Co-reporter:Katerina Kumpan, Amit Nathubhai, Chenlu Zhang, Pauline J. Wood, Matthew D. Lloyd, Andrew S. Thompson, Teemu Haikarainen, Lari Lehtiö, Michael D. Threadgill
Bioorganic & Medicinal Chemistry 2015 23(13) pp: 3013-3032
Publication Date(Web):
DOI:10.1016/j.bmc.2015.05.005
Co-reporter:Helen A. Paine, Amit Nathubhai, Esther C.Y. Woon, Peter T. Sunderland, Pauline J. Wood, Mary F. Mahon, Matthew D. Lloyd, Andrew S. Thompson, Teemu Haikarainen, Mohit Narwal, Lari Lehtiö, Michael D. Threadgill
Bioorganic & Medicinal Chemistry 2015 23(17) pp: 5891-5908
Publication Date(Web):
DOI:10.1016/j.bmc.2015.06.061
Co-reporter:Elvis A. Twum, Timothy J. Woodman, Wenyi Wang and Michael D. Threadgill
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 36) pp:6208-6214
Publication Date(Web):07 Aug 2013
DOI:10.1039/C3OB41386A
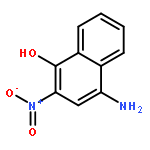
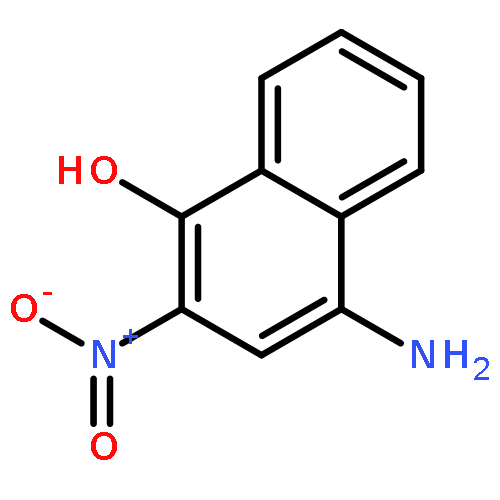
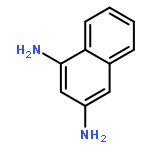
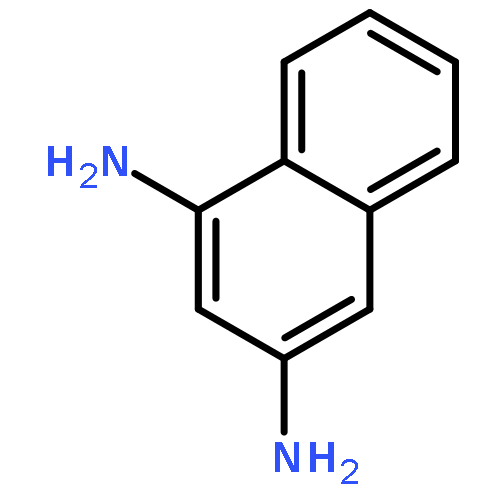
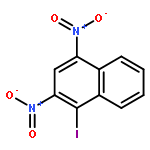
1-Iodonaphthalene-2,4-diamines in trifluoroacetic acid/chloroform give stable Wheland-like tetrahedral cationic species observable by NMR, through an initial intramolecular protonation. Dynamic equilibria allow proton-deuterium exchange of aromatic protons and provide a mechanism for deiodination of 1-iodonaphthalene-2,4-diamines.
Co-reporter:Amit Nathubhai, Pauline J. Wood, Matthew D. Lloyd, Andrew S. Thompson, and Michael D. Threadgill
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 12) pp:1173-1177
Publication Date(Web):October 15, 2013
DOI:10.1021/ml400260b
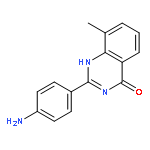
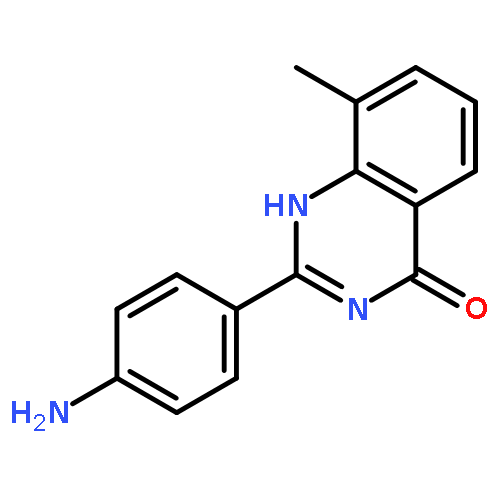
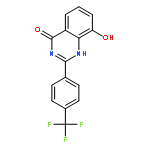
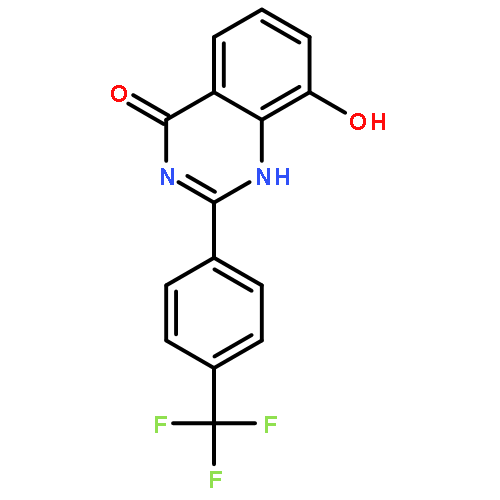
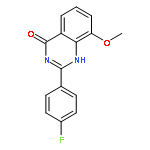
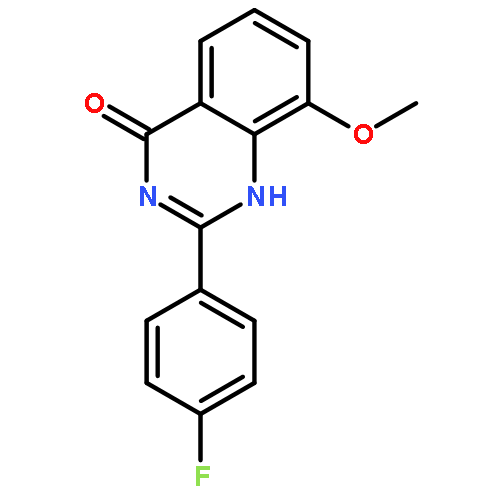
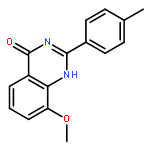
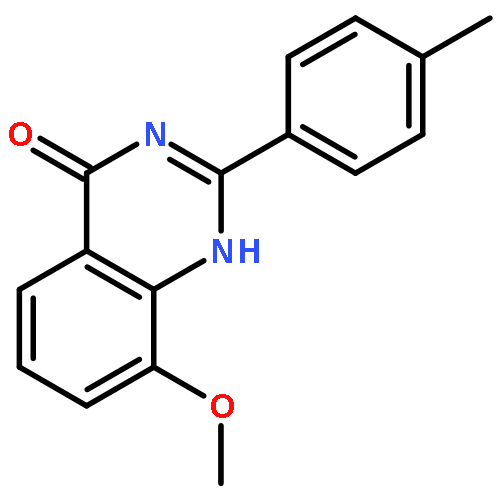
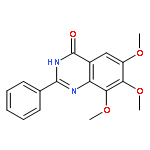
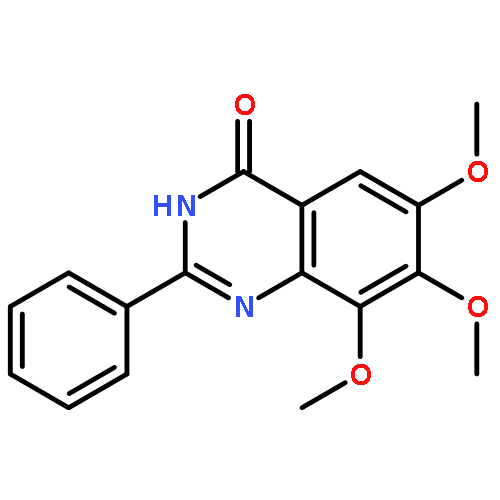
