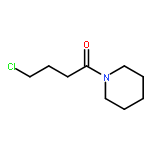
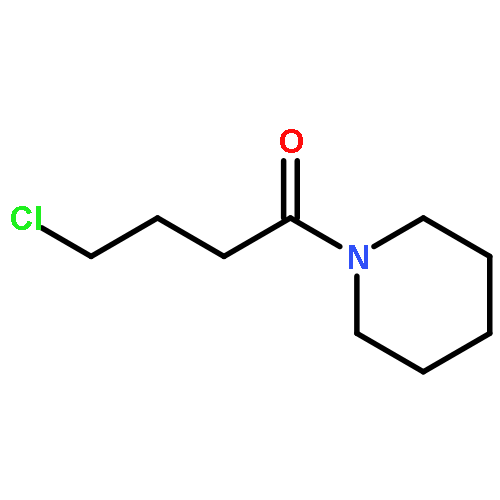
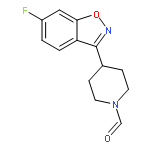
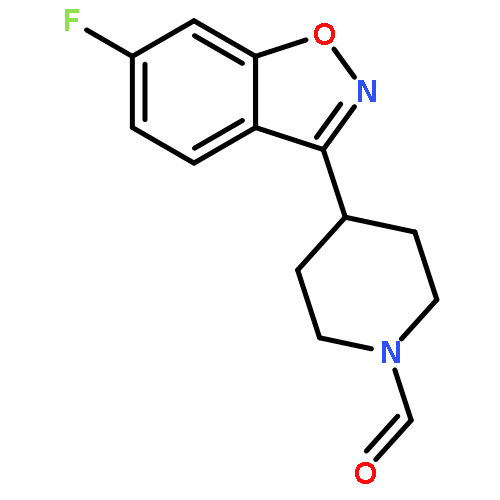
Co-reporter:Elena Sguazzini, Hayden R. Schmidt, Kavita A. Iyer, Andrew C. Kruse, Małgorzata Dukat
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 13(Issue 13) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.bmcl.2017.04.088
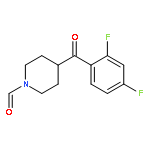
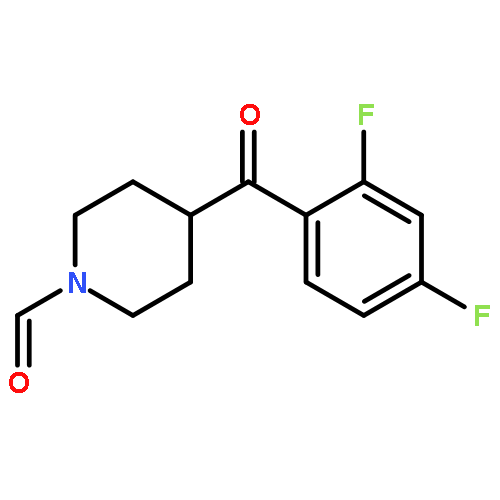
Fenpropimorph (1) is considered a “super high-affinity” σ1 receptor ligand (Ki = 0.005 nM for guinea pig σ1 receptors). Here, we examine the binding of 1 and several of its deconstructed analogs at human σ1 (hσ1) receptors. We monitored their subtype selectivity by determining the binding affinity at σ2 receptors. In addition, we validated an existing pharmacophore model at the molecular level by conducting 3D molecular modeling studies, using the crystal structure of hσ1 receptors, and Hydrophatic INTeractions (HINT) analysis. Our structure affinity relationship studies showed that 1 binds with lower affinity at hσ1 receptors (Ki = 17.3 nM) compared to guinea pig; moreover, we found that none of the fenpropimorph methyl groups is important for its binding at hσ1 receptors, nor is stereochemistry. For example, removal of all methyl groups as seen in 4 resulted in an almost 5-fold higher affinity at hσ1 receptors compared to 1 and 350-fold selectivity versus σ2 receptors. In addition, although the O atom of the morpholine ring does not contribute to affinity at hσ1 receptors (and might even detract from it), it plays role in subtype (σ1 versus σ2 receptor) selectivity.Download high-res image (64KB)Download full-size image
Co-reporter:Xiaolei Pan, Kavita A. Iyer, Hebing Liu, Douglas H. Sweet, Małgorzata Dukat
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 18(Issue 18) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.bmcl.2017.08.008
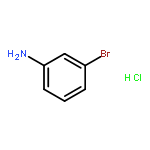
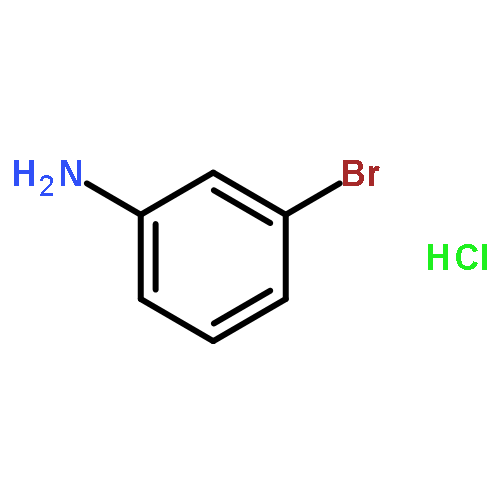
Human organic cation transporters (OCTs) represent an understudied neurotransmitter uptake mechanism for which no selective agents have yet been identified. Several neurotransmitters (e.g. serotonin, norepinephrine) are low-affinity substrates for these transporters, but possess higher affinity for other transporters (e.g. the serotonin or norepinephrine transporters; SERT and NET, respectively). We have identified a new class of OCT inhibitors with a phenylguanidine structural scaffold. Here, we examine the actions of a series of such compounds and report preliminary structure–activity relationships (SARs) – the first dedicated SAR study of OCT3 action. Initial results showed that the presence of a substituent on the phenyl ring, as well as its position, contributes to the phenylguanidines’ inhibitory potency (IC50 values ranging from 2.2 to >450 μM) at hOCT3. There is a trend towards enhanced inhibitory potency of phenylguanidines with increased lipophilic character and the size of the substituent at the phenyl 4-position, with the latter reaching a ceiling effect. The first PiPT-based hOCT3 homology models were generated and are in agreement with our biological data.Download high-res image (200KB)Download full-size image
