalyst operates under mild conditions, neat, and without a base or other additive. These conditions offer remarkable functional group compatibility for applications in organic synthesis, including reactions involving phenols and anilines, which are very difficult to achieve. Mechanistic studies suggest that, unlike other catalysts for this reaction, the redox steps are fast and reversible while imine formation is slow. We perceive that this is the origin of the selectivity realized with these reaction conditions.Keywords: amination; catalysis; dehydrogenation; hydrogen borrowing; imine;
Co-reporter:Xingyue Zhang, Lisa Kam, Ryan Trerise, and Travis J. Williams
Accounts of Chemical Research 2017 Volume 50(Issue 1) pp:
Publication Date(Web):December 29, 2016
DOI:10.1021/acs.accounts.6b00482
ConspectusOne of the greatest challenges in using H2 as a fuel source is finding a safe, efficient, and inexpensive method for its storage. Ammonia borane (AB) is a solid hydrogen storage material that has garnered attention for its high hydrogen weight density (19.6 wt %) and ease of handling and transport. Hydrogen release from ammonia borane is mediated by either hydrolysis, thus giving borate products that are difficult to rereduce, or direct dehydrogenation. Catalytic AB dehydrogenation has thus been a popular topic in recent years, motivated both by applications in hydrogen storage and main group synthetic chemistry. This Account is a complete description of work from our laboratory in ruthenium-catalyzed ammonia borane dehydrogenation over the last 6 years, beginning with the Shvo catalyst and resulting ultimately in the development of optimized, leading catalysts for efficient hydrogen release.We have studied AB dehydrogenation with Shvo’s catalyst extensively and generated a detailed understanding of the role that borazine, a dehydrogenation product, plays in the reaction: it is a poison for both Shvo’s catalyst and PEM fuel cells. Through independent syntheses of Shvo derivatives, we found a protective mechanism wherein catalyst deactivation by borazine is prevented by coordination of a ligand that might otherwise be a catalytic poison. These studies showed how a bidentate N–N ligand can transform the Shvo into a more reactive species for AB dehydrogenation that minimizes accumulation of borazine.Simultaneously, we designed novel ruthenium catalysts that contain a Lewis acidic boron to replace the Shvo -OH proton, thus offering more flexibility to optimize hydrogen release and take on more general problems in hydride abstraction. Our scorpionate-ligated ruthenium species (12) is a best-of-class catalyst for homogeneous dehydrogenation of ammonia borane in terms of its extent of hydrogen release (4.6 wt %), air tolerance, and reusability. Moreover, a synthetically simplified ruthenium complex supported by the inexpensive bis(pyrazolyl)borate ligand is a comparably good catalyst for AB dehydrogenation, among other reactions. In this Account, we present a detailed, concise description of how our work with the Shvo system progressed to the development of our very reactive and flexible dual-site boron-ruthenium catalysts.
Co-reporter:Zhiyao Lu and Travis J. Williams
ACS Catalysis 2016 Volume 6(Issue 10) pp:6670
Publication Date(Web):August 29, 2016
DOI:10.1021/acscatal.6b02101
Di(carbene)-supported nickel species 1 and 2 are efficient catalysts for the room-temperature reduction of CO2 to methanol in the presence of sodium borohydride. The catalysts feature unusual stability, particularly for a base metal catalyst, enabling >1.1 million turnovers of CO2. Moreover, while other systems involve more expensive reducing reagents, sodium borohydride is inexpensive and easily handled. Furthermore, effecting reduction in the presence of water enables direct access to methanol. Preliminary mechanistic data collected are most consistent with a mononuclear nickel active species that mediates rate-determining reduction of a boron formate.Keywords: borohydride; carbon dioxide; catalyst; methanol; nickel