Tankyrases (TNKSs) are poly(ADP-ribose)polymerases (PARPs) that are overexpressed in several clinical cancers. They regulate elongation of telomeres, regulate the Wnt system, and are essential for the function of the mitotic spindle. A set of 2-arylquinazolin-4-ones has been designed and identified as potent and selective TNKS inhibitors, some being more potent and selective than the lead inhibitor XAV939, with IC50 = 3 nM vs. TNKS-2. Methyl was preferred at the 8-position and modest bulk at the 4-position of the 2-phenyl group; electronic effects and H-bonding were irrelevant, but charge in the 4′-substituent must be avoided. Molecular modeling facilitated initial design of the compounds and rationalization of the SAR of binding into the nicotinamide-binding site of the target enzymes. These compounds have potential for further development into anticancer drugs.Keywords: hydrophobic pocket; PARP; quinazolin-4-one; selectivity; Tankyrase;
Co-reporter:Esther C.Y. Woon, Peter T. Sunderland, Helen A. Paine, Matthew D. Lloyd, Andrew S. Thompson, Michael D. Threadgill
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 17) pp:5218-5227
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmc.2013.06.031
Poly(ADP-ribose)polymerase-1 (PARP-1) is an important target for drug design for several therapeutic applications. 5-Aminoisoquinolin-1-one (5-AIQ) is a highly water-soluble lead compound; synthetic routes to 3-substituted analogues were explored. Tandem Hurtley coupling of β-diketones with 2-bromo-3-nitrobenzoic acid, retro-Claisen acyl cleavage and cyclisation gave the corresponding 3-substituted 5-nitroisocoumarins. Treatment with ammonia at high temperature and reduction with tin(II) chloride gave eleven target 3-substituted 5-AIQs, which were all soluble in water (>1% w/v) as their HCl salts. Most were more potent than 5-AIQ as inhibitors of PARP-1 and of PARP-2 in vitro, the most active being 5-amino-3-methylisoquinolin-1-one (PARP-1: IC50 = 0.23 μM vs IC50 = 1.6 μM for 5-AIQ). Some rationalisation of the SAR was achieved through molecular modelling.