Co-reporter:Katie Alix, Shailesh Khatri, Philip D. Mosier, Samantha Casterlow, Dong Yan, Heather L. Nyce, Michael M. White, Marvin K. Schulte, and Małgorzata Dukat
ACS Chemical Neuroscience 2016 Volume 7(Issue 11) pp:1565
Publication Date(Web):August 17, 2016
DOI:10.1021/acschemneuro.6b00196
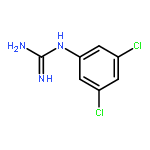
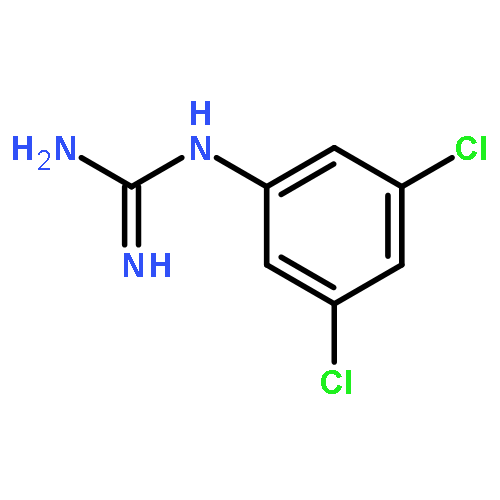
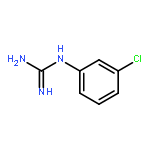
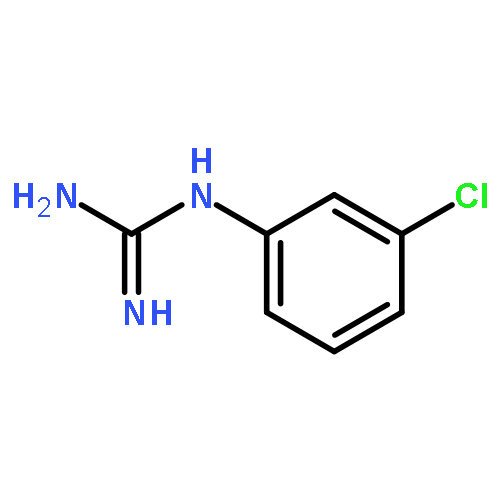
Introduction of minor variations to the substitution pattern of arylguanidine 5-hydroxytryptamine-3 (5-HT3) receptor ligands resulted in a broad spectrum of functionally-active ligands from antagonist to superagonist. For example, (i) introduction of an additional Cl-substituent(s) to our lead full agonist N-(3-chlorophenyl)guanidine (mCPG, 2; efficacy % = 106) yielded superagonists 7–9 (efficacy % = 186, 139, and 129, respectively), (ii) a positional isomer of 2, p-Cl analog 11, displayed partial agonist actions (efficacy % = 12), and (iii) replacing the halogen atom at the meta or para position with an electron donating OCH3 group or a stronger electron withdrawing (i.e., CF3) group resulted in antagonists 13–16. We posit based on combined mutagenesis, crystallographic, and computational analyses that for the 5-HT3 receptor, the arylguanidines that are better able to simultaneously engage the primary and complementary subunits, thus keeping them in close proximity, have greater agonist character while those that are deficient in this ability are antagonists.Keywords: 3D-graphic models; 5-HT3A receptors; Arylguanidines; binding affinities; functional activities; SAR; site-directed mutagenesis
Co-reporter:Małgorzata Dukat, Katie Alix, Jessica Worsham, Shailesh Khatri, Marvin K. Schulte
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 21) pp:5945-5948
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmcl.2013.08.072
2-Amino-6-chloro-3,4-dihydroquinazoline HCl (A6CDQ, 4) binds at 5-HT3 serotonin receptors and displays antidepressant-like action in the mouse tail suspension test (TST). Empirically, 4 was demonstrated to be a 5-HT3 receptor antagonist (two-electrode voltage clamp recordings using frog oocytes; IC50 = 0.26 μM), and one that should readily penetrate the blood–brain barrier (log P = 1.86). 5-HT3 receptor antagonists represent a potential approach to the development of new antidepressants, and 4 is an example of a structurally novel 5-HT3 receptor antagonist that is active in a preclinical antidepressant model (i.e., the mouse TST).4 ED50 = 0.23 mg/kg.
