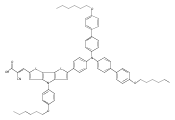
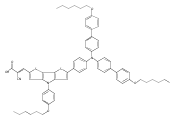
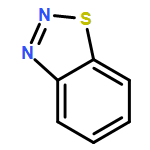
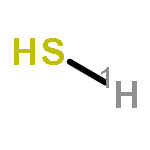
Co-reporter:Shufen Wang;Jiuchuang Yuan;Huixing Li
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 30) pp:19873-19880
Publication Date(Web):2017/08/02
DOI:10.1039/C7CP02153D
In order to study the dynamics of the reaction H(2S) + NaH(X1Σ+) → Na(2S) + H2(X1Σg+), a new potential energy surface (PES) for the ground state of the NaH2 system is constructed based on 35 730 ab initio energy points. Using basis sets of quadruple zeta quality, multireference configuration interaction calculations with Davidson correction were carried out to obtain the ab initio energy points. The neural network method is used to fit the PES, and the root mean square error is very small (0.00639 eV). The bond lengths, dissociation energies, zero-point energies and spectroscopic constants of H2(X1Σg+) and NaH(X1Σ+) obtained on the new NaH2 PES are in good agreement with the experiment data. On the new PES, the reactant coordinate-based time-dependent wave packet method is applied to study the reaction dynamics of H(2S) + NaH(X1Σ+) → Na(2S) + H2(X1Σg+), and the reaction probabilities, integral cross-sections (ICSs) and differential cross-sections (DCSs) are obtained. There is no threshold in the reaction due to the absence of an energy barrier on the minimum energy path. When the collision energy increases, the ICSs decrease from a high value at low collision energy. The DCS results show that the angular distribution of the product molecules tends to the forward direction. Compared with the LiH2 system, the NaH2 system has a larger mass and the PES has a larger well at the H–NaH configuration, which leads to a higher ICS value in the H(2S) + NaH(X1Σ+) → Na(2S) + H2(X1Σg+) reaction. Because the H(2S) + NaH(X1Σ+) → Na(2S) + H2(X1Σg+) reaction releases more energy, the product molecules can be excited to a higher vibrational state.
Co-reporter:Quanjiang Li;Qianqian Ding;Weihua Lin;Jiangcai Wang;Mengtao Sun
RSC Advances (2011-Present) 2017 vol. 7(Issue 20) pp:12170-12178
Publication Date(Web):2017/02/16
DOI:10.1039/C6RA28240G
In this study, we theoretically investigated the Raman and absorption spectra of pyrazine adsorbed on Au5Al5 bimetallic nanoclusters by a time-dependent density functional theory (TD-DFT) method. The surface-enhanced resonance Raman scattering (SERRS) spectra of pyrazine absorbed on different isomers and sites of the Au5Al5 cluster were simulated. The visualization of orbital transitions in electronic transitions was used to analyze the enhancement mechanism of SERRS spectroscopy. Compared with those of isolated pyrazine excited at 598 nm, the SERRS of pyrazine–Au–Au4Al5-a excited at the same incident light can be enhanced on the order of 104, which is a typical charge transfer (CT) resonance excitation and charge transfer from substrate to pyrazine. Due to the fact that the intensity of ultraviolet SERRS can be significantly enhanced to 1.2 × 106 A4 per amu for pyrazine–Au–Au4Al5-a model at 280 nm, the Au5Al5 cluster may be a good candidate for research of the ultraviolet SERRS materials. Other key factors that can change the intensity of SERRS include the resonance excitation wavelength, oscillator strength of the electronic excited state, metal–molecule binding site and structure of the substrate cluster. Hence, the optical properties of complexes can be tuned by varying these factors.
