Co-reporter:Huw M. L. Davies and Daniel Morton
ACS Central Science September 27, 2017 Volume 3(Issue 9) pp:936-936
Publication Date(Web):September 11, 2017
DOI:10.1021/acscentsci.7b00329
C–H functionalization is a very active research field that has attracted the interest of scientists from many disciplines. This Outlook describes the collaborative efforts within the NSF CCI Center for Selective C–H Functionalization (CCHF) to develop catalyst-controlled selective methods to enhance the synthetic potential of C–H functionalization.
Co-reporter:Verena Lehner, Huw M. L. Davies, and Oliver Reiser
Organic Letters September 15, 2017 Volume 19(Issue 18) pp:
Publication Date(Web):August 28, 2017
DOI:10.1021/acs.orglett.7b02009
Rh(II)-catalyzed enantioselective cyclopropanations of furans, providing access to synthetically useful building blocks, are reported. After screening of 10 Rh(II) catalysts, Rh2(S-TCPTTL)4 was identified as a highly efficient and selective catalyst (up to 98% ee, TON 88000, and TOF 24/s) for the cyclopropanation of furans. These cyclopropanes were successfully applied to the enantioselective synthesis of novel paraconic acid derivatives.
Co-reporter:John F. Briones and Huw M. L. Davies
Journal of the American Chemical Society September 11, 2013 Volume 135(Issue 36) pp:13314-13317
Publication Date(Web):August 26, 2013
DOI:10.1021/ja407179c
The reaction of vinyldiazoacetates with enol ethers catalyzed by the binuclear gold complex (R)-DTBMSegphos(AuCl)2 activated by silver hexafluoroantimonate results in a highly enantioselective [3 + 2] cycloaddition. The [3 + 2] cycloaddition proceeds with dynamic kinetic resolution when the enol ether is a 4-substituted 1-(methoxymethylene)cyclohexane. The reaction is initiated by nucleophilic attack of the vinyl ethers at the vinylogous position of the gold vinylcarbene intermediate.
Co-reporter:Igor D. Jurberg and Huw M. L. Davies
Organic Letters October 6, 2017 Volume 19(Issue 19) pp:
Publication Date(Web):September 11, 2017
DOI:10.1021/acs.orglett.7b02436
Two approaches were developed for the conversion of isoxazol-5-ones to 2,3-dihydro-6H-1,3-oxazin-6-ones. The first involves dirhodium-catalyzed reaction of aryl diazoacetates, leading to rhodium carbene intermediates that undergo insertion into the N–O bond of isoxazol-5-ones. The second approach involves conversion of the aryldiazoacetates to the corresponding tosylates, followed by reaction with the isoxazol-5-one in a metal-free one-pot procedure. These studies illustrate an alternative method to achieve a carbene-like transformation without requiring a metal catalyst.
Co-reporter:Daniel Rackl, Chun-Jae Yoo, Christopher W. Jones, and Huw M. L. Davies
Organic Letters June 16, 2017 Volume 19(Issue 12) pp:
Publication Date(Web):June 5, 2017
DOI:10.1021/acs.orglett.7b01073
A tandem reaction system has been developed for the preparation of donor/acceptor-substituted diazo compounds in continuous flow coupled to dirhodium-catalyzed C–H functionalization or cyclopropanation. Hydrazones were oxidized in flow by solid-supported N-iodo-p-toluenesulfonamide potassium salt (PS-SO2NIK) to generate the diazo compounds, which were then purified by passing through a column of molecular sieves/sodium thiosulfate.
Co-reporter:Cecilia Tortoreto, Daniel Rackl, and Huw M. L. Davies
Organic Letters 2017 Volume 19(Issue 4) pp:
Publication Date(Web):February 7, 2017
DOI:10.1021/acs.orglett.6b03681
Thermally induced reactions of donor/acceptor diazo compounds generate carbene intermediates capable of C–H functionalization reactions of alkanes. A variety of C–H insertion products were obtained in moderate to good yields and in certain cases with good site selectivity, favoring the functionalization of the more highly substituted C–H bond.
Co-reporter:Liangbing Fu; David M. Guptill
Journal of the American Chemical Society 2016 Volume 138(Issue 18) pp:5761-5764
Publication Date(Web):April 11, 2016
DOI:10.1021/jacs.6b01941
Enantioselective C–H functionalization of relatively electron-deficient methyl sites was achieved with the combination of 2,2,2-trichloroethyl aryldiazoacetates and tetrakis(triarylcyclopropanecarboxylate) dirhodium catalysts. The substrate scope of the transformation was relatively broad, and C–H functionalization products were furnished with excellent levels of enantioselectivity. As a strategic reaction, crotonate derivatives give 1,6-dicarbonyl compounds, which are useful for further diversification.
Co-reporter:Robert W. Kubiak II, Jeffrey D. Mighion, Sidney M. Wilkerson-Hill, Joshua S. Alford, Tetsushi Yoshidomi, and Huw M. L. Davies
Organic Letters 2016 Volume 18(Issue 13) pp:3118-3121
Publication Date(Web):June 22, 2016
DOI:10.1021/acs.orglett.6b01298
The enantioselective intermolecular sp3 C–H functionalization at the allylic and benzylic positions was achieved using rhodium-catalyzed reactions with 4-phenyl-N-(methanesulfonyl)-1,2,3-triazole. The optimum dirhodium tetracarboxylate catalyst for these reactions was Rh2(S-NTTL)4. The rhodium-bound α-imino carbene intermediates preferentially reacted with tertiary over primary C–H bonds in good yields and moderate levels of enantioselectivity (66–82% ee). This work demonstrates that N-sulfonyltriazoles can be applied to the effective C–H functionalization at sp3 C–H bonds of substrates containing additional functionality.
Co-reporter:Tanja Krainz;Sharon Chow;Natasa Korica;Paul V. Bernhardt;Glen M. Boyle;Peter G. Parsons;Craig M. Williams
European Journal of Organic Chemistry 2016 Volume 2016( Issue 1) pp:41-44
Publication Date(Web):
DOI:10.1002/ejoc.201501271
Abstract
An efficient method to directly access the bicyclo[5.3.0]decane core was achieved by rhodium-catalyzed reaction of a novel donor–acceptor cyclopentenyl diazocarboxylate with a variety of furans. As this motif is commonly found within bioactive antitumor natural products, selected systems were further manipulated and evaluated against cancer cell lines sensitive to protein kinase C (PKC) activation.
Co-reporter:Atsushi D. Yamaguchi; Kathryn M. Chepiga; Junichiro Yamaguchi; Kenichiro Itami
Journal of the American Chemical Society 2015 Volume 137(Issue 2) pp:644-647
Publication Date(Web):January 6, 2015
DOI:10.1021/ja512059d
Syntheses of dictyodendrins A and F have been achieved using a sequential C–H functionalization strategy. The N-alkylpyrrole core is fully functionalized by means of a rhodium(I)-catalyzed C–H arylation at the C3-position, a rhodium(II)-catalyzed double C–H insertion at the C2- and C5-positions, and a Suzuki–Miyaura cross-coupling reaction at the C4-position. The syntheses of dictyodendrins A and F were completed by formal 6π-electrocyclization to generate the pyrrolo[2,3-c]carbazole core of the natural products.
Co-reporter:Elizabeth N. Bess, David M. Guptill, Huw M. L. Davies and Matthew S. Sigman
Chemical Science 2015 vol. 6(Issue 5) pp:3057-3062
Publication Date(Web):18 Mar 2015
DOI:10.1039/C5SC00357A
Achieving selective C–H functionalization is a significant challenge that requires discrimination between many similar C–H bonds. Yet, reaction systems employing Rh2(DOSP)4 and Rh2(BPCP)4 were recently demonstrated to afford high levels of selectivity in the C–H insertion of carbenes into toluene-derived substrates. Herein, we explore the origin of this selectivity through a systematic analysis of substrate and reagent features that alter levels of selectivity from 20:1 to 1:610 for secondary (or tertiary)-to-primary benzylic C–H functionalization of toluene derivatives. Describing this variation using infrared vibrations and point charges, we have developed a mathematical model from which are identified features of the systems that determine levels of site-selectivity and are applied as predictive factors to describe the selectivity behavior of new substrate/reagent combinations.
Co-reporter:Brendan T. Parr and Huw M. L. Davies
Organic Letters 2015 Volume 17(Issue 4) pp:794-797
Publication Date(Web):February 9, 2015
DOI:10.1021/ol503508k
A stereoselective synthesis of cyclohexanes bearing four stereocenters from vinyldiazoacetates and allyl alcohols by a rhodium-carbene initiated domino reaction is described. The reaction cascade features a tandem ylide formation/[2,3]-sigmatropic rearrangement, oxy-Cope rearrangement, and type II carbonyl ene reaction, all of which proceed with a high degree of stereocontrol. The products are routinely isolated with excellent stereocontrol (>97:3 dr, 99% ee).
Co-reporter:Dr. Eric G. Moschetta;Dr. Solymar Negretti;Kathryn M. Chepiga;Dr. Nicholas A. Brunelli;Dr. Ying Labreche;Dr. Yan Feng;Dr. Fateme Rezaei; Ryan P. Lively; William J. Koros; Huw M. L. Davies; Christopher W. Jones
Angewandte Chemie 2015 Volume 127( Issue 22) pp:6570-6574
Publication Date(Web):
DOI:10.1002/ange.201500841
Abstract
Flexible composite polymer/oxide hollow fibers are used as flow reactors for heterogeneously catalyzed reactions in organic synthesis. The fiber synthesis allows for a variety of supported catalysts to be embedded in the walls of the fibers, thus leading to a diverse set of reactions that can be catalyzed in flow. Additionally, the fiber synthesis is scalable (e.g. several reactor beds containing many fibers in a module may be used) and thus they could potentially be used for the large-scale production of organic compounds. Incorporating heterogeneous catalysts in the walls of the fibers presents an alternative to a traditional packed-bed reactor and avoids large pressure drops, which is a crucial challenge when employing microreactors.
Co-reporter:Solymar Negretti, Carolyn M. Cohen, Jane J. Chang, David M. Guptill, Huw M.L. Davies
Tetrahedron 2015 Volume 71(Issue 39) pp:7415-7420
Publication Date(Web):30 September 2015
DOI:10.1016/j.tet.2015.05.045
Highly functionalized cyclopropanecarboxylates were readily prepared by rhodium-catalyzed cyclopropanation of alkenes with aryldiazoacetates and styryldiazoaceates, in which the ester functionality is either trimethylsilylethyl (TMSE) or trichloroethyl (TCE). By having labile protecting groups on the ester, chiral triarylcyclopropane carboxylate ligands were conveniently prepared. The asymmetric induction during cyclopropanation is dependent on the nature of the ester group and the chiral dirhodium tetracarboxylate catalyst. The prolinate catalyst Rh2(S-DOSP)4 was the optimum catalyst for asymmetric intermolecular cyclopropanation of TMSE diazoesters with styrene, while Rh2(R-BPCP)4 was the optimum catalyst for TCE diazoesters.
Co-reporter:Dr. Eric G. Moschetta;Dr. Solymar Negretti;Kathryn M. Chepiga;Dr. Nicholas A. Brunelli;Dr. Ying Labreche;Dr. Yan Feng;Dr. Fateme Rezaei; Ryan P. Lively; William J. Koros; Huw M. L. Davies; Christopher W. Jones
Angewandte Chemie International Edition 2015 Volume 54( Issue 22) pp:6470-6474
Publication Date(Web):
DOI:10.1002/anie.201500841
Abstract
Flexible composite polymer/oxide hollow fibers are used as flow reactors for heterogeneously catalyzed reactions in organic synthesis. The fiber synthesis allows for a variety of supported catalysts to be embedded in the walls of the fibers, thus leading to a diverse set of reactions that can be catalyzed in flow. Additionally, the fiber synthesis is scalable (e.g. several reactor beds containing many fibers in a module may be used) and thus they could potentially be used for the large-scale production of organic compounds. Incorporating heterogeneous catalysts in the walls of the fibers presents an alternative to a traditional packed-bed reactor and avoids large pressure drops, which is a crucial challenge when employing microreactors.
