Co-reporter:Marc van Dijk, Antonius M. ter Laak, Jörg D. Wichard, Luigi Capoferri, Nico P. E. Vermeulen, and Daan P. Geerke
Journal of Chemical Information and Modeling September 25, 2017 Volume 57(Issue 9) pp:2294-2294
Publication Date(Web):August 4, 2017
DOI:10.1021/acs.jcim.7b00222
Cytochrome P450 aromatase (CYP19A1) plays a key role in the development of estrogen dependent breast cancer, and aromatase inhibitors have been at the front line of treatment for the past three decades. The development of potent, selective and safer inhibitors is ongoing with in silico screening methods playing a more prominent role in the search for promising lead compounds in bioactivity-relevant chemical space. Here we present a set of comprehensive binding affinity prediction models for CYP19A1 using our automated Linear Interaction Energy (LIE) based workflow on a set of 132 putative and structurally diverse aromatase inhibitors obtained from a typical industrial screening study. We extended the workflow with machine learning methods to automatically cluster training and test compounds in order to maximize the number of explained compounds in one or more predictive LIE models. The method uses protein–ligand interaction profiles obtained from Molecular Dynamics (MD) trajectories to help model search and define the applicability domain of the resolved models. Our method was successful in accounting for 86% of the data set in 3 robust models that show high correlation between calculated and observed values for ligand-binding free energies (RMSE < 2.5 kJ mol–1), with good cross-validation statistics.
Co-reporter:C. Ruben Vosmeer;Derk P. Kooi;Luigi Capoferri
Journal of Molecular Modeling 2016 Volume 22( Issue 1) pp:
Publication Date(Web):2016 January
DOI:10.1007/s00894-015-2883-y
Recently an iterative method was proposed to enhance the accuracy and efficiency of ligand-protein binding affinity prediction through linear interaction energy (LIE) theory. For ligand binding to flexible Cytochrome P450s (CYPs), this method was shown to decrease the root-mean-square error and standard deviation of error prediction by combining interaction energies of simulations starting from different conformations. Thereby, different parts of protein-ligand conformational space are sampled in parallel simulations. The iterative LIE framework relies on the assumption that separate simulations explore different local parts of phase space, and do not show transitions to other parts of configurational space that are already covered in parallel simulations. In this work, a method is proposed to (automatically) detect such transitions during the simulations that are performed to construct LIE models and to predict binding affinities. Using noise-canceling techniques and splines to fit time series of the raw data for the interaction energies, transitions during simulation between different parts of phase space are identified. Boolean selection criteria are then applied to determine which parts of the interaction energy trajectories are to be used as input for the LIE calculations. Here we show that this filtering approach benefits the predictive quality of our previous CYP 2D6-aryloxypropanolamine LIE model. In addition, an analysis is performed of the gain in computational efficiency that can be obtained from monitoring simulations using the proposed filtering method and by prematurely terminating simulations accordingly.
Co-reporter:C. Ruben Vosmeer, Karin Kiewisch, Karlijn Keijzer, Lucas Visscher and Daan P. Geerke
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 33) pp:17857-17862
Publication Date(Web):21 Jul 2014
DOI:10.1039/C4CP02401J
Recently we reported a combined QM/MM approach to estimate condensed-phase values of atomic polarizabilities for use in (bio)molecular simulation. The setup relies on a MM treatment of the solvent when determining atomic polarizabilities to describe the response of a QM described solute to its external electric field. In this work, we study the effect of using alternative descriptions of the solvent molecules when evaluating atomic polarizabilities of a methanol solute. In a first step, we show that solute polarizabilities are not significantly affected upon substantially increasing the MM dipole moments towards values that are typically reported in literature for water solvent molecules. Subsequently, solute polarization is evaluated in the presence of a QM described solvent (using the frozen-density embedding method). In the latter case, lower oxygen polarizabilities were obtained than when using MM point charges to describe the solvent, due to introduction of Pauli-repulsion effects.
Co-reporter:C. Ruben Vosmeer, Ariën S. Rustenburg, Julia E. Rice, Hans W. Horn, William C. Swope, and Daan P. Geerke
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 10) pp:3839-3853
Publication Date(Web):April 27, 2012
DOI:10.1021/ct300085z
Accounting for electronic polarization effects in biomolecular simulation (by using a polarizable force field) can increase the accuracy of simulation results. However, the use of gas-phase estimates of atomic polarizabilities αi usually leads to overpolarization in condensed-phase systems. In the current work, a combined QM/MM approach has been employed to obtain condensed-phase estimates of atomic polarizabilities for water and methanol (QM) solutes in the presence of (MM) solvents of different polarity. In a next step, the validity of the linear response and isotropy assumptions were evaluated based on the observed condensed-phase distributions of αi values. The observed anisotropy and low average values for the polarizability of methanol’s carbon atom in polar solvents was explained in terms of strong solute–solvent interactions involving its adjacent hydroxyl group. Our QM/MM estimates for atomic polarizabilities were found to be close to values used in previously reported polarizable water and methanol models. Using our estimate for αO of methanol, a single set of polarizable force field parameters was obtained that is directly transferable between environments of different polarity.
Co-reporter:C. Ruben Vosmeer, Karin Kiewisch, Karlijn Keijzer, Lucas Visscher and Daan P. Geerke
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 33) pp:NaN17862-17862
Publication Date(Web):2014/07/21
DOI:10.1039/C4CP02401J
Recently we reported a combined QM/MM approach to estimate condensed-phase values of atomic polarizabilities for use in (bio)molecular simulation. The setup relies on a MM treatment of the solvent when determining atomic polarizabilities to describe the response of a QM described solute to its external electric field. In this work, we study the effect of using alternative descriptions of the solvent molecules when evaluating atomic polarizabilities of a methanol solute. In a first step, we show that solute polarizabilities are not significantly affected upon substantially increasing the MM dipole moments towards values that are typically reported in literature for water solvent molecules. Subsequently, solute polarization is evaluated in the presence of a QM described solvent (using the frozen-density embedding method). In the latter case, lower oxygen polarizabilities were obtained than when using MM point charges to describe the solvent, due to introduction of Pauli-repulsion effects.