Co-reporter:Zhiyao Lu, Ivan Demianets, Rasha Hamze, Nicholas J. Terrile, and Travis J. Williams
ACS Catalysis 2016 Volume 6(Issue 3) pp:2014
Publication Date(Web):January 28, 2016
DOI:10.1021/acscatal.5b02732
We report the synthesis and reactivity of a very robust iridium catalyst for glycerol to lactate conversion. The high reactivity and selectivity of this catalyst enable a sequence for the conversion of biodiesel waste stream to lactide monomers for the preparation of poly(lactic acid). Furthermore, experimental data collected with this system provide a general understanding of its reactive mechanism.Keywords: biodiesel; dehydrogenation; glycerol; iridium; lactic acid
Co-reporter:Xingyue Zhang, Lisa Kam and Travis J. Williams
Dalton Transactions 2016 vol. 45(Issue 18) pp:7672-7677
Publication Date(Web):23 Mar 2016
DOI:10.1039/C6DT00604C
Ammonia borane (AB) has high hydrogen density (19.6 wt%), and can, in principle, release up to 3 equivalents of H2 under mild catalytic conditions. A limited number of catalysts are capable of non-hydrolytic dehydrogenation of AB beyond 2 equivalents of H2 under mild conditions, but none of these is shown directly to derivatise borazine, the product formed after 2 equivalents of H2 are released. We present here a high productivity ruthenium-based catalyst for non-hydrolytic AB dehydrogenation that is capable of borazine dehydrogenation, and thus exhibits among the highest H2 productivity reported to date for anhydrous AB dehydrogenation. At 1 mol% loading, (phen)Ru(OAc)2(CO)2 (1) effects AB dehydrogenation through 2.7 equivalents of H2 at 70 °C, is robust through multiple charges of AB, and is water and air stable. We further demonstrate that catalyst 1 has the ability both to dehydrogenate borazine in isolation and dehydrogenate AB itself. This is important, both because borazine derivatisation is productivity-limiting in AB dehydrogenation and because borazine is a fuel cell poison that is commonly released in H2 production from this medium.
Co-reporter:Xingyue Zhang, Zhiyao Lu, Lena K. Foellmer, and Travis J. Williams
Organometallics 2015 Volume 34(Issue 15) pp:3732-3738
Publication Date(Web):July 29, 2015
DOI:10.1021/acs.organomet.5b00409
We previously reported that quantitative poisoning, a test for homogeneous catalysis, behaves oddly in the dehydrogenation of ammonia borane (AB) by Shvo’s catalyst, whereas the “poison” 1,10-phenanthroline (phen) accelerates catalysis and apparently prevents catalyst deactivation. Thus, we proposed a protective role for phen in the catalysis. Herein we account for the mechanistic origin of this accelerated AB dehydrogenation in the presence of phen and define the relevance boundaries of our prior proposal. In so doing, we present syntheses for novel amine- and pyridine-ligated homologues of the Shvo catalyst and show their catalytic efficacy in AB dehydrogenation. These catalysts are synthetically easy to access, air stable, and rapidly release over 2 equiv of H2. The mechanisms of these reactions are also discussed.
Co-reporter:Vincent Li, Yoo-Jin Ghang, Richard J. Hooley and Travis J. Williams
Chemical Communications 2014 vol. 50(Issue 11) pp:1375-1377
Publication Date(Web):11 Dec 2013
DOI:10.1039/C3CC48389D
The relaxivity of a magnetically responsive Gd complex can be controlled by non-covalent molecular recognition with a water-soluble deep cavitand. Lowered relaxivity is conferred by a self-assembled micellar “off state”, and the contrast can be regenerated by addition of a superior guest.
Co-reporter:Zhiyao Lu and Travis J. Williams
Chemical Communications 2014 vol. 50(Issue 40) pp:5391-5393
Publication Date(Web):24 Dec 2013
DOI:10.1039/C3CC47384H
We report a novel ruthenium bis(pyrazolyl)borate scaffold that enables cooperative reduction reactivity in which boron and ruthenium centers work in concert to effect selective nitrile reduction. The pre-catalyst compound [κ3-(1-pz)2HB(N = CHCH3)]Ru(cymene)+ TfO− (pz = pyrazolyl) was synthesized using readily-available materials through a straightforward route, thus making it an appealing catalyst for a number of reactions.