Co-reporter:Huw M. L. Davies and Joshua S. Alford
Chemical Society Reviews 2014 vol. 43(Issue 15) pp:5151-5162
Publication Date(Web):07 May 2014
DOI:10.1039/C4CS00072B
Metal-stabilized carbenes derived from diazo compounds have become broadly useful reactive intermediates for organic synthesis. This tutorial review will describe the recent advances in using N-sulfonyl-1,2,3-triazoles as precursors for the formation of metal-bound imino carbene intermediates. These intermediates undergo a variety of synthetically useful transformations, which include transannulation reactions to generate new heterocycles, cyclopropanation and subsequent ring expansions, ylide formation with subsequent rearrangements, and C–H functionalization. Furthermore, many of these transformations can be conducted with high levels of enantioselectivity by use of chiral rhodium(II) catalysts.
Co-reporter:Changming Qin
Journal of the American Chemical Society 2014 Volume 136(Issue 27) pp:9792-9796
Publication Date(Web):June 16, 2014
DOI:10.1021/ja504797x
The influence of sterically demanding dirhodium tetracarboxylate catalysts on the site selectivity of C–H functionalization by means of rhodium carbene-induced C–H insertion is described. The established dirhodium tetraprolinate-catalyzed reactions of aryldiazoacetates cause preferential C–H functionalization of secondary C–H bonds as a result of competing steric and electronic effects. The sterically more demanding dirhodium tetrakis(triarylcyclopropanecarboxylate) catalysts, exemplified by dirhodium tetrakis[(R)-(1-(biphenyl)-2,2-diphenylcyclopropanecarboxylate)] [Rh2(R-BPCP)4], favor C–H functionalization of activated primary C–H bonds. Highly site-selective and enantioselective C–H functionalization of a variety of simple substrates containing primary benzylic, allylic, and methoxy C–H bonds was achieved with this catalyst. The utility of this approach has been demonstrated by the late-stage primary C–H functionalization of (−)-α-cedrene and a steroid.
Co-reporter:Ping Lu ; Artur Mailyan ; Zhenhua Gu ; David M. Guptill ; Hengbin Wang ; Huw M. L. Davies ;Armen Zakarian
Journal of the American Chemical Society 2014 Volume 136(Issue 51) pp:17738-17749
Publication Date(Web):November 19, 2014
DOI:10.1021/ja510573v
The evolution of a program directed at the enantioselective total synthesis of maoecrystal V, a highly modified ent-kauranoid, is described. An early stage chiral auxiliary-directed asymmetric C–H functionalization for the construction of a key benzofuran intermediate enabled the first asymmetric synthesis of the natural enantiomer of maoecrystal V, confirming the assigned stereochemistry. A divergent course of the central intramolecular Diels–Alder reaction, which is dependent on the nature of the dienophile, initially led to the development of an unanticipated and previously unknown isomer of maoecrystal V, which we named maoecrystal ZG. In light of the reported selective and potent cytotoxic activity of maoecrystal V, the cytotoxic properties of maoecrystal ZG were also investigated.
Co-reporter:Joshua S. Alford and Huw M. L. Davies
Journal of the American Chemical Society 2014 Volume 136(Issue 29) pp:10266-10269
Publication Date(Web):July 6, 2014
DOI:10.1021/ja5058967
An effective method for aminoacylation of indoles and pyrroles has been achieved. The transformation involves a multicomponent one-pot cascade reaction between indoles or pyrroles, ynol ethers, and sulfonyl azides, creating four different bonds regioselectively through N-sulfonyltriazole intermediates. The oxo-tryptamines and oxo-pyrroloethanamines are generated in moderate to high yields under mild reaction conditions.
Co-reporter:David M. Guptill
Journal of the American Chemical Society 2014 Volume 136(Issue 51) pp:17718-17721
Publication Date(Web):December 4, 2014
DOI:10.1021/ja5107404
A new class of reagents is described for C–H functionalization by means of C–H insertion using donor/acceptor-substituted rhodium(II) carbene intermediates. The 2,2,2-trichloroethyl aryl and heteroaryl diazoacetates, together with the dirhodium triarylcyclopropane carboxylate catalyst Rh2(R-BPCP)4, enabled the enantioselective intermolecular C–H functionalization of a range of methyl ethers with high levels of site selectivity and enantioselectivity.
Co-reporter:Liangbing Fu, Hengbin Wang, and Huw M. L. Davies
Organic Letters 2014 Volume 16(Issue 11) pp:3036-3039
Publication Date(Web):May 20, 2014
DOI:10.1021/ol5011505
A rhodium-catalyzed asymmetric synthesis of β-lactones via intramolecular C–H insertion into the ester group of aryldiazoacetates has been developed. The β-lactones were synthesized in high yields and with high levels of diastereo- and enantioselectivity. Halo and trifluoromethyl substituents at the ortho position of the aryldiazoacetates enhance intramolecular C–H insertions over intermolecular reactions, allowing C–H insertion of even methyl C–H bonds.
Co-reporter:Jillian E. Spangler, Yajing Lian, Sandeep N. Raikar, and Huw M. L. Davies
Organic Letters 2014 Volume 16(Issue 18) pp:4794-4797
Publication Date(Web):September 10, 2014
DOI:10.1021/ol502257d
Treatment of (E)-1-(methoxymethylene)-1,2,3,4-tetrahydronaphthalene with styryl diazoacetates in the presence of catalytic amounts of the dirhodium complex Rh2(S-DOSP)4 provides a highly enantioenriched hexacyclic product with 10 new stereogenic centers. The transformation proceeds by a cascade sequence starting with a double cyclopropanation of a benzene ring, followed by a Cope rearrangement of a divinylcyclopropane and then an intramolecular Diels–Alder cycloaddition.
Co-reporter:Dr. Pablo E. Guzmán;Dr. Yajing Lian ;Dr. Huw M. L. Davies
Angewandte Chemie 2014 Volume 126( Issue 48) pp:13299-13303
Publication Date(Web):
DOI:10.1002/ange.201406440
Abstract
A regio-, diastereo-, and enantioselective [4+3] cycloaddition between vinylcarbenes and dienes has been achieved using the dirhodium tetracarboxylate catalyst [Rh2(S-BTPCP)4]. This methodology provides facile access to 1,4-cycloheptadienes that are regioisomers of those formed from the tandem cyclopropanation/Cope rearrangement reaction of vinylcarbenes with dienes.
Co-reporter:Dr. Pablo E. Guzmán;Dr. Yajing Lian ;Dr. Huw M. L. Davies
Angewandte Chemie International Edition 2014 Volume 53( Issue 48) pp:13083-13087
Publication Date(Web):
DOI:10.1002/anie.201406440
Abstract
A regio-, diastereo-, and enantioselective [4+3] cycloaddition between vinylcarbenes and dienes has been achieved using the dirhodium tetracarboxylate catalyst [Rh2(S-BTPCP)4]. This methodology provides facile access to 1,4-cycloheptadienes that are regioisomers of those formed from the tandem cyclopropanation/Cope rearrangement reaction of vinylcarbenes with dienes.
Co-reporter: Huw M. L. Davies;Dr. Daniel Morton
Angewandte Chemie International Edition 2014 Volume 53( Issue 39) pp:10256-10258
Publication Date(Web):
DOI:10.1002/anie.201406633
Co-reporter: Huw M. L. Davies;Dr. Daniel Morton
Angewandte Chemie 2014 Volume 126( Issue 39) pp:10422-10424
Publication Date(Web):
DOI:10.1002/ange.201406633
Co-reporter:Jillian E. Spangler
Journal of the American Chemical Society 2013 Volume 135(Issue 18) pp:6802-6805
Publication Date(Web):April 22, 2013
DOI:10.1021/ja4025337
Herein we report the synthesis of pyrroloindolines via a catalytic enantioselective formal [3+2] cycloaddition of C(3)-substituted indoles. This methodology utilizes 4-aryl-1-sulfonyl-1,2,3-triazoles as carbenoid precursors and the rhodium(II)-tetracarboxylate catalyst Rh2(S-PTAD)4. A variety of aryl-substituted pyrroloindolines were prepared in good yields and with high levels of enantioinduction.
Co-reporter:Brendan T. Parr ; Samantha A. Green
Journal of the American Chemical Society 2013 Volume 135(Issue 12) pp:4716-4718
Publication Date(Web):March 11, 2013
DOI:10.1021/ja401386z
The synthesis of highly functionalized pyrroles has been achieved by reaction of rhodium-stabilized imino-carbenes with furans. The reaction features an initial [3+2] annulation to form bicyclic hemiaminals, followed by ring opening to generate trisubstituted pyrroles.
Co-reporter:Hengbin Wang ; Gang Li ; Keary M. Engle ; Jin-Quan Yu
Journal of the American Chemical Society 2013 Volume 135(Issue 18) pp:6774-6777
Publication Date(Web):April 20, 2013
DOI:10.1021/ja401731d
The enantioselective synthesis of 2,3-dihydrobenzofurans was achieved by using two sequential C–H functionalization reactions, a rhodium-catalyzed enantioselective intermolecular C–H insertion followed by a palladium-catalyzed C–H activation/C–O cyclization. Further diversification of the 2,3-dihydrobenzofuran structures was possible by a subsequent palladium-catalyzed intermolecular Heck-type sp2 C–H functionalization.
Co-reporter:Changming Qin
Journal of the American Chemical Society 2013 Volume 135(Issue 39) pp:14516-14519
Publication Date(Web):September 12, 2013
DOI:10.1021/ja4069003
Rhodium-catalyzed reaction of vinyldiazoacetates with nitrones results in a formal [3+2]-cycloaddition to generate 2,5-dihydroisoxazoles with high levels of asymmetric induction. The cascade reaction begins with a vinylogous addition event, followed by an iminium addition ring-closure/hydride migration/alkene isomerization cascade. Dirhodium tetrakis(triarylcyclopropanecarboxylates) are the optimum catalysts for this process.
Co-reporter:Hengbin Wang, David M. Guptill, Adrian Varela-Alvarez, Djamaladdin G. Musaev and Huw M. L. Davies
Chemical Science 2013 vol. 4(Issue 7) pp:2844-2850
Publication Date(Web):15 Apr 2013
DOI:10.1039/C3SC50425E
The rhodium-catalyzed reaction of electron-deficient alkenes with substituted aryldiazoacetates and vinyldiazoacetates results in highly stereoselective cyclopropanations. With adamantylglycine derived catalyst Rh2(S-TCPTAD)4, high asymmetric induction (up to 98% ee) can be obtained with a range of substrates. Computational studies suggest that the reaction is facilitated by weak interaction between the carbenoid and the substrate carbonyl but subsequently proceeds via different pathways depending on the nature of the carbonyl. Acrylates and acrylamides result in the formation of cyclopropanation products while the use of unsaturated aldehydes and ketones results in the formation of epoxides.
Co-reporter:Clayton P. Owens, Adrián Varela-Álvarez, Vyacheslav Boyarskikh, Djamaladdin G. Musaev, Huw M. L. Davies and Simon B. Blakey
Chemical Science 2013 vol. 4(Issue 6) pp:2590-2596
Publication Date(Web):19 Apr 2013
DOI:10.1039/C3SC50886B
Recently, a small number of diverse iridium complexes have been shown to catalyze unusual atom transfer C–H functionalization reactions. To further our understanding and enhance the utility of iridium complexes for C–H functionalization, we report the design and synthesis of a family of iridium(III)-bis(oxazolinyl)phenyl complexes. The ability to tune the ligand environment around the metal in these systems is exploited to design complexes with the ability to catalyze the asymmetric insertion of donor/acceptor iridium carbenoids into activated C–H bonds. Low catalyst loadings (0.5 mol%) routinely lead to excellent reaction yields (51–99%) and enantioselectivities (83–99%). Density functional theory calculations provide compelling evidence that in these complexes the carbene binds to the iridium cis to the phenyl group of the bis(oxazolinyl)phenyl ligand. This finding is vital for understanding the observed stereochemical induction and is of particular significance in the field of enantioselective transition metal-catalysed atom transfer reactions utilizing oxazoline–X–oxazoline tridentate ligands, as previously employed stereochemical models for these ligand sets are based on the assumption that reactive ligands and Lewis bases bind trans to the central X ligand.