Co-reporter:Peter T. Sunderland ; Esther C. Y. Woon ; Archana Dhami ; Aoife B. Bergin ; Mary F. Mahon ; Pauline J. Wood ; Louise A. Jones ; Sophie R. Tully ; Matthew D. Lloyd ; Andrew S. Thompson ; Hashim Javaid ; Niall M. B. Martin ;Michael D. Threadgill
Journal of Medicinal Chemistry 2011 Volume 54(Issue 7) pp:2049-2059
Publication Date(Web):March 18, 2011
DOI:10.1021/jm1010918
PARP-2 is a member of the poly(ADP-ribose) polymerase family, with some activities similar to those of PARP-1 but with other distinct roles. Two series of isoquinolin-1-ones were designed, synthesized, and evaluated as selective inhibitors of PARP-2, using the structures of the catalytic sites of the isoforms. A new efficient synthesis of 5-aminoisoquinolin-1-one was developed, and acylation with acyl chlorides gave 5-acylaminoisoquinolin-1-ones. By examination of isoquinolin-1-ones with carboxylates tethered to the 5-position, Heck coupling of 5-iodoisoquinolin-1-one furnished the 5-CH═CHCO2H compound for reduction to the 5-propanoic acid. Alkylation of 5-aminoisoquinolin-1-one under mildly basic conditions, followed by hydrolysis, gave 5-(carboxymethylamino)isoquinolin-1-one, whereas it was alkylated at 2-N with methyl propenoate and strong base. Compounds were assayed in vitro for inhibition of PARP-1 and PARP-2, using FlashPlate and solution-phase assays, respectively. The 5-benzamidoisoquinolin-1-ones were more selective for inhibition of PARP-2, whereas the 5-(ω-carboxyalkyl)isoquinolin-1-ones were less so. 5-Benzamidoisoquinolin-1-one is the most PARP-2-selective compound (IC50(PARP-1)/IC50(PARP-2) = 9.3) to date, in a comparative study.
Co-reporter:Peter T. Sunderland, Archana Dhami, Mary F. Mahon, Louise A. Jones, Sophie R. Tully, Matthew D. Lloyd, Andrew S. Thompson, Hashim Javaid, Niall M. B. Martin and Michael D. Threadgill
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 3) pp:881-891
Publication Date(Web):02 Dec 2010
DOI:10.1039/C0OB00665C
The considerable interest in substituted isoquinolin-1-ones related to 5-aminoisoquinolin-1-one (5-AIQ) as drugs points to a need for an efficient and straightforward synthesis of the 4,5-disubstituted bicycles. Bromination of 5-nitroisoquinolin-1-one gave 4-bromo-5-nitroisoquinolin-1-one but neither this nor 5-amino-4-bromoisoquinolin-1-one would participate in Pd-catalysed couplings. Protection of the lactam as 1-methoxy- and 1-benzyloxy-4-bromo-5-nitroisoquinolines, however, permitted Stille, Suzuki and Buchwald–Hartwig couplings to take place in high yields, insensitive to electronic demands and severe steric bulk in the arylboronic acids. Lithiation of 4-bromo-1-methoxy-5-nitroisoquinoline and quench with iodomethane gave 1-methoxy-4-methyl-5-nitroisoquinoline in low yield. Demethylation of the 1-methoxy-4-substituted-5-nitroisoquinolines with hydrogen bromide gave 4-substituted-5-nitroisoquinolin-1-ones, whereas hydrogenolytic debenzylation was achieved with simultaneous reduction of the 5-nitro group. 5-Amino-4-(4-trifluoromethylphenyl)isoquinolin-1-one was identified as a new potent and selective inhibitor of poly(ADP-ribose)polymerase-2 (PARP-2).
Co-reporter:Amit Nathubhai, Richard Patterson, Timothy J. Woodman, Harriet E. C. Sharp, Miranda T. Y. Chui, Hugo H. K. Chung, Stephanie W. S. Lau, Jun Zheng, Matthew D. Lloyd, Andrew S. Thompson and Michael D. Threadgill
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 17) pp:6089-6099
Publication Date(Web):26 May 2011
DOI:10.1039/C1OB05430A
Dimethylformamide dimethylacetal (DMFDMA) is widely used as a source of electrophilic one-carbon units at the formate oxidation level; however, electrophilic methylation with this reagent is previously unreported. Reaction of anthranilamide with DMFDMA at 150 °C for short periods gives mainly quinazolin-4-one. However, prolonged reaction with dimethylformamide di(primary-alkyl)acetals leads to subsequent alkylation at N3. 3-Substituted anthranilamides give 8-substituted 3-alkylquinazolin-4-ones. Condensation of anthranilamides with dimethylacetamide dimethylacetal provides 2,3-dimethylquinazolin-4-ones. In these reactions, the source of the N3-alkyl group is the O-alkyl group of the orthoamides. By contrast, reaction with the more sterically crowded dimethylformamide di(isopropyl)acetal diverts the alkylation to the oxygen, giving 4-isopropoxyquinazolines, along with N3-methylquinazolin-4-ones where the methyl is derived from N-Me of the orthoamides. Reaction of anthranilamide with the highly sterically demanding dimethylformamide di(t-butyl)acetal gives largely quinazolin-4-one, whereas dimethylformamide di(neopentyl)acetal forms a mixture of quinazolin-4-one and N3-methylquinazolin-4-one. The observations are rationalised in terms of formation of intermediate cationic electrophiles (alkoxymethylidene-N,N-dimethylammonium) by thermal elimination of the corresponding alkoxide from the orthoamides. These are the first observations of orthoamides as direct alkylating agents.