Co-reporter:Man Dong;Wentao Li;Di He
RSC Advances (2011-Present) 2017 vol. 7(Issue 12) pp:7008-7014
Publication Date(Web):2017/01/20
DOI:10.1039/C6RA27765A
The dynamic properties of the title reaction calculated by classical and quantum methods show large deviations from each other, whereas for the barrierless and exothermal reaction two methods should show good agreement. In order to further investigate the reaction mechanism of the title reaction, a global PES for the electronic ground state was constructed. The energy points are calculated by the multireference configuration interaction method with aug-cc-pVQZ and cc-pwCVQZ basis sets for H and Li atoms, respectively. The neural network approach is adopted in the fitting process. The classical and quantum methods are applied in the dynamic calculation based on the new PES. As expected, the dynamic properties obtained by these two methods are in good agreement with each other. In addition, two reaction mechanisms were found. When the energy is below 0.2 eV the insert reaction mechanism is dominant, and this changes to the abstract reaction mechanism as the energy increases.
Co-reporter:Shufen Wang;Di He;Wentao Li
RSC Advances (2011-Present) 2017 vol. 7(Issue 57) pp:35648-35654
Publication Date(Web):2017/07/17
DOI:10.1039/C7RA05223E
A global potential energy surface (PES) of the ground state of the Au+H2 system was constructed using a neural network method with permutation invariant polynomials. More than 40 000 ab initio points were calculated using a multi-reference configuration interaction approach, and these were adopted into a fitting process; the root mean square error was 0.02 eV. The topography of the present PES was compared in detail with the PES reported by Dorta-Urra (J. Chem. Phys., 135, 2011, 091102). The potential well in the present PES is about 0.12 eV deeper than the previous PES. The dynamics calculations based on these two PESs were performed by the time-dependent quantum wave packet method with a second-order split operator. The reaction probability, integral cross section and differential cross section based on these two PESs were calculated in this work. The results obtained from these two PESs were compared with the experimental data. It was clear that the integral cross section calculated by using the present PES is closer to the experimental data than the one obtained from Dorta-Urra PES. In addition, the reaction mechanism of the title reaction changed from an indirect reaction to an abstraction reaction as the collision energy increased.
Co-reporter:Jiuchuang Yuan, Di He and Maodu Chen
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 17) pp:11732-11739
Publication Date(Web):31 Mar 2015
DOI:10.1039/C4CP05352D
A new global potential energy surface (PES) is obtained for the ground electronic state of the LiH2 system based on high-level energies. The energy points are calculated at the multireference configuration interaction level with aug-cc-pVXZ (X = Q, 5) basis sets, and these energies are extrapolated to the complete basis set limit. The neural network method and hierarchical construction scheme are applied in the fitting process and the root mean square error of the fitting result is very small (0.004 eV). The dissociation energies and equilibrium distances for LiH(X1Σ+) and H2(X1Σg+) obtained from the new PES are in good agreement with the experimental data. On the new PES, time-dependent wave packet studies for the H(2S) + LiH(X1Σ+) → Li(2S) + H2(X1Σg+) reaction have been carried out. In this reaction, no threshold is found due to the absence of an energy barrier on the minimum energy path. The calculated integral cross sections are high at low collision energy and will decrease with the increase of the collision energy. The product molecule H2 tends to be forward scattering due to direct reactive collisions, which becomes more evident at higher collision energies.