Co-reporter:Changming Qin and Huw M. L. Davies
Organic Letters 2013 Volume 15(Issue 2) pp:310-313
Publication Date(Web):January 3, 2013
DOI:10.1021/ol303217s
The rhodium-catalyzed reaction of 2-diazo-5-arylpent-4-enoates can be controlled by the appropriate choice of catalyst and catalyst loading to form either 2-arylbicyclo[1.1.0]butane carboxylates or cyclohexene derivatives. Both products are produced in a highly diastereoselective manner, with 2-arylbicyclo[1.1.0]butane carboxylates preferentially formed under low catalyst loadings. When the reaction is catalyzed by Rh2(R-BTPCP)4, the 2-arylbicyclo[1.1.0]butane carboxylates are generated with high levels of asymmetric induction (70–94% ee).
Co-reporter:Kathryn M. Chepiga, Yan Feng, Nicholas A. Brunelli, Christopher W. Jones, and Huw M. L. Davies
Organic Letters 2013 Volume 15(Issue 24) pp:6136-6139
Publication Date(Web):November 19, 2013
DOI:10.1021/ol403006r
A silica-supported dirhodium(II) tetraprolinate catalyst was synthesized in four steps from l-proline and used in a range of enantioselective transformations of donor/acceptor carbenoids. These include cyclopropenation, cyclopropanation, tandem ylide formation/[2,3] sigmatropic rearrangement, and a variety of combined C–H functionalization/Cope rearrangement reactions. The products of these transformations were obtained in yields and levels of enantioselectivity comparable to those obtained with its homogeneous counterpart, Rh2(S-DOSP)4. The silica-supported Rh2(S-DOSP)4 derivative was successfully recycled over five reactions.
Co-reporter:Changming Qin and Huw M. L. Davies
Organic Letters 2013 Volume 15(Issue 24) pp:6152-6154
Publication Date(Web):November 14, 2013
DOI:10.1021/ol403017e
A silver-catalyzed vinylogous fluorination of vinyl diazoacetates to generate γ-fluoro-α,β-unsaturated carbonyls is presented. Application of this method to the fluorination of farnesol and steroid derivatives was achieved.
Co-reporter:David M. Guptill, Carolyn M. Cohen, and Huw M. L. Davies
Organic Letters 2013 Volume 15(Issue 24) pp:6120-6123
Publication Date(Web):November 27, 2013
DOI:10.1021/ol4028978
The rhodium-catalyzed decomposition of 2-(triisopropylsilyl)ethyl aryl- and vinyldiazoacetates results in the stereoselective formation of Z-allylsilanes. The transformation is considered to proceed by silyl-directed intramolecular C–H functionalization to form a β-lactone intermediate followed by a silyl-activated extrusion of carbon dioxide.
Co-reporter:Valerij A. Nikolaev, Murat B. Supurgibekov, Huw M. L. Davies, Joachim Sieler, and Valerija M. Zakharova
The Journal of Organic Chemistry 2013 Volume 78(Issue 9) pp:4239-4244
Publication Date(Web):April 24, 2013
DOI:10.1021/jo302726m
Incorporation of a trifluoromethyl group into the structure of 4-(alkoxycarbonyl)vinyldiazocarbonyl compounds greatly decreases the tendency of the carbenoid intermediates formed during Rh(II)-catalyzed reactions to undergo intermolecular processes. Instead, they are prone to experience intramolecular [1,5]- and [1,3]-electrocyclizations to produce reactive cyclopropenes and furans, and these are capable of further transformations.
Co-reporter:Kathryn M. Chepiga, Changming Qin, Joshua S. Alford, Spandan Chennamadhavuni, Timothy M. Gregg, Jeremy P. Olson, Huw M.L. Davies
Tetrahedron 2013 69(27–28) pp: 5765-5771
Publication Date(Web):
DOI:10.1016/j.tet.2013.04.075
Co-reporter:Brendan T. Parr ; Huw M. L. Davies
Angewandte Chemie International Edition 2013 Volume 52( Issue 38) pp:10044-10047
Publication Date(Web):
DOI:10.1002/anie.201304310
Co-reporter:Brendan T. Parr ; Huw M. L. Davies
Angewandte Chemie 2013 Volume 125( Issue 38) pp:10228-10231
Publication Date(Web):
DOI:10.1002/ange.201304310
Co-reporter:Huw M. L. Davies and Yajing Lian
Accounts of Chemical Research 2012 Volume 45(Issue 6) pp:923
Publication Date(Web):May 11, 2012
DOI:10.1021/ar300013t
The development of methods for the stereoselective functionalization of sp3 C–H bonds is a challenging undertaking. This Account describes the scope of the combined C–H functionalization/Cope rearrangement (CHCR), a reaction that occurs between rhodium-stabilized vinylcarbenoids and substrates containing allylic C–H bonds. Computational studies have shown that the CHCR reaction is initiated by a hydride transfer to the carbenoid from an allyl site on the substrate, which is then rapidly followed by C–C bond formation between the developing rhodium-bound allyl anion and the allyl cation. In principle, the reaction can proceed through four distinct orientations of the vinylcarbenoid and the approaching substrate. The early examples of the CHCR reaction were all highly diastereoselective, consistent with a reaction proceeding via a chair transition state with the vinylcarbenoid adopting an s-cis conformation. Recent computational studies have revealed that other transition state orientations are energetically accessible, and these results have guided the development of highly stereoselective CHCR reactions that proceed through a boat transition state with the vinylcarbenoid in an s-cis configuration.The CHCR reaction has broad applications in organic synthesis. In some new protocols, the CHCR reaction acts as a surrogate to some of the classic synthetic strategies in organic chemistry. The CHCR reaction has served as a synthetic equivalent of the Michael reaction, the vinylogous Mukaiyama aldol reaction, the tandem Claisen rearrangement/Cope rearrangement, and the tandem aldol reaction/siloxy-Cope rearrangement. In all of these cases, the products are generated with very high diastereocontrol. With a chiral dirhodium tetracarboxylate catalyst such as Rh2(S-DOSP)4 or Rh2(S-PTAD)4, researchers can achieve very high levels of asymmetric induction. Applications of the CHCR reaction include the effective enantiodifferentiation of racemic dihydronaphthalenes and the total synthesis of several natural products: (−)-colombiasin A, (−)-elisapterosin B, and (+)-erogorgiaene. By combining the CHCR reaction into a further cascade sequence, we and other researchers have achieved the asymmetric synthesis of 4-substituted indoles, a new class of monoamine reuptake inhibitors.
Co-reporter:Zhanjie Li ; Brendan T. Parr
Journal of the American Chemical Society 2012 Volume 134(Issue 26) pp:10942-10946
Publication Date(Web):June 13, 2012
DOI:10.1021/ja303023n
The tandem ylide formation/[2,3]-sigmatropic rearrangement between donor/acceptor rhodium carbenoids and chiral allyl alcohols is a convergent C–C bond forming process, which generates two vicinal stereogenic centers. Any of the four possible stereoisomers can be selectively synthesized by appropriate combination of the chiral catalyst Rh2(DOSP)4 and the chiral alcohol.
Co-reporter:Zhanjie Li ; Vyacheslav Boyarskikh ; Jørn H. Hansen ; Jochen Autschbach ; Djamaladdin G. Musaev
Journal of the American Chemical Society 2012 Volume 134(Issue 37) pp:15497-15504
Publication Date(Web):August 27, 2012
DOI:10.1021/ja3061529
Rhodium-catalyzed reactions of tertiary propargylic alcohols with methyl aryl- and styryldiazoacetates result in tandem reactions, consisting of oxonium ylide formation followed by [2,3]-sigmatropic rearrangement. This process competes favorably with the standard O–H insertion reaction of carbenoids. The resulting allenes are produced with high enantioselectivity (88–98% ee) when the reaction is catalyzed by the dirhodium tetraprolinate complex, Rh2(S-DOSP)4. Kinetic resolution is possible when racemic tertiary propargylic alcohols are used as substrates. Under the kinetic resolution conditions, the allenes are formed with good diastereoselectivity and enantioselectivity (up to 6.1:1 dr, 88–93% ee), and the unreacted alcohols are enantioenriched to 65–95% ee. Computational studies reveal that the high asymmetric induction is obtained via an organized transition state involving a two-point attachment: ylide formation between the alcohol oxygen and the carbenoid and hydrogen bonding of the alcohol to a carboxylate ligand. The 2,3-sigmatropic rearrangement proceeds through initial cleavage of the O–H bond to generate an intermediate with close-lying open-shell singlet, triplet, and closed-shell singlet electronic states. This intermediate would have significant diradical character, which is consistent with the observation that the 2,3-sigmatropic rearrangement is favored with donor/acceptor carbenoids and more highly functionalized propargylic alcohols.
Co-reporter:Austin G. Smith
Journal of the American Chemical Society 2012 Volume 134(Issue 44) pp:18241-18244
Publication Date(Web):October 25, 2012
DOI:10.1021/ja3092399
A highly asymmetric vinylogous addition of acyclic silyl enol ethers to siloxyvinyldiazoacetate is described. The reaction features a diastereoselective 1,4-siloxy group migration event. Products are obtained in up to 97% ee. When more sterically crowded silyl enol ethers are employed, an enantioselective formal [3+2] cycloaddition becomes the dominant reaction pathway. Control experiments reveal the (Z)-olefin geometry to be critical for high levels of enantiocontrol.
Co-reporter:John F. Briones and Huw M. L. Davies
Journal of the American Chemical Society 2012 Volume 134(Issue 29) pp:11916-11919
Publication Date(Web):July 9, 2012
DOI:10.1021/ja304506g
Highly enantioselective cyclopropenation of internal alkynes with aryldiazoacetates was achieved using the binuclear gold catalyst (S)-xylylBINAP(AuCl)2, activated by silver hexafluoroantimonate.
Co-reporter:Daniel Morton, Allison R. Dick, Debashis Ghosh and Huw M. L. Davies
Chemical Communications 2012 vol. 48(Issue 47) pp:5838-5840
Publication Date(Web):24 Apr 2012
DOI:10.1039/C2CC31973J
The preparation and reactivity of steroidal vinyldiazo compounds is reported, providing a convenient, substituent tolerant, chemo- and stereoselective entry into 4- and 6-substituted androgen analogues from a common precursor. Under dirhodium catalysis, O–H insertion occurs at the carbenoid site, leading to 4-substituted steroids, but under silver catalysis, O–H insertion occurs at the vinylogous position, leading to 6-substituted steroids.
Co-reporter:Stephanie R. Hansen, Jillian E. Spangler, Jørn H. Hansen, and Huw M. L. Davies
Organic Letters 2012 Volume 14(Issue 17) pp:4626-4629
Publication Date(Web):August 24, 2012
DOI:10.1021/ol3020754
Synthetically useful transformations arise from the thermal decomposition of aryldiazoacetates in the presence of primary and secondary amines without the use of a metal catalyst. Thermally generated, free donor/acceptor carbenes directly undergo N–H insertion with amines through selective aza-ylide formation to afford a variety of α-amino esters in 53–96% yields.
Co-reporter:Joshua S. Alford and Huw M. L. Davies
Organic Letters 2012 Volume 14(Issue 23) pp:6020-6023
Publication Date(Web):November 15, 2012
DOI:10.1021/ol3029127
Warming of 4-phthalimido-N-mesyl-1,2,3-triazole in the presence of alkenes followed by silica gel induced hydrolysis results in a highly diastereoselective and catalyst-free entry to N-phthalimidocyclopropanecarboxaldehydes.
Co-reporter:Dr. Damien Valette;Dr. Yajing Lian;John P. Haydek;Dr. Kenneth I. Hardcastle ;Dr. Huw M. L. Davies
Angewandte Chemie 2012 Volume 124( Issue 34) pp:8764-8767
Publication Date(Web):