Co-reporter:Xinping Wu, Anna C. Dawsey, Buddhima N. Siriwardena-Mahanama, Matthew J. Allen, Travis J. Williams
Journal of Fluorine Chemistry 2014 Volume 168() pp:177-183
Publication Date(Web):December 2014
DOI:10.1016/j.jfluchem.2014.09.018
•The first use of fluoroalkyl moieties to accentuate the relaxivity of a gadolinium agent.•This effect above is specific to a phosphonate-bearing Gd agent.•The origin of the effect primarily involves the agent's solution tumbling rate.Responsive magnetic resonance imaging (MRI) contrast agents, those that change their relaxivity according to environmental stimuli, have promise as next generation imaging probes in medicine. While several of these are known based on covalent modification of the contrast agents, fewer are known based on controlling non-covalent interactions. We demonstrate here accentuated relaxivity of a T1-shortening contrast agent, Gd-DOTP5− based on non-covalent, hydrogen bonding of Gd-DOTP5− with a novel fluorous amphiphile. By contrast to the phosphonate-containing Gd-DOTP5− system, the relaxivity of the analogous clinically approved contrast agent, Gd-DOTA− is unaffected by the same fluorous amphiphile under similar conditions.Mechanistic studies show that placing the fluorous amphiphile in proximity of the gadolinium center in Gd-DOTP5− caused an increase in τm (bound-water residence lifetime or the inverse of water exchange rate, τm = 1/kex) and an increase in τR (rotational correlation time), with τR being the factor driving enhanced relaxivity. Further, these effects were not observed when Gd-DOTA− was treated with the same fluorous amphiphile. Thus, Gd-DOTP5− and Gd-DOTA− respond to the fluorous amphiphile differently, presumably because the former binds to the amphiphile with higher affinity. (DOTP = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraphosphonic acid; DOTA = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid).
Co-reporter:Jeff A. Celaje, Megan K. Pennington-Boggio, Robinson W. Flaig, Michael G. Richmond, and Travis J. Williams
Organometallics 2014 Volume 33(Issue 8) pp:2019-2026
Publication Date(Web):April 7, 2014
DOI:10.1021/om500173j
The syntheses of novel dimethylbis(2-pyridyl)borate nickel(II) complexes 4 and 6 are reported. These complexes were unambiguously characterized by X-ray analysis. In dichloromethane solvent, complex 4 undergoes a unique square-planar to square-planar rotation around the nickel(II) center, for which activation parameters of ΔH⧧ = 12.2(1) kcal mol–1 and ΔS⧧ = 0.8(5) eu were measured via NMR inversion recovery experiments. Complex 4 was also observed to isomerize via a relatively slow ring flip: ΔH⧧ = 15.0(2) kcal mol–1; and ΔS⧧ = −4.2(7) eu. DFT studies support the experimentally measured rotation activation energy (cf. calculated ΔH⧧ = 11.1 kcal mol–1) as well as the presence of a high-energy triplet intermediate (ΔH = 8.8 kcal mol–1).
Co-reporter:Megan K. Pennington-Boggio, Brian L. Conley, Michael G. Richmond, Travis J. Williams
Polyhedron 2014 Volume 84() pp:24-31
Publication Date(Web):14 December 2014
DOI:10.1016/j.poly.2014.05.042
Rhodium(I) and Iridium(I) borate complexes of the structure [Me2B(2-py)2]ML2 (L2 = (tBuNC)2, (CO)2, (C2H4)2, cod, dppe) were prepared and structurally characterized (cod = 1,5-cyclooctadiene; dppe = 1,2-diphenylphosphinoethane). Each contains a boat-configured chelate ring that participates in a boat-to-boat ring flip. Computational evidence shows that the ring flip proceeds through a transition state that is near planarity about the chelate ring.We observe an empirical, quantitative correlation between the barrier of this ring flip and the π acceptor ability of the ancillary ligand groups on the metal. The ring flip barrier correlates weakly to the Tolman and Lever ligand parameterization schemes, apparently because these combine both σ and π effects while we propose that the ring flip barrier is dominated by π bonding. This observation is consistent with metal–ligand π interactions becoming temporarily available only in the near-planar transition state of the chelate ring flip and not the boat-configured ground state. Thus, this is a first-of-class observation of metal–ligand π bonding governing conformational dynamics.Graphical abstractRhodium(I) and Iridium(I) borate complexes [Me2B(2-py)2]ML2 (L2 = (tBuNC)2, (CO)2, (C2H4)2, cod, dppe) have a boat-configured chelate ring that participates in a boat-to-boat ring flip. The barrier of this ring flip is correlated to the π acceptor ability of the ancillary ligand groups on the metal.