Co-reporter:Tiangang Yang, Long Huang, Tao Wang, Chunlei Xiao, Yurun Xie, Zhigang Sun, Dongxu Dai, Maodu Chen, Donghui Zhang, and Xueming Yang
The Journal of Physical Chemistry A 2015 Volume 119(Issue 50) pp:12284-12290
Publication Date(Web):August 24, 2015
DOI:10.1021/acs.jpca.5b06395
The reaction of fluorine atom with vibrationally excited H2 at v = 1 has been studied using a high resolution crossed molecular beam apparatus at collision energies of 0.52 and 0.90 kcal/mol. Product HF rotational state-resolved differential cross sections (DCSs) were measured at v′ = 2, 3, 4 levels. The product angular distributions are predominantly backward scattered except for a small forward signal of HF(v′ = 4) at 0.90 kcal/mol. At the collision energy of 0.52 kcal/mol, the forward scattering peak of the HF(v′ = 2) product, which arises in F + H2(v = 0) reaction from the Feshbach resonances, disappears in F + H2(v = 1) reaction. Oscillatory structures do not appear in the backward direction of the scattering as the collision energy increases from 0.4 to 2.0 kcal/mol, indicating there are no explicit reaction resonances in the F + H2(v = 1, j = 0) → HF + H reaction in the studied energy range. Quantum dynamics calculations on a highly accurate potential energy surface are in good agreement with the experimental results and reveal that the reaction occurs via likely a direct abstraction mechanism, not via long-lived reactive resonances.
Co-reporter:Xiuming Zhao and Maodu Chen
RSC Advances 2014 vol. 4(Issue 108) pp:63596-63602
Publication Date(Web):18 Nov 2014
DOI:10.1039/C4RA10141C
The Raman and absorption spectra of a Ag2–PATP–Au2 junction adsorbed on graphene and boron-doped graphene were investigated by using density functional theory (DFT) and time-dependent DFT methods. The interactions between the graphene and junction result in charge transfer (CT) from the graphene to the junction due to their different work functions. This CT leads to charge redistribution on the junction, and then the changes of static polarizabilities, which directly influence the enhancement of normal Raman spectra. The absorption spectra show that the graphene and boron-doped graphene induce some CT excited states in the visible and infrared regions. When the energy of incident light is close to the energy of these CT excited states, these electronic transitions will be excited, which leads to the enhancement of pre-resonant Raman scattering (pre-RRS) spectra. In pre-RRS spectra, the B-doped model has stronger Raman intensities, since it produces more CT excited states with intense oscillator strength near the incident light than the graphene model. The non-totally symmetric modes (b2) are strongly enhanced as well as the totally symmetric modes (a1), indicating the contribution of Herzberg–Teller (HT) scattering. The charge difference densities (CDDs) method was employed to directly visualize the CT from the graphene sheet to the molecule.
Co-reporter:Huixing Li, Yuanzuo Li and Maodu Chen
RSC Advances 2014 vol. 4(Issue 101) pp:57916-57922
Publication Date(Web):30 Oct 2014
DOI:10.1039/C4RA10896E
A group of sensitizers P1–P7, as well as the reference molecule XS54, were theoretically designed and investigated, and the properties of photo-induced charge transfer were characterized by density functional theory (DFT) and time-dependent density functional theory (TD-DFT). Research shows that P5 and P6 possess a broad spectral response, and exhibit strong electron migration ability along the conjugated bridge. The benzothiadiazole (BTD) unit being placed near the accepter unit can effectively extend the spectral range and improve the electron delocalization. Our theoretical design promotes the deeper understanding of dye-sensitized solar cells that absorb over a broad visible region.
Co-reporter:Jiuchuang Yuan, Dahai Cheng and Maodu Chen
RSC Advances 2014 vol. 4(Issue 68) pp:36189-36195
Publication Date(Web):04 Aug 2014
DOI:10.1039/C4RA06297C
Time-dependent wave packet (TDWP) and quasiclassical trajectory (QCT) calculations are carried out for the Au (2S) + HD (X1∑+g) reaction. The reaction probabilities, integral cross sections (ICSs) and differential cross sections (DCSs) of the two reactive channels (AuH + D/AuD + H) are calculated. The ICS results indicate that the dominant reactive channel changes from AuD + H to AuH + D with the collision energy increasing. As is known from the distributions of the complex lifetime, this change mainly results from the difference between the direct reactive mechanisms in the two channels. With collision energy increasing, the growth of direct reactive collisions in AuH + D channel is much larger than that in the AuD + H channel. This is explained by the fact that, in the direct reactive mechanism with a small impact parameter, the D atom with a larger mass can get away easily from the potential well, and the H atom is likely to be captured by the Au atom. To compare vector properties of the two reactive channels, a study of the stereodynamics for the title reaction is also carried out by the QCT method. Due to more direct reactive collisions, product rotational polarization of the AuH + D channel is more obvious than that of the AuD + H channel.