Co-reporter:Shawquia Young;Minna Vainio;Mika Scheinin;Ma&x142;gorzata Dukat
Basic & Clinical Pharmacology & Toxicology 2010 Volume 107( Issue 2) pp:690-697
Publication Date(Web):
DOI:10.1111/j.1742-7843.2010.00563.x
Abstract: Previously, we reported that antinociceptive synergism of a 5-HT3/α2-adrenoceptor ligand MD-354 (m-chlorophenylguanidine) and clonidine combination occurs, in part, through a 5-HT3 receptor antagonist mechanism. In the present investigation, a possible role for α2-adrenoceptors was examined. Mechanistic studies using yohimbine (a subtype non-selective α2-adrenoceptor antagonist), BRL 44408 (a preferential α2A-adrenoceptor antagonist) and imiloxan (a preferential α2B/C-adrenoceptor antagonist) on the antinociceptive actions of a MD-354/clonidine combination were conducted. Subcutaneous pre-treatment with all three antagonists inhibited the antinociceptive synergism of MD-354 and clonidine in the mouse tail-flick assay in a dose-dependent manner (AD50 = 0.33, 2.1, and 0.17 mg/kg, respectively). Enhancement of clonidine antinociception by MD-354 did not potentiate clonidine’s locomotor suppressant activity in a mouse locomotor assay. When [ethyl-3H]RS-79948-197 was used as radioligand, MD-354 displayed almost equal affinity to α2A- and α2B-adrenoceptors (Ki = 110 and 220 nM) and showed lower affinity at α2C-adrenoceptors (Ki = 4,700 nM). MD-354 had no subtype-selectivity for the α2-adrenoceptor subtypes as an antagonist in functional [35S]GTPγS binding assays. MD-354 was a weak partial agonist at α2A-adrenoceptors. Overall, in addition to the 5-HT3 receptor component, the present investigation found MD-354 to be a weak partial α2A-adrenoceptor agonist that enhances clonidine’s thermal antinociceptive actions through an α2-adrenoceptor-mediated mechanism without augmenting sedation.
Co-reporter:Małgorzata Dukat;Anna Wesołowska;Genevieve Alley;Shawquia Young
Psychopharmacology 2010 Volume 210( Issue 4) pp:547-557
Publication Date(Web):2010 July
DOI:10.1007/s00213-010-1857-0
(−)Nicotine produces antinociceptive effects in rodents. meta-Chlorophenylguanidine (MD-354), an analgesia-enhancing agent, binds at 5-HT3 and α2-adrenoceptors and potentiates the antinociceptive effects of an “inactive” dose of clonidine. The present study examined the actions of MD-354 on (−)nicotine-induced antinociception.Mouse tail-flick and other assays were employed.In the tail-flick assay, (−)nicotine (ED50 = 1.66 mg/kg) but not MD-354 produced dose-related antinociceptive effects. Administered in combination with (−)nicotine (2.5 mg/kg), MD-354 (AD50 = 3.4 mg/kg) did not potentiate, but effectively antagonized the antinociceptive actions of (−)nicotine. In a mouse hot-plate assay, MD-354 failed to modify (−)nicotine responses. In combination with a locomotor activity-suppressing dose of (−)nicotine, MD-354 (up to 17 mg/kg) failed to antagonize (−)nicotine-induced hypolocomotion. In a rat drug discrimination paradigm using (−)nicotine as training drug, MD-354 produced saline-appropriate responding; in combination with the training dose of (−)nicotine, MD-354 failed to antagonize the nicotine cue.MD-354 selectively antagonizes the antinociceptive actions of (−)nicotine in the tail-flick, but not in the hot-plate assay, or either the motor effects, or discriminative stimulus effects of (−)nicotine. The most parsimonious explanation is that MD-354 might act as a negative allosteric modulator of α7 nACh receptors, and radioligand binding and functional data are provided to support this conclusion.

![Guanidine, [4-chloro-3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/366500/366498-46-6.png)
![Guanidine, [4-chloro-3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/366500/366498-46-6_b.png)