Co-reporter:Anna C. Dawsey, Kathryn L. Hathaway, Susie Kim, and Travis J. Williams
Journal of Chemical Education 2013 Volume 90(Issue 7) pp:922-925
Publication Date(Web):March 18, 2013
DOI:10.1021/ed3006902
Dotarem and Magnevist, two clinically available magnetic resonance imaging (MRI) contrast agents, were assessed in a high school science classroom with respect to which is the better contrast agent. Magnevist, the more efficacious contrast agent, has negative side effects because its gadolinium center can escape from its ligand. However, Dotarem, though a less efficacious contrast agent, is a safer drug choice. After the experiment, students are confronted with the FDA warning on Magnevist, which enabled a discussion of drug efficacy versus safety. We describe a laboratory experiment in which NMR spin lattice relaxation rate measurements are used to quantify the relaxivities of the active ingredients of Dotarem and Magnevist. The spin lattice relaxation rate gives the average amount of time it takes the excited nucleus to relax back to the original state. Students learn by constructing molar relaxivity curves based on inversion recovery data sets that Magnevist is more relaxive than Dotarem. This experiment is suitable for any analytical chemistry laboratory with access to NMR.Keywords: Analytical Chemistry; Applications of Chemistry; Drugs/Pharmaceutical; First-Year Undergraduate/General; Hands-On Learning/Manipulative; High School/Introductory Chemistry; Laboratory Instruction; NMR Spectroscopy; Second-Year Undergraduate;
Co-reporter:Vincent Li, Andy Y. Chang, Travis J. Williams
Tetrahedron 2013 69(36) pp: 7741-7745
Publication Date(Web):
DOI:10.1016/j.tet.2013.05.092
Co-reporter:Anna C. Dawsey, Vincent Li, Kimberly C. Hamilton, Jianmei Wang and Travis J. Williams
Dalton Transactions 2012 vol. 41(Issue 26) pp:7994-8002
Publication Date(Web):29 Feb 2012
DOI:10.1039/C2DT00025C
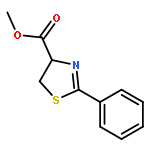
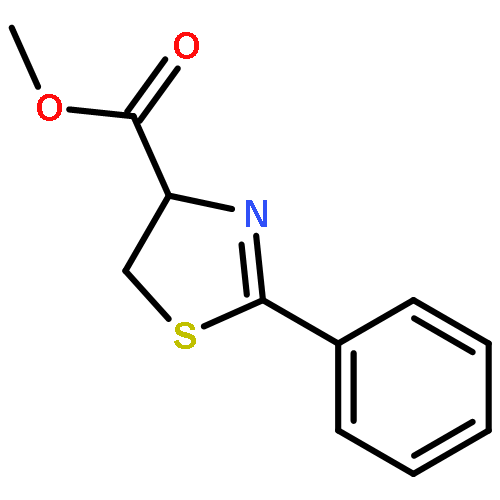
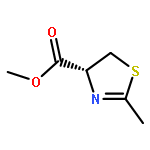
We report herein convenient, aerobic conditions for the oxidation of thiazolines to thiazoles and data regarding the oxidation mechanism. These reactions feature operationally simple and environmentally benign conditions and proceed in good yield to afford the corresponding azoles, thus enabling the inexpensive preparation of valuable molecular building blocks. Incorporation of a novel diimine-ligated copper catalyst, [(MesDABMe)CuII(OH2)3]2+ [−OTf]2, provides increased reaction efficiency in many cases. In other cases copper-free conditions involving a stoichiometric quantity of base affords superior results.
Co-reporter:Xinping Wu, Emine Boz, Amy M. Sirkis, Andy Y. Chang, Travis J. Williams
Journal of Fluorine Chemistry 2012 Volume 135() pp:292-302
Publication Date(Web):March 2012
DOI:10.1016/j.jfluchem.2011.12.011
We report herein convenient procedures for the use of highly fluorinated α,ω-diols (e.g. 1) as building blocks for the rapid assembly of amphiphilic materials containing a fluorous phase region. We describe expedient conversion of the parent diols to both symmetrically and asymmetrically substituted amphiphiles via the installation of an intermediate trifluoromethanesulfonyl ester. These sulfonate esters are versatile and easily manipulated intermediates, which can be readily converted to a variety of nitrogen, halogen, and carbon groups. Moreover, we show that for guanidine-terminated fluorous amphiphiles, these molecules can bind phosphonic acid groups in aqueous media. Thus, these materials offer a new strategy for decorating phosphorylated biomolecules with fluorine-rich coatings.Graphical abstractWe report convenient procedures for the use of highly fluorinated α,ω-diols as building blocks for the rapid assembly of amphiphilic materials containing a fluorous phase region.Highlights► Fluorinated α,ω-diols converted to amphiphilic materials through convenient procedures. ► Conversion can be symmetrical and asymmetrical. ► Guanidine-terminated fluorous amphiphiles can bind to phosphonic acid groups in aqueous media, which offers a new strategy for decorating phosphorylated biomolecules with fluorine-rich coating.