![2-Cyclohexen-1-one, 3,5,5-trimethyl-4-[(1E)-3-oxo-1-butenyl]-](http://img.cochemist.com/ccimg/79800/79734-43-3.png)
![2-Cyclohexen-1-one, 3,5,5-trimethyl-4-[(1E)-3-oxo-1-butenyl]-](http://img.cochemist.com/ccimg/79800/79734-43-3_b.png)
![12-methylbenzo[a]anthracene-3,9-diol](http://img.cochemist.com/ccimg/80200/80150-02-3.png)
![12-methylbenzo[a]anthracene-3,9-diol](http://img.cochemist.com/ccimg/80200/80150-02-3_b.png)














![(8r,9s,10r,13s,14s,16s,17r)-16,17-dihydroxy-10,13-dimethyl-1,2,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-3-one](http://img.cochemist.com/ccimg/17600/17528-90-4.png)
![(8r,9s,10r,13s,14s,16s,17r)-16,17-dihydroxy-10,13-dimethyl-1,2,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-3-one](http://img.cochemist.com/ccimg/17600/17528-90-4_b.png)


![2-[2,6-dichloro-3-(hydroxymethyl)anilino]benzoic Acid](http://img.cochemist.com/ccimg/67400/67318-61-0.png)
![2-[2,6-dichloro-3-(hydroxymethyl)anilino]benzoic Acid](http://img.cochemist.com/ccimg/67400/67318-61-0_b.png)


![2-[3-(hydroxymethyl)-2-methylanilino]benzoic Acid](http://img.cochemist.com/ccimg/5200/5129-20-4.png)
![2-[3-(hydroxymethyl)-2-methylanilino]benzoic Acid](http://img.cochemist.com/ccimg/5200/5129-20-4_b.png)




![Benzo[a]pyrene-7,8-diol](http://img.cochemist.com/ccimg/57400/57303-99-8.png)
![Benzo[a]pyrene-7,8-diol](http://img.cochemist.com/ccimg/57400/57303-99-8_b.png)
![3-Buten-2-one,4-[(1S)-2,6,6-trimethyl-2-cyclohexen-1-yl]-, (3E)-](http://img.cochemist.com/ccimg/14400/14398-36-8.png)
![3-Buten-2-one,4-[(1S)-2,6,6-trimethyl-2-cyclohexen-1-yl]-, (3E)-](http://img.cochemist.com/ccimg/14400/14398-36-8_b.png)




![BENZO[A]PYREN-10-OL](http://img.cochemist.com/ccimg/56900/56892-31-0.png)
![BENZO[A]PYREN-10-OL](http://img.cochemist.com/ccimg/56900/56892-31-0_b.png)









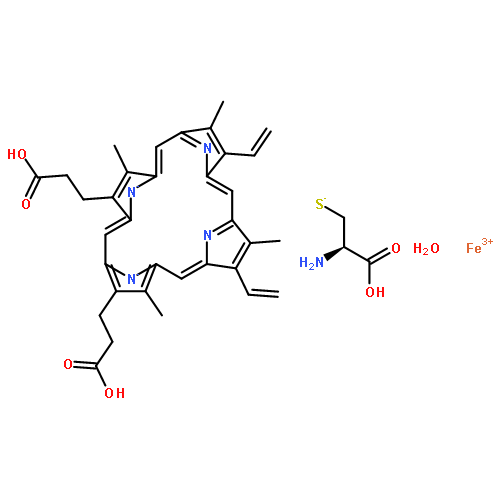
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)
![BENZO[A]PYREN-2-OL](http://img.cochemist.com/ccimg/56900/56892-30-9.png)
![BENZO[A]PYREN-2-OL](http://img.cochemist.com/ccimg/56900/56892-30-9_b.png)
![BENZO[A]ANTHRACENE-3,9-DIOL](http://img.cochemist.com/ccimg/56700/56614-97-2.png)
![BENZO[A]ANTHRACENE-3,9-DIOL](http://img.cochemist.com/ccimg/56700/56614-97-2_b.png)
![ETHYL 5-BROMO-1H-PYRROLO[2,3-B]PYRIDINE-2-CARBOXYLATE](http://img.cochemist.com/ccimg/38000/37994-82-4.png)
![ETHYL 5-BROMO-1H-PYRROLO[2,3-B]PYRIDINE-2-CARBOXYLATE](http://img.cochemist.com/ccimg/38000/37994-82-4_b.png)