Co-reporter:Anne Beauchard, Elvis A. Twum, Matthew D. Lloyd, Michael D. Threadgill
Tetrahedron Letters 2011 Volume 52(Issue 41) pp:5311-5314
Publication Date(Web):12 October 2011
DOI:10.1016/j.tetlet.2011.08.017
Glutamine (Gln) is often a difficult amino acid to incorporate during solution-phase peptide synthesis, owing to poor solubility and unwanted dehydrations as side-reactions. Current approaches to solving these problems are highly atom-inefficient. Nα-Cbz-β-cyanomethyl-l-Ala is readily accessible by dehydration of Cbz-l-Gln. β-Cyanomethyl-l-Ala can be incorporated into short peptides easily by conventional methods. The nitrile is stable to the hydrogenolysis conditions used to remove Cbz and to acidic deprotection but is quantitatively hydrated to the γ-carboxamide of l-Gln with hydrogen peroxide. Thus β-cyanomethyl-l-Ala may represent a new, soluble, perfectly atom-efficient synthon for l-Gln.β-Cyanomethyl-l-Ala is a masked form of l-Gln for use in solution-phase peptide synthesis.
Co-reporter:Anna-Marie Lord ; Mary F. Mahon ; Matthew D. Lloyd ;Michael D. Threadgill
Journal of Medicinal Chemistry 2009 Volume 52(Issue 3) pp:868-877
Publication Date(Web):December 31, 2008
DOI:10.1021/jm8013629
Poly(ADP-ribose)polymerase-1 is an important target enzyme in drug design; inhibitors have a wide variety of therapeutic activities. A series of quinoline-8-carboxamides was designed to maintain the required pharmacophore conformation through an intramolecular hydrogen bond. 3-Substituted quinoline-8-carboxamides were synthesized by Pd-catalyzed couplings (Suzuki, Sonogashira, Stille) to 3-iodoquinoline-8-carboxamide, an efficient process that introduces diversity in the final step. 2-Substituted quinoline-8-carboxamides were prepared by selective Pd-catalyzed couplings at the 2-position of 2,8-dibromoquinoline, followed by lithium−bromine exchange of the intermediate 2-(alkyl/aryl)-8-bromoquinolines and reaction with trimethylsilyl isocyanate. The intramolecular hydrogen bond was confirmed by X-ray and by NMR. The SAR of the 3-substituted compounds for inhibition of human recombinant PARP-1 activity showed a requirement for a small narrow group. Substituents in the 2-position increased potency, with the most active 2-methylquinoline-8-carboxamide having IC50 = 500 nM (IC50 = 1.8 μM for 5-aminoisoquinolin-1-one (5-AIQ, a standard water-soluble inhibitor)).
Co-reporter:Anne Beauchard, Victoria A. Phillips, Matthew D. Lloyd, Michael D. Threadgill
Tetrahedron 2009 65(39) pp: 8176-8184
Publication Date(Web):
DOI:10.1016/j.tet.2009.07.085
Co-reporter:Archana Dhami, Mary F. Mahon, Matthew D. Lloyd, Michael D. Threadgill
Tetrahedron 2009 65(24) pp: 4751-4765
Publication Date(Web):
DOI:10.1016/j.tet.2009.04.007
Co-reporter:Ghadeer A.R.Y. Suaifan, Tawfiq Arafat, Michael D. Threadgill
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 10) pp:3474-3488
Publication Date(Web):15 May 2007
DOI:10.1016/j.bmc.2007.03.005
The pseudoprolines S-Dmo (5,5-dimethyl-4-oxaproline) and R-Dmt (5,5-dimethyl-4-thiaproline) have been used to study the effects of forcing a fully cis conformation in peptides. Synthesis of peptides containing these (which have the same configuration as l-Pro) is straightforward. However, synthesis of peptides containing S-Dmt is difficult, owing to the rapid cyclisation of l-Aaa-S-Dmt amides and esters to form the corresponding diketopiperazines (DKP); thus the intermediacy of l-Aaa-S-Dmt amides and esters must be avoided in the synthetic sequence. Peptides containing the l-Gln-l-Val-D(S)-Dmt motif are particularly difficult, owing to the insolubility of coupling partners containing Gln. Introduction of Gln as N-Boc-pyroglutamate overcame the latter difficulty and the dipeptide active ester BocPygValOC6F5 coupled in good yield with S-DmtOH. BocPygVal-S- DmtNH(CH2)2C6H4NO2 was converted quantitatively to BocGlnVal-S-DmtNH(CH2)2C6H4NO2 with ammonia, demonstrating the utility of this approach. Two peptide derivatives (CbzSerLysLeuGlnVal-S-DmtNH(CH2)2C6H4NO2 and CbzSerSerLysLeuGlnVal-S- DmtNH(CH2)2C6H4NO2) were assembled, using these new methods of coupling a dipeptide acid active ester with S-DmtOH and introduction of Gln as Pyg, followed by conventional peptide couplings. The presence of the Val caused these peptides to be cleaved very slowly by prostate-specific antigen (PSA) at Leu ↓ Gln, rather than the expected Gln ↓ Val.