Co-reporter:Megan K. Pennington-Boggio, Brian L. Conley, Travis J. Williams
Journal of Organometallic Chemistry 2012 716() pp: 6-10
Publication Date(Web):
DOI:10.1016/j.jorganchem.2012.05.017
Co-reporter:Zhiyao Lu, Brian L. Conley, and Travis J. Williams
Organometallics 2012 Volume 31(Issue 19) pp:6705-6714
Publication Date(Web):August 30, 2012
DOI:10.1021/om300562d
We propose a mechanistic model for three-stage dehydrogenation of ammonia–borane (AB) catalyzed by Shvo’s cyclopentadienone-ligated ruthenium complex. We provide evidence for a plausible mechanism for catalyst deactivation and the transition from fast catalysis to slow catalysis and relate those findings to the invention of a second-generation catalyst that does not suffer from the same deactivation chemistry. The primary mechanism of catalyst deactivation is borazine-mediated hydroboration of the ruthenium species that is the active oxidant in the fast catalysis case. This transition is characterized by a change in the rate law for the reaction and changes in the apparent resting state of the catalyst. Also, in this slow catalysis situation, we see an additional intermediate in the sequence of boron, nitrogen species, aminodiborane. This occurs with concurrent generation of NH3, which itself does not strongly affect the rate of AB dehydrogenation.
Co-reporter:Brian L. Conley ; Denver Guess
Journal of the American Chemical Society 2011 Volume 133(Issue 36) pp:14212-14215
Publication Date(Web):August 9, 2011
DOI:10.1021/ja2058154
We describe an efficient homogeneous ruthenium catalyst for the dehydrogenation of ammonia borane (AB). This catalyst liberates more than 2 equiv of H2 and up to 4.6 system wt % H2 from concentrated AB suspensions under air. Importantly, this catalyst is robust, delivering several cycles of dehydrogenation at high [AB] without loss of catalytic activity, even with exposure to air and water.
Co-reporter:Travis J. Williams, Allan D. Kershaw, Vincent Li, and Xinping Wu
Journal of Chemical Education 2011 Volume 88(Issue 5) pp:665-669
Publication Date(Web):March 11, 2011
DOI:10.1021/ed1006822
A convenient laboratory experiment is described in which NMR magnetization transfer by inversion recovery is used to measure the kinetics and thermochemistry of amide bond rotation. The experiment utilizes Varian spectrometers with the VNMRJ 2.3 software, but can be easily adapted to any NMR platform. The procedures and sample data sets in this article will enable instructors to use inversion recovery as a laboratory activity in applied NMR classes and provide research students with a convenient template with which to acquire inversion recovery data on research samples.Keywords: Analytical Chemistry; Computer-Based Learning; Equilibrium; Graduate Education/Research; Hands-On Learning/Manipulatives; Instrumental Methods; Kinetics; Laboratory Instruction; NMR Spectroscopy; Upper-Division Undergraduate;
Co-reporter:Brian L. Conley, Megan K. Pennington-Boggio, Emine Boz and Travis J. Williams
Chemical Reviews 2010 Volume 110(Issue 4) pp:2294
Publication Date(Web):January 22, 2010
DOI:10.1021/cr9003133
Co-reporter:Brian L. Conley
Journal of the American Chemical Society 2010 Volume 132(Issue 6) pp:1764-1765
Publication Date(Web):January 20, 2010
DOI:10.1021/ja909858a
A boron-pendant ruthenium species forms a unique agostic methyl bridge between the boron and ruthenium atoms in the presence of a ligating solvent, acetonitrile. NMR inversion−recovery experiments enabled the activation and equilibrium thermochemistry for formation of the agostic bridge to be measured. The mechanism for bridge formation involves displacement of an acetonitrile ligand; thus, this is a rare example of a case where an agostic C−H ligand competitively displaces another tightly binding ligand from a coordinatively saturated complex. Characterization of this complex gives unique insights into the development of C−H activation catalysis based on this ligand−metal bifunctional motif.