Co-reporter:Dahai Cheng, Jiuchuang Yuan, and Maodu Chen
The Journal of Physical Chemistry A 2014 Volume 118(Issue 1) pp:55-61
Publication Date(Web):December 12, 2013
DOI:10.1021/jp410868v
Time-dependent wave packet (TDWP) and quasiclassical trajectory (QCT) calculations have been carried out for the reaction S(3P) + HD(X1Σg+) at the lowest 13A″ state with both rotational and vibrational excitations of reactant HD. The calculated integral cross sections from QCT agree fairly well with the TDWP calculations. The reaction probability results from TDWP show that the reaction displays a strong tendency to the SD channel. When the reactant HD is vibrationally excited, both channels are promoted apparently. The vibration of the HD bond tends to reduce the difference of reactivity between the two channels. The detailed state-to-state differential cross sections (DCSs) are calculated. These distributions show some significant characters of the barrier-type reactions. At the same time, the scattering width of product SD has a certain relationship with its rotation excitation. For the vector properties, P(θr), P(ϕr), and P(θr,ϕr) distributions are calculated by QCT, and the increased collision energy weakens the rotational polarization of the SD molecule.
Co-reporter:Huixing Li, Yuanzuo Li and Maodu Chen
RSC Advances 2013 vol. 3(Issue 30) pp:12133-12139
Publication Date(Web):01 May 2013
DOI:10.1039/C3RA41816B
The absorption spectra and excited states of four triphenylamine-based organic dye molecules (TC1, TC2, TC3 and TC4) for dye sensitized solar cells (DSSC) have been theoretically investigated with density function theory (DFT) and time-dependent density function theory (TDDFT). In order to simulate the optical properties of the real DSSCs, sensitizer/titanium dioxide cluster interface systems TC4-(TiO2)n (n = 1–8) are also modeled. It is found that different adsorption sites and sizes of the clusters will exert critical effects on the molecular orbital energy levels of the complexes, and ultimately on their light-harvesting capabilities. In all the tested sensitizer/titanium dioxide cluster interface systems, the binding of the organic dye molecule to the 3-fold-coordinated titanium of the cluster (TiO2)6 could most benefit the widening of the spectrum response region and the interactions between the sensitizer and the cluster. From the intuitive physical pictures given by the three-dimensional (3D) real-space analysis methods of transition and charge difference densities, one reveals the photo-induced charge transfer microcosmic process of the sampled organic sensitizer/titanium dioxide cluster system TC4-(TiO2)6.
Co-reporter:Huixing Li
Journal of Molecular Modeling 2013 Volume 19( Issue 12) pp:5317-5325
Publication Date(Web):2013 December
DOI:10.1007/s00894-013-2024-4
The electronic structures of three D-A-π-A indoline dyes (WS-2, WS-6, and WS-11) used in dye-sensitized solar cells (DSSCs) were studied by performing quantum chemistry calculations. The coplanarity of the A-π-A segment and distinct noncoplanarity of the indoline donor part of each dye were confirmed by checking the calculated geometric parameters. The relationships between molecular modifications and the optical properties of the dyes were derived in terms of the partial density of states, absorption spectrum, frontier molecular orbital, and excited-state charge transfer. 3D real-space analysis of the transition density (TD) and charge difference density (CDD) was also performed to further investigate the excited-state features of the molecular systems, as they provide visualized physical pictures of the charge separation and transfer. It was found that modifying the alkyl chain of the bridge unit near the acceptor unit is an efficient way to decrease dye aggregation and improve DSSC efficiency. Inserting a hexylthiophene group next to the donor unit leads to a complicated molecular structure and a decrease in the charge-transfer ability of the system, which has an unfavorable impact on DSSC performance.