![Benzo[a]pyren-1-ol](http://img.cochemist.com/ccimg/13400/13345-23-8.png)
![Benzo[a]pyren-1-ol](http://img.cochemist.com/ccimg/13400/13345-23-8_b.png)
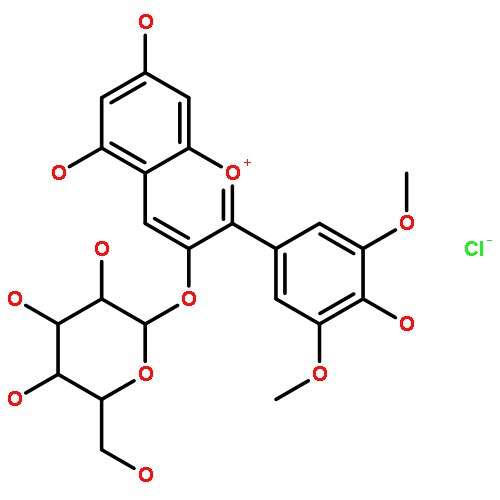
![1-Benzopyrylium,3-[[6-O-(6-deoxy-a-L-mannopyranosyl)-b-D-glucopyranosyl]oxy]-5,7-dihydroxy-2-(4-hydroxy-3-methoxyphenyl)-,chloride (1:1)](http://img.cochemist.com/ccimg/27600/27539-32-8.png)
![1-Benzopyrylium,3-[[6-O-(6-deoxy-a-L-mannopyranosyl)-b-D-glucopyranosyl]oxy]-5,7-dihydroxy-2-(4-hydroxy-3-methoxyphenyl)-,chloride (1:1)](http://img.cochemist.com/ccimg/27600/27539-32-8_b.png)
![Benzo[a]pyren-8-ol](http://img.cochemist.com/ccimg/13400/13345-26-1.png)
![Benzo[a]pyren-8-ol](http://img.cochemist.com/ccimg/13400/13345-26-1_b.png)

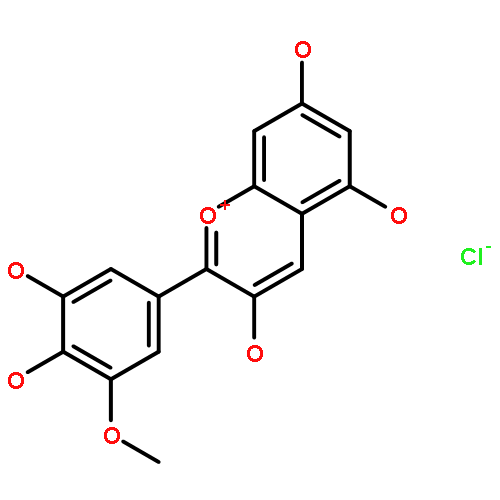


![Benzo[pqr]tetraphen-3-ol](http://img.cochemist.com/ccimg/13400/13345-21-6.png)
![Benzo[pqr]tetraphen-3-ol](http://img.cochemist.com/ccimg/13400/13345-21-6_b.png)




![7,14-Methano-2H,6H-dipyrido[1,2-a:1',2'-e][1,5]diazocine,dodecahydro-, (7S,7aR,14S,14aR)-](http://img.cochemist.com/ccimg/500/446-95-7.png)
![7,14-Methano-2H,6H-dipyrido[1,2-a:1',2'-e][1,5]diazocine,dodecahydro-, (7S,7aR,14S,14aR)-](http://img.cochemist.com/ccimg/500/446-95-7_b.png)




![(2S,8R,9S,10R,13S,14S,17S)-2,17-DIHYDROXY-10,13-DIMETHYL-1,2,6,7,8,9,11,12,14,15,16,17-DODECAHYDROCYCLOPENTA[A]PHENANTHREN-3-ONE](http://img.cochemist.com/ccimg/10400/10390-14-4.png)
![(2S,8R,9S,10R,13S,14S,17S)-2,17-DIHYDROXY-10,13-DIMETHYL-1,2,6,7,8,9,11,12,14,15,16,17-DODECAHYDROCYCLOPENTA[A]PHENANTHREN-3-ONE](http://img.cochemist.com/ccimg/10400/10390-14-4_b.png)






![3-Buten-2-one,4-[(1R)-2,6,6-trimethyl-2-cyclohexen-1-yl]-, (3E)-](http://img.cochemist.com/ccimg/24200/24190-29-2.png)
![3-Buten-2-one,4-[(1R)-2,6,6-trimethyl-2-cyclohexen-1-yl]-, (3E)-](http://img.cochemist.com/ccimg/24200/24190-29-2_b.png)