Co-reporter:Christophe Frixa, Mary F. Mahon, Andrew S. Thompson and Michael D. Threadgill
Organic & Biomolecular Chemistry 2003 vol. 1(Issue 2) pp:306-317
Publication Date(Web):19 Dec 2002
DOI:10.1039/B209534C
Selective delivery of 10B to tumours is one of the major remaining problems in boron neutron capture therapy (BNCT) of cancer. Porphyrins are selectively accumulated in tumours. Thus two series of carborane-carrying porphyrins were constructed, with additional functionality for attachment of uncharged potentially water-solubilising polyethers. 3-(1,2-Dicarbaclosododecaboran(12)-1-ylmethoxy)benzaldehyde was prepared by protection of the aldehyde of 3-(prop-2-ynyloxy)benzaldehyde as a dithioacetal, treatment with decaborane(14) and deprotection. Condensation with a 3-nitrophenyldipyrromethane gave a separable mixture of meso-(3-nitrophenyl)-meso-(3-carboranylmethoxyphenyl)porphyrins, resulting from extensive scrambling at the porphyrinogen stage. Similarly, condensation of 3-(1,2-dicarbaclosododecaboran(12)-1-yl)benzaldehyde with this dipyrromethane gave an analogous mixture of meso-(3-nitrophenyl)-meso-(3-carboranylphenyl)porphyrins. In this second series, the two regioisomeric bis(nitrophenyl)bis(carboranylphenyl)porphyrins could only be distinguished by X-ray crystallography, their NMR spectra being identical. The nitro groups of the mono(nitrophenyl)porphyrins and the bis(nitrophenyl)porphyrins were reduced to the corresponding amines with tin(II) chloride and the monoamines were coupled with a ω-methoxy poly(ethylene glycol) chloroformate of mean MW 600 to give the MeOPEGylated tricarboranyl porphyrins.
Co-reporter:Claire L.M. Goodyer, Edwin C. Chinje, Mohammed Jaffar, Ian J. Stratford, Michael D. Threadgill
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 19) pp:4189-4206
Publication Date(Web):15 September 2003
DOI:10.1016/S0968-0896(03)00451-6
Inhibition of the isoforms of nitric oxide synthase (NOS) has important applications in therapy of several diseases, including cancer. Using 1400W [N-(3-aminomethylbenzyl)acetamidine], thiocitrulline and Nδ-(4,5-dihydrothiazol-2-yl)ornithine as lead compounds, series of N-benzyl- and N-phenyl-2-amino-4,5-dihydrothiazoles and thioureas were designed as inhibitors of NOS. Ring-substituted benzyl and phenyl isothiocyanates were synthesised by condensation of the corresponding amines with thiophosgene and addition of ammonia gave the corresponding thioureas in high yields. The substituted 2-amino-4,5-dihydrothiazoles were approached by two routes. Treatment of simple benzylamines with 2-methylthio-4,5-dihydrothiazole at 180 °C afforded the corresponding 2-benzylamino-4,5-dihydrothiazoles. For less nucleophilic amines and those carrying more thermally labile substituents, the 4,5-dihydrothiazoles were approached by acid-catalysed cyclisation of N-(2-hydroxyethyl)thioureas. This cyclisation was shown to proceed by an SN2-like process. Modest inhibitory activity was shown by most of the thioureas and 4,5-dihydrothiazoles, with N-(3-aminomethylphenyl)thiourea (IC50=13 μM vs rat neuronal NOS and IC50=23 μM vs rat inducible NOS) and 2-(3-aminomethylphenylamino)-4,5-dihydrothiazole (IC50=13 μM vs rat neuronal NOS and IC50=19 μM vs human inducible NOS) being the most potent. Several thioureas and 4,5-dihydrothiazoles were found to stimulate the activity of human inducible NOS in a time-dependent manner.Compounds inhibit or stimulate NOS activity, depending on R3 and R4.
Co-reporter:Claire L.M. Goodyer, Edwin C. Chinje, Mohammed Jaffar, Ian J. Stratford, Michael D. Threadgill
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 21) pp:3679-3680
Publication Date(Web):3 November 2003
DOI:10.1016/j.bmcl.2003.08.018
Treatment of Nα-Cbz-Nε-(2-hydroxyethylaminothiocarbonyl)-l-lysine N-(2-hydroxyethyl)amide with boiling hydrochloric acid gave Nε-(4,5-dihydrothiazol-2-yl)-l-lysine. This was a weak and non-isoform selective inhibitor of NOS, whereas Nε-aminothiocarbonyl-l-lysine and its methyl ester were potent, with IC50=13 and 18 μM, respectively, against human iNOS and IC50=3 and 8 μM, respectively, against rat nNOS. Time dependence was observed for inhibition of nNOS by the ester.Compounds inhibit NOS activity; the methyl ester (R=Me) shows time-dependence in inhibition of rat nNOS.