DOI:10.1002/ange.201204047
Co-reporter:Dr. Damien Valette;Dr. Yajing Lian;John P. Haydek;Dr. Kenneth I. Hardcastle ;Dr. Huw M. L. Davies
Angewandte Chemie International Edition 2012 Volume 51( Issue 34) pp:8636-8639
Publication Date(Web):
DOI:10.1002/anie.201204047
Co-reporter:Huw M. L. Davies and Daniel Morton
Chemical Society Reviews 2011 vol. 40(Issue 4) pp:1857-1869
Publication Date(Web):01 Mar 2011
DOI:10.1039/C0CS00217H
This tutorial review presents a description of the controlling elements of intermolecular C–H functionalization by means of C–H insertion by donor/acceptor rhodium carbenes. These rhodium carbenes, readily derived from the combination of diazo compounds with dirhodium(II) catalysts, are sufficiently reactive to undergo a wide range of C–H insertions. They are also capable of highly selective reactions, controlled by a combination of steric and electronic factors. An overview of the structural factors that influence site selectivity will be given, followed by a description of the exceptional diastereo- and enantioselectivity that can be achieved. Several examples will be shown of how this methodology can be applied to streamline the synthesis of natural products and pharmaceutical targets.
Co-reporter:Jørn H. Hansen ; Timothy M. Gregg ; Stephanie R. Ovalles ; Yajing Lian ; Jochen Autschbach
Journal of the American Chemical Society 2011 Volume 133(Issue 13) pp:5076-5085
Publication Date(Web):March 8, 2011
DOI:10.1021/ja111408v
The combined C−H activation/Cope rearrangement (CHCR) is an effective C−H functionalization process that has been used for the asymmetric synthesis of natural products and pharmaceutical building blocks. Up until now, a detailed understanding of this process was lacking. Herein, we describe a combination of theoretical and experimental studies that have resulted in a coherent description of the likely mechanism of the reaction. Density functional studies on the reactions of rhodium vinylcarbenoids at allylic C−H sites demonstrate that the CHCR proceeds through a concerted, but highly asynchronous, hydride-transfer/C−C bond-forming event. Even though most of the previously known examples of this process are highly diastereoselective, the calculations demonstrate that other transition-states and stereochemical outcomes might be possible by appropriate modifications of the reagents, and this was confirmed experimentally. The calculations also indicate that there is a potential energy surface bifurcation between CHCR and the competing direct C−H insertion.
Co-reporter:Changming Qin ; Vyacheslav Boyarskikh ; Jørn H. Hansen ; Kenneth I. Hardcastle ; Djamaladdin G. Musaev
Journal of the American Chemical Society 2011 Volume 133(Issue 47) pp:19198-19204
Publication Date(Web):November 2, 2011
DOI:10.1021/ja2074104
Dirhodium tetrakis-(R)-(1-(4-bromophenyl)-2,2-diphenylcyclopropanecarboxylate) (Rh2(R-BTPCP)4) was found to be an effective chiral catalyst for enantioselective reactions of aryl- and styryldiazoacetates. Highly enantioselective cyclopropanations, tandem cyclopropanation/Cope rearrangements and a combined C–H functionalization/Cope rearrangement were achieved using Rh2(R-BTPCP)4 as catalyst. The advantages of Rh2(R-BTPCP)4 include its ease of synthesis, its tolerance to the size of the ester group in the styryldiazoacetates, and its compatibility with dichloromethane as solvent. Computational studies suggest that the catalyst adopts a D2-symmetric arrangement, but when the carbenoid binds to the catalyst, two of the p-bromophenyl groups on the ligands rotate outward to make room for the carbenoid and the approach of the substrate to the carbenoid.
Co-reporter:Yajing Lian
Journal of the American Chemical Society 2011 Volume 133(Issue 31) pp:11940-11943
Publication Date(Web):July 8, 2011
DOI:10.1021/ja2051155
Vinyl ethers selectively undergo the combined C–H functionalization/Cope rearrangement reaction via an s-cis/boat transition state. With chiral dirhodium catalysts, products are generated in a highly diastereoselective and enantioselective fashion. This reaction can be considered as a surrogate to the traditional vinylogous Mukaiyama aldol reaction. Effective kinetic resolution has been achieved, leading to the recovery of a cyclic vinyl ether with axial chirality of high enantiomeric purity.
Co-reporter:Brendan T. Parr, Zhanjie Li and Huw M. L. Davies
Chemical Science 2011 vol. 2(Issue 12) pp:2378-2382
Publication Date(Web):02 Sep 2011
DOI:10.1039/C1SC00434D
A domino sequence has been developed between vinyldiazoacetates and racemic allyl alcohols, involving five distinct steps. The sequence generates highly functionalized cyclopentanes with four new stereogenic centers as single diastereomers in 64–92% ee. The first step is a rhodium-catalyzed oxygen ylide formation, which is then followed by a [2,3]-sigmatropic rearrangement, an oxy-Cope rearrangement, a keto/enol tautomerization, and then finally a carbonyl ene reaction. With appropriate substrates, a further silyl deprotection and a 6-exo-trigcyclization can be added to the domino process.
Co-reporter:Jørn H. Hansen and Huw M. L. Davies
Chemical Science 2011 vol. 2(Issue 3) pp:457-461
Publication Date(Web):22 Nov 2010
DOI:10.1039/C0SC00422G
Silver(I)-catalyzed reactions of vinyldiazoacetates have been shown to involve the intermediacy of silver-carbenoid species. The silver vinylcarbenoid displays a strong preference for electrophilic reactivity at the vinyl terminus in O–H insertion reactions.
Co-reporter:Hengbin Wang, Justin R. Denton, and Huw M. L. Davies
Organic Letters 2011 Volume 13(Issue 16) pp:4316-4319
Publication Date(Web):July 19, 2011
DOI:10.1021/ol2016548
A triple cascade process was developed for the rapid synthesis of polycyclic benzo-fused dihydrofurans. The first step is a rhodium-catalyzed cyclopropanation of α-aryldiazoketones with alkenes. This is followed by a silver-catalyzed ring expansion to dihydrofurans, which then undergo a gold-catalyzed cyclization to form benzo-fused dihydrofurans.
Co-reporter:John F. Briones, Huw M.L. Davies
Tetrahedron 2011 67(24) pp: 4313-4317
Publication Date(Web):
DOI:10.1016/j.tet.2011.04.029
Co-reporter:Jørn H. Hansen;Brendan T. Parr;Dr. Philip Pelphrey;Dr. Quihui Jin;Dr. Jochen Autschbach; Huw M. L. Davies
Angewandte Chemie International Edition 2011 Volume 50( Issue 11) pp:2544-2548
Publication Date(Web):
DOI:10.1002/anie.201004923
Co-reporter:Yajing Lian;Dr. Kenneth I. Hardcastle ;Dr. Huw M. L. Davies
Angewandte Chemie International Edition 2011 Volume 50( Issue 40) pp:9370-9373
Publication Date(Web):
DOI:10.1002/anie.201103568
Co-reporter:Jørn H. Hansen;Brendan T. Parr;Dr. Philip Pelphrey;Dr. Quihui Jin;Dr. Jochen Autschbach; Huw M. L. Davies
Angewandte Chemie 2011 Volume 123( Issue 11) pp:2592-2596
Publication Date(Web):
DOI:10.1002/ange.201004923
Co-reporter:Yajing Lian;Dr. Kenneth I. Hardcastle ;Dr. Huw M. L. Davies
Angewandte Chemie 2011 Volume 123( Issue 40) pp:9542-9545
Publication Date(Web):
DOI:10.1002/ange.201103568
Co-reporter:Yajing Lian ; Laura C. Miller ; Stephen Born ; Richmond Sarpong
Journal of the American Chemical Society 2010 Volume 132(Issue 35) pp:12422-12425
Publication Date(Web):August 12, 2010
DOI:10.1021/ja103916t
The tandem cyclopropanation/Cope rearrangement between bicyclic dienes and siloxyvinyldiazoacetate, catalyzed by the dirhodium catalyst Rh2(R-PTAD)4, effectively accomplishes enantiodivergent [4 + 3] cycloadditions. The reaction proceeds by a cyclopropanation followed by a Cope rearrangement of the resulting divinylcyclopropane. This methodology was applied to the synthesis of (+)-barekoxide (1) and (−)-barekol (2).
Co-reporter:John F. Briones ; Jørn Hansen ; Kenneth I. Hardcastle ; Jochen Autschbach
Journal of the American Chemical Society 2010 Volume 132(Issue 48) pp:17211-17215
Publication Date(Web):November 16, 2010
DOI:10.1021/ja106509b
Dirhodium tetrakis((S)-N-(dodecylbenzenesulfonyl)prolinate) (Rh2(S-DOSP)4) is an effective catalyst for highly enantioselective cyclopropenation reactions between terminal alkynes and arylvinyldiazoacetates. The resulting vinylcyclopropenes can undergo rhodium-catalyzed regioselective rearrangement to cyclopentadienes. Computational studies indicate that the high enantioselectivity of the process is governed by the specific orientation of the alkyne during its approach to the carbenoid through a relatively late transition state. The specific orientation occurs due to the presence of a hydrogen bonding interaction between the alkyne hydrogen and a carboxylate ligand on the dirhodium catalyst.
Co-reporter:Philip Pelphrey, Jørn Hansen and Huw M. L. Davies
Chemical Science 2010 vol. 1(Issue 2) pp:254-257
Publication Date(Web):11 Jun 2010
DOI:10.1039/C0SC00109K
Rhodium(II)-catalyzed reactions of donor/acceptor carbenoids can be achieved with catalyst loadings as low as 6 × 10−7 mol equivalents when the reactions are conducted in the absence of solvent. ReactIR studies demonstrate that donor/acceptor carbenoids are ideal for achieving high catalyst turnover numbers due to the rapid reactions of their diazo precursors and the enhanced selectivity of this class of carbenoids.
Co-reporter:Yajing Lian and Huw M. L. Davies
Organic Letters 2010 Volume 12(Issue 5) pp:924-927
Publication Date(Web):February 1, 2010
DOI:10.1021/ol9028385
An unusual rhodium carbenoid approach for introduction of 4-substituted (Z)-pent-2-enoates into sterically encumbered pyrroles and indoles is described. These studies show that (Z)-vinylcarbenoids have a greater tendency than (E)-vinylcarbenoids to react at the vinylogous position of the carbenoid rather than at the carbenoid center.
Co-reporter:Huw M. L. Davies and Justin R. Denton
Chemical Society Reviews 2009 vol. 38(Issue 11) pp:3061-3071
Publication Date(Web):30 Sep 2009
DOI:10.1039/B901170F
The metal catalyzed reactions of diazo compounds have been broadly used in organic synthesis. The resulting metal–carbenoid intermediates are capable of undergoing a range of unconventional reactions, and due to their high energy, they are ideal for initiating cascade sequences leading to the rapid generation of structural complexity. This tutorial review will give an overview of the most versatile reactions of donor/acceptor carbenoids, an exciting class of intermediates capable of highly selective reactions. This will include cyclopropanation, [4 + 3] cycloaddition, and C–H functionalization methodologies. The application of this chemistry to the synthesis of a range of natural products will be described.
Co-reporter:Zhanjie Li
Journal of the American Chemical Society 2009 Volume 132(Issue 1) pp:396-401
Publication Date(Web):December 8, 2009
DOI:10.1021/ja9075293
The rhodium-catalyzed reaction of racemic allyl alcohols with methyl phenyldiazoacetate or methyl styryldiazoacetate results in a two-step process, an initial oxonium ylide formation followed by a [2,3]-sigmatropic rearrangement. This process competes favorably with the more conventional O−H insertion chemistry as long as donor/acceptor carbenoids and highly substituted allyl alcohols are used as substrates. When the reactions are catalyzed by Rh2(S-DOSP)4, tertiary α-hydroxycarboxylate derivatives with two adjacent quaternary centers are produced with high enantioselectivity (85−98% ee).
Co-reporter:Yajing Lian
Journal of the American Chemical Society 2009 Volume 132(Issue 2) pp:440-441
Publication Date(Web):December 21, 2009
DOI:10.1021/ja9078094