Co-reporter:Xiaohong Zhao, Maodu Chen
Chemical Physics Letters 2011 Volume 512(1–3) pp:35-39
Publication Date(Web):16 August 2011
DOI:10.1016/j.cplett.2011.07.003
Abstract
The singlet and triplet excited state as well as the influence of an iodine on the steady-spectra of 10-hydroxybenzo[h]quinoline (HBQ) and 7,9-diiodo-10-hydroxybenzo[h]quinoline (DIHBQ) were investigated using theoretical approach. For the HBQ and DIHBQ, only enol conformation exists in the singlet excited state, whereas there are two hydrogen bonded conformations in the triplet excited state: enol- and keto-tautomer conformation. The enol phosphorescence peak of HBQ which has not been detected in experiments is at 669 nm. The phosphorescence spectrum of DIHBQ (λmax = 692 nm) may be the enol phosphorescence, which was assigned as keto-tautomer phosphorescence in the previous study.
Co-reporter:Shasha Liu, Yuanzuo Li, Xiuming Zhao, Xiaoxia Liu, Maodu Chen
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2011 Volume 82(Issue 1) pp:205-212
Publication Date(Web):November 2011
DOI:10.1016/j.saa.2011.07.035
We investigate surface-enhanced Raman scattering (SERS) spectra of pyridine–Agn (n = 2–8) complexes by density functional theory (DFT) and time-dependent DFT (TDDFT) methods. In simulated normal Raman scattering (NRS) spectra, profiles of pyridine–Agn (n = 2–8) complexes are analogical with that of isolated pyridine. Nevertheless, calculated pre-SERS spectra are strongly dependent on electronic transition states of new complexes. Wavelengths at 335 nm, 394.8 nm, 316.9 nm and 342.6 nm, which are nearly resonant with pure charge transfer excitation states, are adopted as incident light when simulating pre-SERS spectra for pyridine–Agn (n = 2–8) complexes, respectively. We obtain enhancement factors from 103 to 105 in pre-SERS spectra compared with corresponding NRS spectra. The obvious increase in Raman intensities mainly result from charge transfer resonance Raman enhancement. A charge difference densities (CDDs) methodology is adopted in describing chemical enhancement mechanism. This methodology aims at visualizing charge transfer from Agn (n = 2–8) clusters to pyridine on resonant electronic transition, which is one of the most direct evidences for chemical enhancement mechanism.Graphical abstractWe investigate the surface-enhanced Raman scattering spectra of pyridine–Agn (n = 2–8) complexes by density functional theory and time-dependent DFT methods. Wavelength at 335 nm, 394.8 nm, 316.6 nm and 346.2 nm, which are nearly resonant with the pure charge transfer excitation states, are adopted as incident light when simulating the pre-SERS spectra for pyridine–Agn (n = 2–8) complexes, respectively. We obtain the enhancement factors about 103 to 105 in pre-SERS spectra. The obvious increase in Raman intensities mainly result from charge transfer resonance Raman enhancement.Highlights► The enhancement factors are from 103 to 105 in pre-SERS spectra. ► The obvious enhancements result from charge transfer resonance Raman enhancement. ► Charge difference densities methodology is used in describing chemical enhancement mechanism.