Co-reporter:Brian L. Conley and Travis J. Williams
Chemical Communications 2010 vol. 46(Issue 26) pp:4815-4817
Publication Date(Web):27 May 2010
DOI:10.1039/C003157G
Shvo's cyclopentadienone-ligated ruthenium complex is an efficient catalyst for the liberation of exactly two molar equivalents of hydrogen from ammonia-borane, a prospective hydrogen storage medium. The mechanism for the dehydrogenation features a ruthenium hydride resting state from which dihydrogen loss is the rate-determining step.
Co-reporter:Megan K. Thorson;Kortney L. Klinkel;Jianmei Wang
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 2) pp:295-302
Publication Date(Web):
DOI:10.1002/ejic.200800975
Abstract
Cyclopentadienone-ligated ruthenium complexes, such as Shvo's catalyst, are known to oxidize reversibly alcohols to the corresponding carbonyl compounds. The mechanism of this reaction has been the subject of some controversy, but it is generally believed to proceed through concerted transfer of proton and hydride, respectively, to the cyclopentadienone ligand and the ruthenium center. In this paper we further study the hydride transfer process as an example of a coordinatively directed hydride abstraction by adding quantitative understanding to some features of this mechanism that are not well understood. We find that an oxidant as weak as acetone can be used to re-oxidize the intermediate ruthenium hydride without catalyst re-oxidation becoming rate-limiting. Furthermore, C–H cleavage is a significantly electrophilic event, as demonstrated by a Hammett reaction parameter of ρ = –0.89. We then describe how the application of our mechanistic insights obtained from the study have enabled us to extend the ligand-directed hydride abstraction strategy to include a rare example of an iron(0) oxidation catalyst.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Brian L. Conley and Travis J. Williams
Chemical Communications 2010 - vol. 46(Issue 26) pp:NaN4817-4817
Publication Date(Web):2010/05/27
DOI:10.1039/C003157G
Shvo's cyclopentadienone-ligated ruthenium complex is an efficient catalyst for the liberation of exactly two molar equivalents of hydrogen from ammonia-borane, a prospective hydrogen storage medium. The mechanism for the dehydrogenation features a ruthenium hydride resting state from which dihydrogen loss is the rate-determining step.
Co-reporter:Anna C. Dawsey, Vincent Li, Kimberly C. Hamilton, Jianmei Wang and Travis J. Williams
Dalton Transactions 2012 - vol. 41(Issue 26) pp:NaN8002-8002
Publication Date(Web):2012/02/29
DOI:10.1039/C2DT00025C
We report herein convenient, aerobic conditions for the oxidation of thiazolines to thiazoles and data regarding the oxidation mechanism. These reactions feature operationally simple and environmentally benign conditions and proceed in good yield to afford the corresponding azoles, thus enabling the inexpensive preparation of valuable molecular building blocks. Incorporation of a novel diimine-ligated copper catalyst, [(MesDABMe)CuII(OH2)3]2+ [−OTf]2, provides increased reaction efficiency in many cases. In other cases copper-free conditions involving a stoichiometric quantity of base affords superior results.
Co-reporter:Zhiyao Lu and Travis J. Williams
Chemical Communications 2014 - vol. 50(Issue 40) pp:NaN5393-5393
Publication Date(Web):2013/12/24
DOI:10.1039/C3CC47384H
We report a novel ruthenium bis(pyrazolyl)borate scaffold that enables cooperative reduction reactivity in which boron and ruthenium centers work in concert to effect selective nitrile reduction. The pre-catalyst compound [κ3-(1-pz)2HB(N = CHCH3)]Ru(cymene)+ TfO− (pz = pyrazolyl) was synthesized using readily-available materials through a straightforward route, thus making it an appealing catalyst for a number of reactions.