An effective Rh2(S-DOSP)4-catalyzed asymmetric cyclopentannulation of indolyl rings has been developed. Depending on the substitution pattern of the indole, two distinct regioisomeric products can be generated. These studies demonstrate that rhodium-catalyzed reactions of donor/acceptor carbenoids proceeding by means of zwitterionic intermediates can be carried out with very high asymmetric induction.
Co-reporter:James R. Manning, Tammy Sexton, Steven R. Childers, Huw M.L. Davies
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 1) pp:58-61
Publication Date(Web):1 January 2009
DOI:10.1016/j.bmcl.2008.11.022
A series of enantiomerically pure 1-naphthyl and 4-indolyl arylalkylamines were prepared and evaluated for their binding affinities to the monoamine transporters. The two series of enantiomers displayed considerable differences in binding selectivity between the monoamine transporters, leading to the design of (S)-4-(3,4-dichlorophenyl)-4-(1H-indol-4-yl)-N-methylbutan-1-amine as a potent inhibitor for the dopamine and serotonin transporters.The enantioselective synthesis and evaluation of novel diarylalkylamines are reported.
Co-reporter:Dominic L. Ventura, Zhanjie Li, Michael G. Coleman, Huw M.L. Davies
Tetrahedron 2009 65(16) pp: 3052-3061
Publication Date(Web):
DOI:10.1016/j.tet.2008.11.059
Co-reporter:Joshua S. Alford ; Jillian E. Spangler
Journal of the American Chemical Society () pp:
Publication Date(Web):July 26, 2013
DOI:10.1021/ja405043g
A highly effective synthesis of 2,3-fused pyrroles from cyclic ketones has been achieved. The transformation includes a rhodium-catalyzed reaction of 4-alkenyl-1-sulfonyl-1,2,3-triazoles featuring an unusual 4π electrocyclization. The methodology was further extended to the synthesis of indoles using a one-pot reaction starting from 1-ethynylcyclohexenes.
Co-reporter:John F. Briones
Journal of the American Chemical Society () pp:
Publication Date(Web):August 26, 2013
DOI:10.1021/ja407179c
The reaction of vinyldiazoacetates with enol ethers catalyzed by the binuclear gold complex (R)-DTBMSegphos(AuCl)2 activated by silver hexafluoroantimonate results in a highly enantioselective [3 + 2] cycloaddition. The [3 + 2] cycloaddition proceeds with dynamic kinetic resolution when the enol ether is a 4-substituted 1-(methoxymethylene)cyclohexane. The reaction is initiated by nucleophilic attack of the vinyl ethers at the vinylogous position of the gold vinylcarbene intermediate.
Co-reporter:John F. Briones
Organic Letters () pp:
Publication Date(Web):June 27, 2011
DOI:10.1021/ol201503j
Silver triflate was found to be an efficient catalyst for the cyclopropenation of internal alkynes using donor-/acceptor-substituted diazo compounds as carbenoid precursors. Highly substituted cyclopropenes, which cannot be synthesized directly via rhodium(II)-catalyzed carbenoid chemistry, can now be readily accessed.
Co-reporter:Daniel Morton, Allison R. Dick, Debashis Ghosh and Huw M. L. Davies
Chemical Communications 2012 - vol. 48(Issue 47) pp:NaN5840-5840
Publication Date(Web):2012/04/24
DOI:10.1039/C2CC31973J
The preparation and reactivity of steroidal vinyldiazo compounds is reported, providing a convenient, substituent tolerant, chemo- and stereoselective entry into 4- and 6-substituted androgen analogues from a common precursor. Under dirhodium catalysis, O–H insertion occurs at the carbenoid site, leading to 4-substituted steroids, but under silver catalysis, O–H insertion occurs at the vinylogous position, leading to 6-substituted steroids.
Co-reporter:Jørn H. Hansen and Huw M. L. Davies
Chemical Science (2010-Present) 2011 - vol. 2(Issue 3) pp:NaN461-461
Publication Date(Web):2010/11/22
DOI:10.1039/C0SC00422G
Silver(I)-catalyzed reactions of vinyldiazoacetates have been shown to involve the intermediacy of silver-carbenoid species. The silver vinylcarbenoid displays a strong preference for electrophilic reactivity at the vinyl terminus in O–H insertion reactions.
Co-reporter:Brendan T. Parr, Zhanjie Li and Huw M. L. Davies
Chemical Science (2010-Present) 2011 - vol. 2(Issue 12) pp:NaN2382-2382
Publication Date(Web):2011/09/02
DOI:10.1039/C1SC00434D
A domino sequence has been developed between vinyldiazoacetates and racemic allyl alcohols, involving five distinct steps. The sequence generates highly functionalized cyclopentanes with four new stereogenic centers as single diastereomers in 64–92% ee. The first step is a rhodium-catalyzed oxygen ylide formation, which is then followed by a [2,3]-sigmatropic rearrangement, an oxy-Cope rearrangement, a keto/enol tautomerization, and then finally a carbonyl ene reaction. With appropriate substrates, a further silyl deprotection and a 6-exo-trigcyclization can be added to the domino process.
Co-reporter:Huw M. L. Davies and Daniel Morton
Chemical Society Reviews 2011 - vol. 40(Issue 4) pp:NaN1869-1869
Publication Date(Web):2011/03/01
DOI:10.1039/C0CS00217H
This tutorial review presents a description of the controlling elements of intermolecular C–H functionalization by means of C–H insertion by donor/acceptor rhodium carbenes. These rhodium carbenes, readily derived from the combination of diazo compounds with dirhodium(II) catalysts, are sufficiently reactive to undergo a wide range of C–H insertions. They are also capable of highly selective reactions, controlled by a combination of steric and electronic factors. An overview of the structural factors that influence site selectivity will be given, followed by a description of the exceptional diastereo- and enantioselectivity that can be achieved. Several examples will be shown of how this methodology can be applied to streamline the synthesis of natural products and pharmaceutical targets.
Co-reporter:Huw M. L. Davies and Joshua S. Alford
Chemical Society Reviews 2014 - vol. 43(Issue 15) pp:NaN5162-5162
Publication Date(Web):2014/05/07
DOI:10.1039/C4CS00072B
Metal-stabilized carbenes derived from diazo compounds have become broadly useful reactive intermediates for organic synthesis. This tutorial review will describe the recent advances in using N-sulfonyl-1,2,3-triazoles as precursors for the formation of metal-bound imino carbene intermediates. These intermediates undergo a variety of synthetically useful transformations, which include transannulation reactions to generate new heterocycles, cyclopropanation and subsequent ring expansions, ylide formation with subsequent rearrangements, and C–H functionalization. Furthermore, many of these transformations can be conducted with high levels of enantioselectivity by use of chiral rhodium(II) catalysts.
Co-reporter:Elizabeth N. Bess, David M. Guptill, Huw M. L. Davies and Matthew S. Sigman
Chemical Science (2010-Present) 2015 - vol. 6(Issue 5) pp:NaN3062-3062
Publication Date(Web):2015/03/18
DOI:10.1039/C5SC00357A
Achieving selective C–H functionalization is a significant challenge that requires discrimination between many similar C–H bonds. Yet, reaction systems employing Rh2(DOSP)4 and Rh2(BPCP)4 were recently demonstrated to afford high levels of selectivity in the C–H insertion of carbenes into toluene-derived substrates. Herein, we explore the origin of this selectivity through a systematic analysis of substrate and reagent features that alter levels of selectivity from 20:1 to 1:610 for secondary (or tertiary)-to-primary benzylic C–H functionalization of toluene derivatives. Describing this variation using infrared vibrations and point charges, we have developed a mathematical model from which are identified features of the systems that determine levels of site-selectivity and are applied as predictive factors to describe the selectivity behavior of new substrate/reagent combinations.
Co-reporter:Huw M. L. Davies and Justin R. Denton
Chemical Society Reviews 2009 - vol. 38(Issue 11) pp:NaN3071-3071
Publication Date(Web):2009/09/30
DOI:10.1039/B901170F
The metal catalyzed reactions of diazo compounds have been broadly used in organic synthesis. The resulting metal–carbenoid intermediates are capable of undergoing a range of unconventional reactions, and due to their high energy, they are ideal for initiating cascade sequences leading to the rapid generation of structural complexity. This tutorial review will give an overview of the most versatile reactions of donor/acceptor carbenoids, an exciting class of intermediates capable of highly selective reactions. This will include cyclopropanation, [4 + 3] cycloaddition, and C–H functionalization methodologies. The application of this chemistry to the synthesis of a range of natural products will be described.
Co-reporter:Philip Pelphrey, Jørn Hansen and Huw M. L. Davies
Chemical Science (2010-Present) 2010 - vol. 1(Issue 2) pp:NaN257-257
Publication Date(Web):2010/06/11
DOI:10.1039/C0SC00109K
Rhodium(II)-catalyzed reactions of donor/acceptor carbenoids can be achieved with catalyst loadings as low as 6 × 10−7 mol equivalents when the reactions are conducted in the absence of solvent. ReactIR studies demonstrate that donor/acceptor carbenoids are ideal for achieving high catalyst turnover numbers due to the rapid reactions of their diazo precursors and the enhanced selectivity of this class of carbenoids.
Co-reporter:Hengbin Wang, David M. Guptill, Adrian Varela-Alvarez, Djamaladdin G. Musaev and Huw M. L. Davies
Chemical Science (2010-Present) 2013 - vol. 4(Issue 7) pp:NaN2850-2850
Publication Date(Web):2013/04/15
DOI:10.1039/C3SC50425E
The rhodium-catalyzed reaction of electron-deficient alkenes with substituted aryldiazoacetates and vinyldiazoacetates results in highly stereoselective cyclopropanations. With adamantylglycine derived catalyst Rh2(S-TCPTAD)4, high asymmetric induction (up to 98% ee) can be obtained with a range of substrates. Computational studies suggest that the reaction is facilitated by weak interaction between the carbenoid and the substrate carbonyl but subsequently proceeds via different pathways depending on the nature of the carbonyl. Acrylates and acrylamides result in the formation of cyclopropanation products while the use of unsaturated aldehydes and ketones results in the formation of epoxides.
Co-reporter:Clayton P. Owens, Adrián Varela-Álvarez, Vyacheslav Boyarskikh, Djamaladdin G. Musaev, Huw M. L. Davies and Simon B. Blakey
Chemical Science (2010-Present) 2013 - vol. 4(Issue 6) pp:NaN2596-2596
Publication Date(Web):2013/04/19
DOI:10.1039/C3SC50886B
Recently, a small number of diverse iridium complexes have been shown to catalyze unusual atom transfer C–H functionalization reactions. To further our understanding and enhance the utility of iridium complexes for C–H functionalization, we report the design and synthesis of a family of iridium(III)-bis(oxazolinyl)phenyl complexes. The ability to tune the ligand environment around the metal in these systems is exploited to design complexes with the ability to catalyze the asymmetric insertion of donor/acceptor iridium carbenoids into activated C–H bonds. Low catalyst loadings (0.5 mol%) routinely lead to excellent reaction yields (51–99%) and enantioselectivities (83–99%). Density functional theory calculations provide compelling evidence that in these complexes the carbene binds to the iridium cis to the phenyl group of the bis(oxazolinyl)phenyl ligand. This finding is vital for understanding the observed stereochemical induction and is of particular significance in the field of enantioselective transition metal-catalysed atom transfer reactions utilizing oxazoline–X–oxazoline tridentate ligands, as previously employed stereochemical models for these ligand sets are based on the assumption that reactive ligands and Lewis bases bind trans to the central X ligand.