Co-reporter:Xiaohong Zhao, Yufang Liu, Lichuan Zhou, Yuanzuo Li, Maodu Chen
Journal of Luminescence 2010 Volume 130(Issue 8) pp:1431-1436
Publication Date(Web):August 2010
DOI:10.1016/j.jlumin.2010.03.007
The time-dependent density functional theory (TDDFT) method was carried out to investigate the excited state intramolecular proton transfer (ESIPT) process of 3-hydroxy-2-(pyridin-2-yl)-4H-chromen-4-one (1a). 1a has two tautomeric forms: one is 1a(O), which is induced by intramolecular hydrogen bond O–H⋯O=C, and the other one is 1a(N), which is caused by intramolecular hydrogen bond O–H⋯N. From excited state to tautomer excited state coming from ESIPT, the hydroxyl hydrogen breaks away and the dissociated hydrogen adsorbed on pyridinic nitrogen or carbonyl oxygen formed new intramolecular HB and the corresponding bond length and bond angle varied greatly. In comparison, a similar process of proton transfer for 1a(N)H+ protonated 1a(N) from ground state to excited state was obtained. This detailed proton transfer mechanism was provided by molecular orbitals analysis and it may be applied to molecular switch and organic Lewis acid/base. We investigated the excited state proton transfer mechanism of the four molecules through the theoretical method for the first time and gave unambiguous geometry of excited state.
Co-reporter:Wenqin Zhang, Yuanzuo Li, Xuesong Xu, Maodu Chen
Chemical Physics 2010 Volume 367(2–3) pp:115-119
Publication Date(Web):8 February 2010
DOI:10.1016/j.chemphys.2009.11.006
Abstract
The isotope effect on integral cross section and product rotational polarization has been investigated for title reactions D+ + H2 and H+ + D2 by quasiclassical trajectory method. The calculated results show the isotope effect on the integral cross section is weak at low collision energy but remarkable at high collision energy. However, the rotational polarization of the product HD molecule is sensitive to the mass factor at low collision energy of 0.524 eV. The initial vibrational excitation plays a more important role in the anisotropic distribution of the product angular momentum vector for the reaction D+ + H2 than that for the reaction H+ + D2.
Co-reporter:Xiaohong Zhao, Shasha Liu, Yuanzuo Li, Maodu Chen
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2010 Volume 75(Issue 2) pp:794-798
Publication Date(Web):February 2010
DOI:10.1016/j.saa.2009.11.057
The chemical mechanism of Normal Raman Scattering (NRS) and pre-surface enhanced Raman scattering (pre-SERS) spectra for Pyrazine–Ag2 complex, Ag2–Pyrazine–Ag2 junction and Ag2–Pyrazine–Au2 junction were investigated with density functional theory (DFT) and charge difference densities (CDDs) for the first time. The NRS intensities of the above three structures enhanced obviously relative to isolated Pyrazine and the enhancement mechanism was confirmed to be static chemical enhancement. The pre-SERS intensities of the above three structures enhanced evidently compared to corresponding NRS intensities, and the enhancement mechanism was confirmed to charge transfer (CT) resonance Raman enhancement. The largest enhanced orders of NRS and pre-SERS intensities among the three structures were up to 103 and 105, respectively. Compared the intensity of pre-SERS with corresponding intensity of NRS spectra, the enhancement effect of Pyrazine–Ag2 complex was larger than the others. Intramolecular and intermolecular CT on resonant electronic transition were described by CDDs.
Co-reporter:Yuanzuo Li, Huixing Li, Xiuming Zhao and Maodu Chen
The Journal of Physical Chemistry A 2010 Volume 114(Issue 26) pp:6972-6977
Publication Date(Web):June 16, 2010
DOI:10.1021/jp103204q
The absorption spectra of dianionic tetrocyanoethylene and dicationic tetrathiafulvalene dimers have been studied theoretically with the time-dependent density functional theory and the recently proposed Coulomb-attenuated model. The nature of the excited states was further explored by means of the two-dimensional (2D) site (transition density matrix) and three-dimensional (3D) cube (transition density and charge difference density) representations. By use of the 3D transition density and charge difference density, we visualized the orientation of transition dipole moment and also explained charge-transfer characteristics occurring in the dianionic/dicationic π-dimers system. It is found that for the dianionic/dicationic π-dimers system there exist two kinds of charge-transfer patterns for the mainly excited states, the intermolecular charge transfer and the mixture of intramolecular charge transfer coupled with intermolecular charge transfer. Meanwhile, the coupling effect of excition and the oscillation of electron−hole pairs between the monomers have been revealed with 2D site representation of transition density matrix, which also indicates the electron−hole coherence upon photon excitation.