Co-reporter:Christophe Frixa, Martin Scobie, Steven J. Black, Andrew S. Thompson and Michael D. Threadgill
Chemical Communications 2002 (Issue 23) pp:2876-2877
Publication Date(Web):31 Oct 2002
DOI:10.1039/B209339A
The structure of the 2:1 complex between β-cyclodextrin and 1-phenyl-1,2-dicarba-closo-dodecaborane(12) is demonstrated by NOE and NOESY spectroscopy; this complex is remarkably refractory.
Co-reporter:Sandra Ferrer, Declan P. Naughton, Ifat Parveen and Michael D. Threadgill
Organic & Biomolecular Chemistry 2002 (Issue 3) pp:335-340
Publication Date(Web):03 Jan 2002
DOI:10.1039/B109776H
Regioselective methods were investigated to prepare N- and O-alkylated isoquinolin-1-ones efficiently. The predicted regioselective alkylation of the nitrogen with (hetero)benzyl halides was complemented using (hetero)benzylic Mitsunobu electrophiles to alkylate predominantly at the oxygen. A series of drug-delivery conjugates was prepared demonstrating control over the site of alkylation. The Mitsunobu reaction provides a new approach to 1-alkoxyisoquinolines that were unavailable via previous harsher synthetic methods.
Co-reporter:Ifat Parveen;Michael D. Threadgill
Journal of Labelled Compounds and Radiopharmaceuticals 2000 Volume 43(Issue 9) pp:883-889
Publication Date(Web):17 AUG 2000
DOI:10.1002/1099-1344(200008)43:9<883::AID-JLCR372>3.0.CO;2-4
Curcumin (E,E-1,7-di(4-hydroxy-3-methoxyphenyl)-3-hydroxyhepta-1,3,6-trien-5-one) is an investigational cancer chemopreventive agent. Lithium-bromine exchange of 1-bromo-4-(1-ethoxyethyl)-3-methoxybenzene with butyl lithium, quench with O2HCNMe2 and acidic work-up gave 4-hydroxy-3-methoxybenz-[2H]-aldehyde ([2H]-vanillin) in 27% yield (based on O2HCNMe2). Condensation with pentane-2,4-dione then gave E,E-1,7-di(4-hydroxy-3-methoxyphenyl)-3-hydroxy-1,7-di-[2H]-hepta-1,3,6-trien-5 one ([2H2]-curcumin). Adaptation of the procedures and use of OH14CNMe2 provided [14C]-curcumin (specific radioactivity 12·7 MBq mmol−1) via [14C]-vanillin. Copyright © 2000 John Wiley & Sons, Ltd.
Co-reporter:Ifat Parveen, Declan P Naughton, William J.D Whish, Michael D Threadgill
Bioorganic & Medicinal Chemistry Letters 1999 Volume 9(Issue 14) pp:2031-2036
Publication Date(Web):19 July 1999
DOI:10.1016/S0960-894X(99)00306-6
5-Chloromethyl-1-methyl-2-nitroimidazole reacted efficiently with the anion derived from 5-bromo-isoquinolin-1-one to give 5-bromo-2-((1-methyl-2-nitroimidazol-5-yl)methyl)isoquinolin-1-one. Biomimetic reduction effected release of the 5-bromoisoquinolin-1-one. The 2-nitroimidazol-5-ylmethyl unit thus has potential for development as a general prodrug system for selective drug delivery to hypoxic tissues.Graphic
Co-reporter:Peter T. Sunderland, Archana Dhami, Mary F. Mahon, Louise A. Jones, Sophie R. Tully, Matthew D. Lloyd, Andrew S. Thompson, Hashim Javaid, Niall M. B. Martin and Michael D. Threadgill
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 3) pp:NaN891-891
Publication Date(Web):2010/12/02
DOI:10.1039/C0OB00665C
The considerable interest in substituted isoquinolin-1-ones related to 5-aminoisoquinolin-1-one (5-AIQ) as drugs points to a need for an efficient and straightforward synthesis of the 4,5-disubstituted bicycles. Bromination of 5-nitroisoquinolin-1-one gave 4-bromo-5-nitroisoquinolin-1-one but neither this nor 5-amino-4-bromoisoquinolin-1-one would participate in Pd-catalysed couplings. Protection of the lactam as 1-methoxy- and 1-benzyloxy-4-bromo-5-nitroisoquinolines, however, permitted Stille, Suzuki and Buchwald–Hartwig couplings to take place in high yields, insensitive to electronic demands and severe steric bulk in the arylboronic acids. Lithiation of 4-bromo-1-methoxy-5-nitroisoquinoline and quench with iodomethane gave 1-methoxy-4-methyl-5-nitroisoquinoline in low yield. Demethylation of the 1-methoxy-4-substituted-5-nitroisoquinolines with hydrogen bromide gave 4-substituted-5-nitroisoquinolin-1-ones, whereas hydrogenolytic debenzylation was achieved with simultaneous reduction of the 5-nitro group. 5-Amino-4-(4-trifluoromethylphenyl)isoquinolin-1-one was identified as a new potent and selective inhibitor of poly(ADP-ribose)polymerase-2 (PARP-2).