![Silane, [[1-(1-cyclopenten-1-yl)ethenyl]oxy]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/163900/163811-59-4.png)
![Silane, [[1-(1-cyclopenten-1-yl)ethenyl]oxy]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/163900/163811-59-4_b.png)
![Acetaldehyde, [tris(1-methylethyl)silyl]-](http://img.cochemist.com/ccimg/162300/162247-31-6.png)
![Acetaldehyde, [tris(1-methylethyl)silyl]-](http://img.cochemist.com/ccimg/162300/162247-31-6_b.png)
![Silane, (1,1-dimethylethyl)[(3-methoxyphenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/154000/153974-48-2.png)
![Silane, (1,1-dimethylethyl)[(3-methoxyphenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/154000/153974-48-2_b.png)
![Isoxazole, 3-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-5-methyl-](http://img.cochemist.com/ccimg/153200/153168-60-6.png)
![Isoxazole, 3-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-5-methyl-](http://img.cochemist.com/ccimg/153200/153168-60-6_b.png)











![Phenol, 4-[1-[(trimethylsilyl)oxy]-1-propenyl]-, acetate, (Z)-](/data/chemimg/1112400/140902-15-4.png)
![Phenol, 4-[1-[(trimethylsilyl)oxy]-1-propenyl]-, acetate, (Z)-](/data/chemimg/1112400/140902-15-4_b.png)