Co-reporter:Vincent Li, Yoo-Jin Ghang, Richard J. Hooley and Travis J. Williams
Chemical Communications 2014 - vol. 50(Issue 11) pp:NaN1377-1377
Publication Date(Web):2013/12/11
DOI:10.1039/C3CC48389D
The relaxivity of a magnetically responsive Gd complex can be controlled by non-covalent molecular recognition with a water-soluble deep cavitand. Lowered relaxivity is conferred by a self-assembled micellar “off state”, and the contrast can be regenerated by addition of a superior guest.
Co-reporter:Xingyue Zhang, Lisa Kam and Travis J. Williams
Dalton Transactions 2016 - vol. 45(Issue 18) pp:NaN7677-7677
Publication Date(Web):2016/03/23
DOI:10.1039/C6DT00604C
Ammonia borane (AB) has high hydrogen density (19.6 wt%), and can, in principle, release up to 3 equivalents of H2 under mild catalytic conditions. A limited number of catalysts are capable of non-hydrolytic dehydrogenation of AB beyond 2 equivalents of H2 under mild conditions, but none of these is shown directly to derivatise borazine, the product formed after 2 equivalents of H2 are released. We present here a high productivity ruthenium-based catalyst for non-hydrolytic AB dehydrogenation that is capable of borazine dehydrogenation, and thus exhibits among the highest H2 productivity reported to date for anhydrous AB dehydrogenation. At 1 mol% loading, (phen)Ru(OAc)2(CO)2 (1) effects AB dehydrogenation through 2.7 equivalents of H2 at 70 °C, is robust through multiple charges of AB, and is water and air stable. We further demonstrate that catalyst 1 has the ability both to dehydrogenate borazine in isolation and dehydrogenate AB itself. This is important, both because borazine derivatisation is productivity-limiting in AB dehydrogenation and because borazine is a fuel cell poison that is commonly released in H2 production from this medium.

![1H-Indole-3-ethanamine, N-[1-(4-methylphenyl)ethyl]-](http://img.cochemist.com/ccimg/917100/917011-84-8.png)
![1H-Indole-3-ethanamine, N-[1-(4-methylphenyl)ethyl]-](http://img.cochemist.com/ccimg/917100/917011-84-8_b.png)
![1H-Indole-3-ethanamine, N-[1-(4-fluorophenyl)ethyl]-](http://img.cochemist.com/ccimg/917100/917011-44-0.png)
![1H-Indole-3-ethanamine, N-[1-(4-fluorophenyl)ethyl]-](http://img.cochemist.com/ccimg/917100/917011-44-0_b.png)


![Pyridine, 2-[[bis(1,1-dimethylethyl)phosphino]methyl]-](http://img.cochemist.com/ccimg/494200/494199-72-3.png)