Co-reporter:Amit Nathubhai, Richard Patterson, Timothy J. Woodman, Harriet E. C. Sharp, Miranda T. Y. Chui, Hugo H. K. Chung, Stephanie W. S. Lau, Jun Zheng, Matthew D. Lloyd, Andrew S. Thompson and Michael D. Threadgill
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 17) pp:NaN6099-6099
Publication Date(Web):2011/05/26
DOI:10.1039/C1OB05430A
Dimethylformamide dimethylacetal (DMFDMA) is widely used as a source of electrophilic one-carbon units at the formate oxidation level; however, electrophilic methylation with this reagent is previously unreported. Reaction of anthranilamide with DMFDMA at 150 °C for short periods gives mainly quinazolin-4-one. However, prolonged reaction with dimethylformamide di(primary-alkyl)acetals leads to subsequent alkylation at N3. 3-Substituted anthranilamides give 8-substituted 3-alkylquinazolin-4-ones. Condensation of anthranilamides with dimethylacetamide dimethylacetal provides 2,3-dimethylquinazolin-4-ones. In these reactions, the source of the N3-alkyl group is the O-alkyl group of the orthoamides. By contrast, reaction with the more sterically crowded dimethylformamide di(isopropyl)acetal diverts the alkylation to the oxygen, giving 4-isopropoxyquinazolines, along with N3-methylquinazolin-4-ones where the methyl is derived from N-Me of the orthoamides. Reaction of anthranilamide with the highly sterically demanding dimethylformamide di(t-butyl)acetal gives largely quinazolin-4-one, whereas dimethylformamide di(neopentyl)acetal forms a mixture of quinazolin-4-one and N3-methylquinazolin-4-one. The observations are rationalised in terms of formation of intermediate cationic electrophiles (alkoxymethylidene-N,N-dimethylammonium) by thermal elimination of the corresponding alkoxide from the orthoamides. These are the first observations of orthoamides as direct alkylating agents.
Co-reporter:Elvis A. Twum, Timothy J. Woodman, Wenyi Wang and Michael D. Threadgill
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 36) pp:NaN6214-6214
Publication Date(Web):2013/08/07
DOI:10.1039/C3OB41386A
1-Iodonaphthalene-2,4-diamines in trifluoroacetic acid/chloroform give stable Wheland-like tetrahedral cationic species observable by NMR, through an initial intramolecular protonation. Dynamic equilibria allow proton-deuterium exchange of aromatic protons and provide a mechanism for deiodination of 1-iodonaphthalene-2,4-diamines.



![1H-INDOLE-2-CARBOXYLIC ACID, 5-[2-(DIMETHYLAMINO)ETHOXY]-](http://img.cochemist.com/ccimg/557800/557772-39-1.png)
![1H-INDOLE-2-CARBOXYLIC ACID, 5-[2-(DIMETHYLAMINO)ETHOXY]-](http://img.cochemist.com/ccimg/557800/557772-39-1_b.png)









![Pyridine, 4-[2-(trimethylsilyl)ethynyl]-](/data/chemimg/467800/133810-35-2.png)
![Pyridine, 4-[2-(trimethylsilyl)ethynyl]-](/data/chemimg/467800/133810-35-2_b.png)

![1,3-Propanedione, 1,3-bis[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/129800/129700-40-9.png)
![1,3-Propanedione, 1,3-bis[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/129800/129700-40-9_b.png)
![3-PHENYLPYRANO[4,3-C]PYRIDIN-1-ONE](http://img.cochemist.com/ccimg/118200/118160-04-6.png)
![3-PHENYLPYRANO[4,3-C]PYRIDIN-1-ONE](http://img.cochemist.com/ccimg/118200/118160-04-6_b.png)