Co-reporter:Xiaohong Zhao and Maodu Chen
The Journal of Physical Chemistry A 2010 Volume 114(Issue 29) pp:7786-7790
Publication Date(Web):June 30, 2010
DOI:10.1021/jp101867u
Excited state charge transfer coupled excited state double proton transfer (ESCT/ESDPT) reaction in methanol (MeOH) for 3-cyano-7-azaindole(3-CNAI), 5-cyano-7-azaindole(5-CNAI), and 3,5-dicyano-7-azaindole(3,5-CNAI) were investigated using time-dependent density functional theory (TDDFT) method for the first time. The intermolecular hydrogen bonds of 3-CNAI-MeOH, 5-CNAI-MeOH, and 3,5-CNAI-MeOH complexes are demonstrated to be strengthened in the excited state and weakened in tautomer excited state, which indicates that reverse proton transfer reaction is not easy to take place. Due to the formation of intermolecular hydrogen bond, the absorption and excited state fluorescence spectra of the above three complexes are red-shifted in comparison with those of isolated molecules. The tautomer excited state fluorescence spectra that are induced by ESDPT reaction are also red-shifted relative to the excited state fluorescence for the above complexes. In addition, the sites where cyano group absorbed on 7-azaindole induces a large discrepancy of electron density distribution in excited state. Frontier molecular orbitals reflect that HOMO and LUMO orbitals of proton transfer PT-3-CNAI-MeOH, PT-5-CNAI-MeOH, and PT-3,5-CNAI-MeOH complexes are different with HOMO and LUMO orbitals of 3-CNAI-MeOH, 5-CNAI-MeOH, and 3,5-CNAI-MeOH complexes, respectively.
Co-reporter:Jian-Min Duan, Xu-Feng Li, Li Yao, Shi Pan, Mao-Du Chen
Optics Communications 2009 Volume 282(Issue 19) pp:4005-4008
Publication Date(Web):1 October 2009
DOI:10.1016/j.optcom.2009.06.050
Local field surface plasmon excitation of pair arrays of silver nanospheres was studied using three-dimensional finite-difference time-domain method. The near-field enhancement was associated with the radius of nanosphere and the incident wavelength, the highest of which always appeared in the penultimate gaps, regardless of the number of the pairs. The surface plasmon resonance could be controlled and tuned by radius of nanosphere and incident wavelength.
Co-reporter:Wenqin Zhang, Shulin Cong, Cuihua Zhang, Xuesong Xu and Maodu Chen
The Journal of Physical Chemistry A 2009 Volume 113(Issue 16) pp:4192-4197
Publication Date(Web):March 16, 2009
DOI:10.1021/jp8105716
Theoretical studies of the dynamics of the abstraction reaction, H′ + HBr (v=0,j=0) → H′H + Br, have been performed with quasiclassical trajectory method (QCT) on a new ab initio potential energy surface (Y. Kurosaki and T. Takayanagi, private communication). The calculated QCT cross sections are in good agreement with earlier quantum wave packet results over most of the collision energy range from 0.1 to 2.6 eV, and the state-resolved rotational distributions of the product H′H molecule are quantitatively consistent with the experimental results. Comparisons of the QCT-calculated rotational-state-resolved cross sections on different potential energy surfaces show that the characteristics of the potential energy surface in the region far away from the minimum energy path have a large influence on the title abstraction reaction dynamics, and the indirect reactions that do not follow the minimum energy path have little influence on the differential cross sections (DCS). The DCSs are mainly governed by the direct reactions that do follow the minimum energy path, at both low and high collision energies. The degree of the rotational alignment of the product H′H molecule is strong at high collision energies, which means that the influence of the indirect reactions on the product rotational alignment is negligible, whereas the distribution of P(ϕr) is sensitive to the indirect reactions at high collision energies. With increasing collision energy, the polarization of the product rotational angular momentum decreases and the molecular rotation of the product prefers an in-plane reaction mechanism rather than the out-of-plane mechanism.