![Pyridine, 2-[[bis(1,1-dimethylethyl)phosphino]methyl]-](http://img.cochemist.com/ccimg/494200/494199-72-3_b.png)
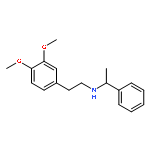
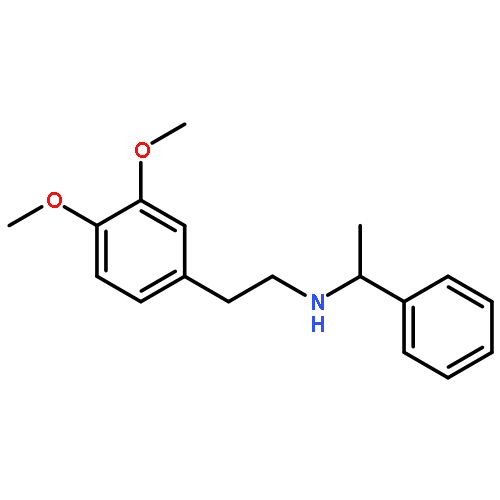


![Phenol, 4-[2-[(phenylmethyl)amino]ethyl]-](http://img.cochemist.com/ccimg/52500/52447-50-4.png)
![Phenol, 4-[2-[(phenylmethyl)amino]ethyl]-](http://img.cochemist.com/ccimg/52500/52447-50-4_b.png)



















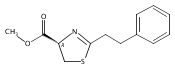

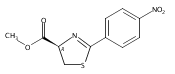






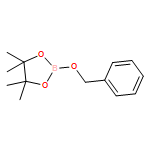
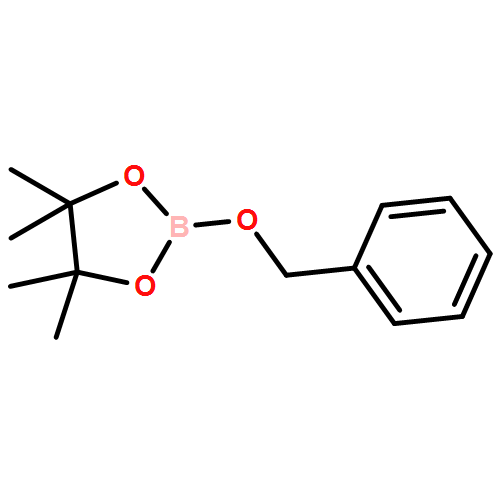

![Gadolinium 2,2',2''-[10-(carboxymethyl)-1,4,7,10-tetraazacyclodod Ecane-1,4,7-triyl]triacetate - 1-deoxy-1-(methylamino)-d-glucitol (1:1:1)](http://img.cochemist.com/ccimg/93000/92943-93-6.png)
![Gadolinium 2,2',2''-[10-(carboxymethyl)-1,4,7,10-tetraazacyclodod Ecane-1,4,7-triyl]triacetate - 1-deoxy-1-(methylamino)-d-glucitol (1:1:1)](http://img.cochemist.com/ccimg/93000/92943-93-6_b.png)

![3,7,7-TRIMETHYLBICYCLO[4.1.0]HEPT-1-ENE](http://img.cochemist.com/ccimg/74900/74806-04-5.png)
![3,7,7-TRIMETHYLBICYCLO[4.1.0]HEPT-1-ENE](http://img.cochemist.com/ccimg/74900/74806-04-5_b.png)








![(3S,3aS,9aS,9bS)-3,6,9-Trimethyl-3,3a,4,5,9a,9b-hexahydroazuleno[ 4,5-b]furan-2,7-dione](http://img.cochemist.com/ccimg/18000/17946-87-1.png)
![(3S,3aS,9aS,9bS)-3,6,9-Trimethyl-3,3a,4,5,9a,9b-hexahydroazuleno[ 4,5-b]furan-2,7-dione](http://img.cochemist.com/ccimg/18000/17946-87-1_b.png)
![[1,6]Dioxacyclododecino[2,3,4-gh]pyrrolizine-2,7-dione,3-ethylidene-3,4,5,6,9,11,13,14,14a,14b-decahydro-6-hydroxy-6-(hydroxymethyl)-5-methyl-,(3E,5R,6S,14aR,14bR)-](http://img.cochemist.com/ccimg/15600/15503-87-4.png)
![[1,6]Dioxacyclododecino[2,3,4-gh]pyrrolizine-2,7-dione,3-ethylidene-3,4,5,6,9,11,13,14,14a,14b-decahydro-6-hydroxy-6-(hydroxymethyl)-5-methyl-,(3E,5R,6S,14aR,14bR)-](http://img.cochemist.com/ccimg/15600/15503-87-4_b.png)
![2H-1-Benzopyran-2-one,7-[[(2E,6R)-6,7-dihydroxy-3,7-dimethyl-2-octen-1-yl]oxy]-](http://img.cochemist.com/ccimg/15000/14957-38-1.png)
![2H-1-Benzopyran-2-one,7-[[(2E,6R)-6,7-dihydroxy-3,7-dimethyl-2-octen-1-yl]oxy]-](http://img.cochemist.com/ccimg/15000/14957-38-1_b.png)





![Acetic acid,2-[[3-[2-(diethylamino)ethyl]-4-methyl-2-oxo-2H-1-benzopyran-7-yl]oxy]-, ethylester](http://img.cochemist.com/ccimg/900/804-10-4.png)
![Acetic acid,2-[[3-[2-(diethylamino)ethyl]-4-methyl-2-oxo-2H-1-benzopyran-7-yl]oxy]-, ethylester](http://img.cochemist.com/ccimg/900/804-10-4_b.png)