![2-Benzofurancarboxamide,N-[2-[[(7bR,8aS)-4,5,8,8a-tetrahydro-7-methyl-4-oxocyclopropa[c]pyrrolo[3,2-e]indol-2(1H)-yl]carbonyl]-1H-indol-5-yl]-](http://img.cochemist.com/ccimg/110400/110314-48-2.png)
![2-Benzofurancarboxamide,N-[2-[[(7bR,8aS)-4,5,8,8a-tetrahydro-7-methyl-4-oxocyclopropa[c]pyrrolo[3,2-e]indol-2(1H)-yl]carbonyl]-1H-indol-5-yl]-](http://img.cochemist.com/ccimg/110400/110314-48-2_b.png)






![4(1H)-Quinazolinone, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/83900/83800-83-3.png)
![4(1H)-Quinazolinone, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/83900/83800-83-3_b.png)
![Benzo[1,2-b:4,3-b']dipyrrole-3(2H)-carboxamide,7-[[1,6-dihydro-4-hydroxy-5-methoxy-7-[[(7bR,8aS)-4,5,8,8a-tetrahydro-7-methyl-4-oxocyclopropa[c]pyrrolo[3,2-e]indol-2(1H)-yl]carbonyl]benzo[1,2-b:4,3-b']dipyrrol-3(2H)-yl]carbonyl]-1,6-dihydro-4-hydroxy-5-methoxy-](http://img.cochemist.com/ccimg/69900/69866-21-3.png)
![Benzo[1,2-b:4,3-b']dipyrrole-3(2H)-carboxamide,7-[[1,6-dihydro-4-hydroxy-5-methoxy-7-[[(7bR,8aS)-4,5,8,8a-tetrahydro-7-methyl-4-oxocyclopropa[c]pyrrolo[3,2-e]indol-2(1H)-yl]carbonyl]benzo[1,2-b:4,3-b']dipyrrol-3(2H)-yl]carbonyl]-1,6-dihydro-4-hydroxy-5-methoxy-](http://img.cochemist.com/ccimg/69900/69866-21-3_b.png)
![Benzamide, N-[2-(aminocarbonyl)phenyl]-4-methoxy-](http://img.cochemist.com/ccimg/55400/55390-89-1.png)
![Benzamide, N-[2-(aminocarbonyl)phenyl]-4-methoxy-](http://img.cochemist.com/ccimg/55400/55390-89-1_b.png)
![2-[(4-nitrobenzoyl)amino]benzamide](http://img.cochemist.com/ccimg/53000/52910-89-1.png)
![2-[(4-nitrobenzoyl)amino]benzamide](http://img.cochemist.com/ccimg/53000/52910-89-1_b.png)
![Benzamide, N-[2-(aminocarbonyl)phenyl]-4-methyl-](http://img.cochemist.com/ccimg/53000/52910-88-0.png)
![Benzamide, N-[2-(aminocarbonyl)phenyl]-4-methyl-](http://img.cochemist.com/ccimg/53000/52910-88-0_b.png)




![Benzamide,N-[2-(aminocarbonyl)phenyl]-](http://img.cochemist.com/ccimg/18600/18543-22-1.png)
![Benzamide,N-[2-(aminocarbonyl)phenyl]-](http://img.cochemist.com/ccimg/18600/18543-22-1_b.png)






![Pyrido[4,3-d]pyrimidin-4(1H)-one, 2-phenyl-](http://img.cochemist.com/ccimg/17100/17037-08-0.png)
![Pyrido[4,3-d]pyrimidin-4(1H)-one, 2-phenyl-](http://img.cochemist.com/ccimg/17100/17037-08-0_b.png)
![Pyrido[2,3-d]pyrimidin-4(1H)-one, 2-phenyl-](http://img.cochemist.com/ccimg/16100/16081-87-1.png)
![Pyrido[2,3-d]pyrimidin-4(1H)-one, 2-phenyl-](http://img.cochemist.com/ccimg/16100/16081-87-1_b.png)
![1-(2,4-DIMETHYLPHENYL)-1H-PYRAZOLO[3,4-D]PYRIMIDIN-4-OL](http://img.cochemist.com/ccimg/7500/7455-77-8.png)
![1-(2,4-DIMETHYLPHENYL)-1H-PYRAZOLO[3,4-D]PYRIMIDIN-4-OL](http://img.cochemist.com/ccimg/7500/7455-77-8_b.png)



![Benzo[d][1,3]dioxole-5-carbonitrile](http://img.cochemist.com/ccimg/4500/4421-09-4.png)
![Benzo[d][1,3]dioxole-5-carbonitrile](http://img.cochemist.com/ccimg/4500/4421-09-4_b.png)







![6-Benzyl-2-phenyl-5,6,7,8-tetrahydro-3H-pyrido[4,3-d]pyrimidin-4-one](http://img.cochemist.com/ccimg/1100/1047-48-9.png)
![6-Benzyl-2-phenyl-5,6,7,8-tetrahydro-3H-pyrido[4,3-d]pyrimidin-4-one](http://img.cochemist.com/ccimg/1100/1047-48-9_b.png)
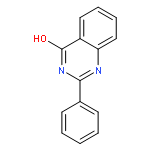
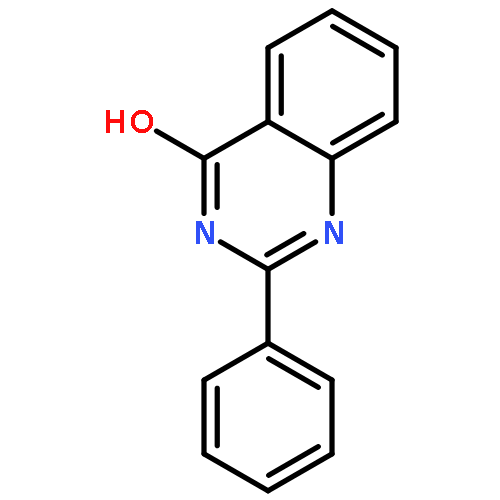
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)