dimethyl-](http://img.cochemist.com/ccimg/134600/134531-82-1.png)
dimethyl-](http://img.cochemist.com/ccimg/134600/134531-82-1_b.png)
dimethyl-](http://img.cochemist.com/ccimg/133500/133464-90-1.png)
dimethyl-](http://img.cochemist.com/ccimg/133500/133464-90-1_b.png)








![Silane, (1,1-dimethylethyl)dimethyl[[(1E)-1-phenyl-1-propenyl]oxy]-](http://img.cochemist.com/ccimg/122200/122124-42-9.png)
![Silane, (1,1-dimethylethyl)dimethyl[[(1E)-1-phenyl-1-propenyl]oxy]-](http://img.cochemist.com/ccimg/122200/122124-42-9_b.png)








![[(1S)-1-methylbutyl] methanesulfonate](http://img.cochemist.com/ccimg/120000/119992-81-3.png)
![[(1S)-1-methylbutyl] methanesulfonate](http://img.cochemist.com/ccimg/120000/119992-81-3_b.png)


![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/118000/117932-70-4.png)
![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/118000/117932-70-4_b.png)


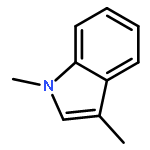
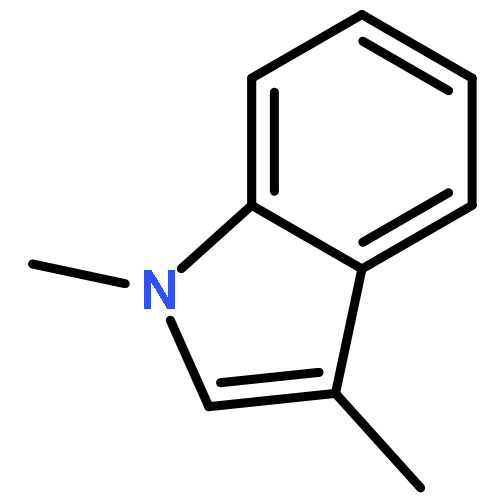
![1H-Indole, 4-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/113000/112970-65-7.png)
![1H-Indole, 4-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/113000/112970-65-7_b.png)



![Silane, [(3-butyl-1-cyclopenten-1-yl)oxy]trimethyl-](http://img.cochemist.com/ccimg/108700/108643-82-9.png)
![Silane, [(3-butyl-1-cyclopenten-1-yl)oxy]trimethyl-](http://img.cochemist.com/ccimg/108700/108643-82-9_b.png)






![Silane, (1,1-dimethylethyl)[(4-methoxyphenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/101900/101803-60-5.png)
![Silane, (1,1-dimethylethyl)[(4-methoxyphenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/101900/101803-60-5_b.png)
![Silane, (1,1-dimethylethyl)dimethyl[(3-phenyl-2-propenyl)oxy]-, (E)-](http://img.cochemist.com/ccimg/100100/100009-29-8.png)
![Silane, (1,1-dimethylethyl)dimethyl[(3-phenyl-2-propenyl)oxy]-, (E)-](http://img.cochemist.com/ccimg/100100/100009-29-8_b.png)













![5-Iodo-2-methylbenzo[d]thiazole](http://img.cochemist.com/ccimg/90500/90414-61-2.png)
![5-Iodo-2-methylbenzo[d]thiazole](http://img.cochemist.com/ccimg/90500/90414-61-2_b.png)


dimethyl-](http://img.cochemist.com/ccimg/90300/90219-45-7.png)
dimethyl-](http://img.cochemist.com/ccimg/90300/90219-45-7_b.png)




![SILANE, [[(1Z)-1,2-DIPHENYLETHENYL]OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/88100/88089-52-5.png)
![SILANE, [[(1Z)-1,2-DIPHENYLETHENYL]OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/88100/88089-52-5_b.png)
![Benzenamine, N-[(4-methoxyphenyl)methylene]-, N-oxide, (Z)-](http://img.cochemist.com/ccimg/87300/87289-90-5.png)
![Benzenamine, N-[(4-methoxyphenyl)methylene]-, N-oxide, (Z)-](http://img.cochemist.com/ccimg/87300/87289-90-5_b.png)









![Silane, trimethyl[(3-methyl-1-cyclopenten-1-yl)oxy]-](http://img.cochemist.com/ccimg/81900/81834-51-7.png)
![Silane, trimethyl[(3-methyl-1-cyclopenten-1-yl)oxy]-](http://img.cochemist.com/ccimg/81900/81834-51-7_b.png)
![SILANE, TRIMETHYL[[(2E)-1-METHYLENE-2-BUTENYL]OXY]-](http://img.cochemist.com/ccimg/81900/81802-34-8.png)
![SILANE, TRIMETHYL[[(2E)-1-METHYLENE-2-BUTENYL]OXY]-](http://img.cochemist.com/ccimg/81900/81802-34-8_b.png)








![SILANE, (1,1-DIMETHYLETHYL)[[1-(4-METHOXYPHENYL)ETHENYL]OXY]DIMETHYL-](http://img.cochemist.com/ccimg/80700/80676-78-4.png)
![SILANE, (1,1-DIMETHYLETHYL)[[1-(4-METHOXYPHENYL)ETHENYL]OXY]DIMETHYL-](http://img.cochemist.com/ccimg/80700/80676-78-4_b.png)






![SILANE, DIMETHYLPHENYL[(2Z)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/79300/79294-17-0.png)
![SILANE, DIMETHYLPHENYL[(2Z)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/79300/79294-17-0_b.png)









![SILANE, [(1,3-DIMETHOXY-1,3-BUTADIENYL)OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/74300/74272-66-5.png)
![SILANE, [(1,3-DIMETHOXY-1,3-BUTADIENYL)OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/74300/74272-66-5_b.png)



![1-[2-(4-METHYLPHENYL)ETHYNYL]NAPHTHALENE](http://img.cochemist.com/ccimg/72800/72735-70-7.png)
![1-[2-(4-METHYLPHENYL)ETHYNYL]NAPHTHALENE](http://img.cochemist.com/ccimg/72800/72735-70-7_b.png)


![Silane, trimethyl[(1-phenyl-1-propenyl)oxy]-, (E)-](http://img.cochemist.com/ccimg/71300/71268-59-2.png)
![Silane, trimethyl[(1-phenyl-1-propenyl)oxy]-, (E)-](http://img.cochemist.com/ccimg/71300/71268-59-2_b.png)


![Silane, trimethyl[(3-phenyl-1-cyclopenten-1-yl)oxy]-](http://img.cochemist.com/ccimg/69900/69849-77-0.png)
![Silane, trimethyl[(3-phenyl-1-cyclopenten-1-yl)oxy]-](http://img.cochemist.com/ccimg/69900/69849-77-0_b.png)


![Silane, (1,1-dimethylethyl)dimethyl[(1-phenylethenyl)oxy]-](http://img.cochemist.com/ccimg/66400/66324-10-5.png)
![Silane, (1,1-dimethylethyl)dimethyl[(1-phenylethenyl)oxy]-](http://img.cochemist.com/ccimg/66400/66324-10-5_b.png)




![Silane, triethyl[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/63600/63522-98-5.png)
![Silane, triethyl[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/63600/63522-98-5_b.png)
![SILANE, [(1E)-1,3-BUTADIENYLOXY]TRIMETHYL-](http://img.cochemist.com/ccimg/63400/63383-46-0.png)
![SILANE, [(1E)-1,3-BUTADIENYLOXY]TRIMETHYL-](http://img.cochemist.com/ccimg/63400/63383-46-0_b.png)











![Silane, [(3,4-dimethoxyphenyl)methoxy]trimethyl-](http://img.cochemist.com/ccimg/61100/61040-75-3.png)
![Silane, [(3,4-dimethoxyphenyl)methoxy]trimethyl-](http://img.cochemist.com/ccimg/61100/61040-75-3_b.png)




![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 3-[(2-aminoethyl)thio]-6-[(1R)-1-hydroxyethyl]-7-oxo-, (5R,6S)-](http://img.cochemist.com/ccimg/60000/59995-64-1.png)
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 3-[(2-aminoethyl)thio]-6-[(1R)-1-hydroxyethyl]-7-oxo-, (5R,6S)-](http://img.cochemist.com/ccimg/60000/59995-64-1_b.png)








![Benzene, [(1Z)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/58000/57918-63-5.png)
![Benzene, [(1Z)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/58000/57918-63-5_b.png)






![Benzenamine, 4-nitro-N-[(2E)-3-phenyl-2-propenylidene]-](http://img.cochemist.com/ccimg/56200/56110-09-9.png)
![Benzenamine, 4-nitro-N-[(2E)-3-phenyl-2-propenylidene]-](http://img.cochemist.com/ccimg/56200/56110-09-9_b.png)




![SILANE, [(4-METHYL-1-CYCLOPENTENE-1,2-DIYL)BIS(OXY)]BIS[TRIMETHYL-](http://img.cochemist.com/ccimg/54900/54851-47-7.png)
![SILANE, [(4-METHYL-1-CYCLOPENTENE-1,2-DIYL)BIS(OXY)]BIS[TRIMETHYL-](http://img.cochemist.com/ccimg/54900/54851-47-7_b.png)















![2-[DIMETHYL(PHENYL)SILYL]ETHANOL](http://img.cochemist.com/ccimg/41900/41885-43-2.png)
![2-[DIMETHYL(PHENYL)SILYL]ETHANOL](http://img.cochemist.com/ccimg/41900/41885-43-2_b.png)
![Benzene,[(1E)-3-(trimethylsilyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/40600/40595-34-4.png)
![Benzene,[(1E)-3-(trimethylsilyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/40600/40595-34-4_b.png)















































![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-methoxy-](/data/chemimg/926100/16034-99-4.png)
![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-methoxy-](/data/chemimg/926100/16034-99-4_b.png)
![Benzene, [(1Z)-2-methoxyethenyl]-](http://img.cochemist.com/ccimg/14400/14371-19-8.png)
![Benzene, [(1Z)-2-methoxyethenyl]-](http://img.cochemist.com/ccimg/14400/14371-19-8_b.png)











![Silane, [1-cyclopentene-1,2-diylbis(oxy)]bis[trimethyl-](http://img.cochemist.com/ccimg/6900/6838-66-0.png)
![Silane, [1-cyclopentene-1,2-diylbis(oxy)]bis[trimethyl-](http://img.cochemist.com/ccimg/6900/6838-66-0_b.png)












![2-methoxy-N-[(E)-phenylmethylidene]aniline](http://img.cochemist.com/ccimg/5900/5877-56-5.png)
![2-methoxy-N-[(E)-phenylmethylidene]aniline](http://img.cochemist.com/ccimg/5900/5877-56-5_b.png)






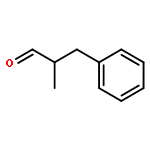
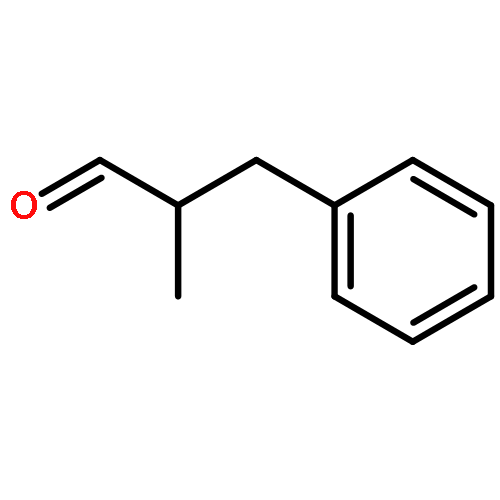

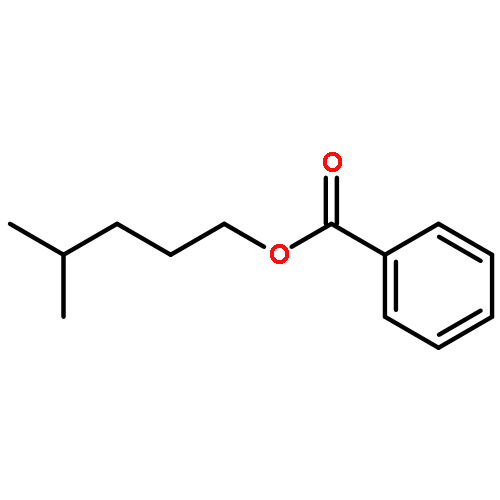


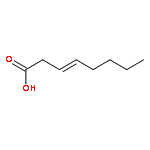
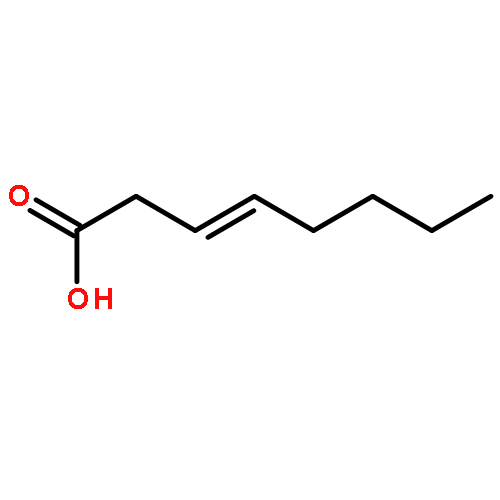


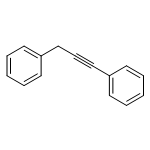
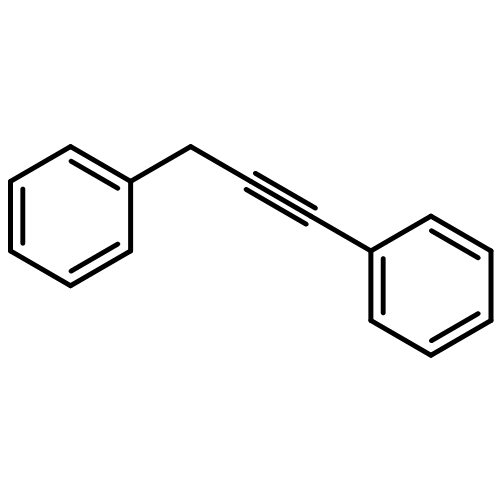





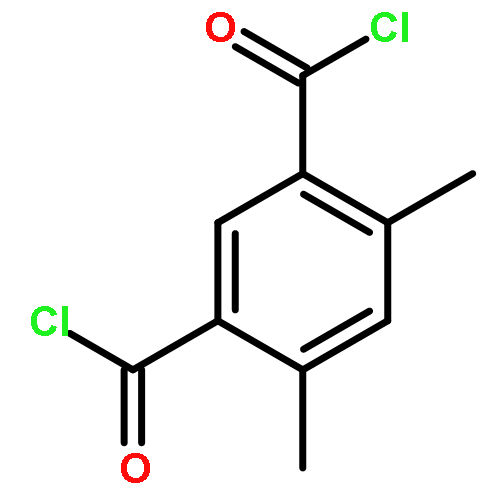






![Benzene, [(1E)-2-methoxyethenyl]-](http://img.cochemist.com/ccimg/4200/4110-75-2.png)
![Benzene, [(1E)-2-methoxyethenyl]-](http://img.cochemist.com/ccimg/4200/4110-75-2_b.png)





















































































![Silane, trimethyl[[(1Z)-1-phenyl-1-butenyl]oxy]-](http://img.cochemist.com/ccimg/120900/120869-79-6.png)
![Silane, trimethyl[[(1Z)-1-phenyl-1-butenyl]oxy]-](http://img.cochemist.com/ccimg/120900/120869-79-6_b.png)




![3-BUTENOIC ACID, 2-DIAZO-3-[(TRIMETHYLSILYL)OXY]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/80000/79975-81-8.png)
![3-BUTENOIC ACID, 2-DIAZO-3-[(TRIMETHYLSILYL)OXY]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/80000/79975-81-8_b.png)





![Silane, (1,1-dimethylethyl)dimethyl[(2-methyl-1-cyclopenten-1-yl)oxy]-](http://img.cochemist.com/ccimg/68100/68081-17-4.png)
![Silane, (1,1-dimethylethyl)dimethyl[(2-methyl-1-cyclopenten-1-yl)oxy]-](http://img.cochemist.com/ccimg/68100/68081-17-4_b.png)
![SILANE, TRIMETHYL[[(1Z)-1-PHENYL-1-PROPENYL]OXY]-](http://img.cochemist.com/ccimg/66400/66323-99-7.png)
![SILANE, TRIMETHYL[[(1Z)-1-PHENYL-1-PROPENYL]OXY]-](http://img.cochemist.com/ccimg/66400/66323-99-7_b.png)

































![Silane, (1,1-dimethylethyl)dimethyl[[(2E)-1-methylene-2-butenyl]oxy]-](http://img.cochemist.com/ccimg/160100/160036-53-3.png)
![Silane, (1,1-dimethylethyl)dimethyl[[(2E)-1-methylene-2-butenyl]oxy]-](http://img.cochemist.com/ccimg/160100/160036-53-3_b.png)













![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0.png)
![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0_b.png)






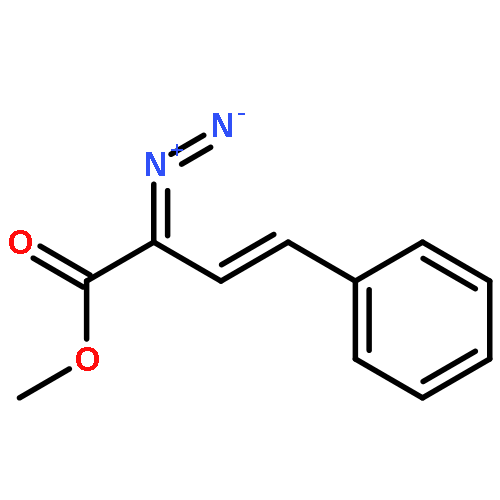
![3-Butenoic acid, 2-diazo-4-[4-(trifluoromethyl)phenyl]-, methyl ester, (E)-](http://img.cochemist.com/ccimg/188000/187950-52-3.png)
![3-Butenoic acid, 2-diazo-4-[4-(trifluoromethyl)phenyl]-, methyl ester, (E)-](http://img.cochemist.com/ccimg/188000/187950-52-3_b.png)



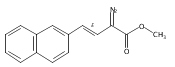
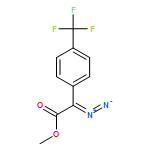

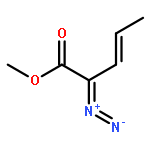





![Tetrakis[(R)-(+)-N-(p-dodecylphenylsulfonyl)prolinato]dirhodium (II) Rh2(R- DOSP)4](http://img.cochemist.com/ccimg/178900/178879-60-2.png)
![Tetrakis[(R)-(+)-N-(p-dodecylphenylsulfonyl)prolinato]dirhodium (II) Rh2(R- DOSP)4](http://img.cochemist.com/ccimg/178900/178879-60-2_b.png)
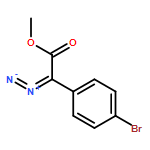

![Rhodium, tetrakis[m-[(aS)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909389-99-7.png)
![Rhodium, tetrakis[m-[(aS)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909389-99-7_b.png)

![Rhodium, tetrakis[m-[1-[(4-dodecylphenyl)sulfonyl]-L-prolinato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/179200/179162-34-6.png)
![Rhodium, tetrakis[m-[1-[(4-dodecylphenyl)sulfonyl]-L-prolinato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/179200/179162-34-6_b.png)