Co-reporter:Shasha Liu, Xiaohong Zhao, Yuanzuo Li, Maodu Chen, Mengtao Sun
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2009 Volume 73(Issue 2) pp:382-387
Publication Date(Web):15 July 2009
DOI:10.1016/j.saa.2009.02.036
Density functional theory (DFT) and time-dependent DFT (TDDFT) methods have been used to investigate the adsorption site effect of Raman scattering for neutral and charged pyridine–Ag4 complexes. The calculated results show that the SERS spectra are strongly dependent on adsorption site and the configuration of new complexes. The normal Raman spectra of neutral and charged pyridine–Ag4 complexes are similar with that of isolated pyridine but with an enhancement factor below 10 times. This enhancement is ascribed to ground state chemical enhancement. The pre-surface-enhanced Raman scattering (SERS) spectra were calculated at 1256 nm, 769 nm and 744.3 nm, which are nearly resonant with the charge transfer excited states S2 for neutral and charged pyridine–Ag4 complexes, respectively. We obtain the enhancement factor about 104 to 105 in pre-SERS spectra which is mainly caused by charge transfer resonance Raman enhancement. The three-dimensional cube representation is also applied to describe the photoinduced CT, which are considered as direct evidence of chemical enhancement, between pyridine and two isomers of Ag4 clusters.
Co-reporter:Yuanzuo Li, Shasha Liu, Maodu Chen, Fengcai Ma
Journal of Photochemistry and Photobiology A: Chemistry 2009 Volume 205(2–3) pp:139-144
Publication Date(Web):25 June 2009
DOI:10.1016/j.jphotochem.2009.04.019
Photoinduced charge transfer (CT) and excited state properties in the mixed coaggregates of 1,3,5-triphenyl-2-pyrazoline (TPP) and 1,4-dicyanonaphthalene (DCN) are investigated theoretically, using time-dependent density functional theory (TD-DFT) as well as the two-dimensional (2D) site (transition density matrix) and three-dimensional (3D) cube (transition density and charge difference density) representations. The calculated results indicate that a strong absorption band stems from the S0 → S4 transition. There are electron–hole coherences between TPP and DCN monomers, which are shown by 2D site representation. Direct visual evidence revealed by 3D cube representations indicates that photoinduced CT mechanism for the mixed coaggregates of TPP and DCN is the mixture of intermolecular and intramolecular CT in the vertical absorption. Some phenyl group of TPP monomer not only serves as the electronic donor in the intramolecular CT, but also in the intermolecular CT process.
Co-reporter:Zhi-hong ZHANG, Mao-du CHEN, Shu-lin CONG
Chemical Research in Chinese Universities 2008 Volume 24(Issue 2) pp:223-225
Publication Date(Web):March 2008
DOI:10.1016/S1005-9040(08)60046-X
Abstract
The Ca+CH3I°CaI+CH3 reaction system has been studied with the quasi-classical trajectory method on the extended Lond-Eyring-Polanyi-Sato(LEPS) potential energy surface. At collision energy Ecol = 10.78 kJ/mol, the calculated results show that the Cal vibrational population peaks are located at v=2. The calculated cross section decreases slowly with the collision energy increasing. The angle product distributions tend toward backward scattering. The calculated < P2(J'·K)> values deviate slightly from -0.5 and decrease with increasing collision energy. The Quasiclassical trajectory calculation(QCT) results are in reasonable agreement with experimental data. Moreover, the dynamics of the reaction has been discussed.
Co-reporter:Yuanzuo Li, Shasha Liu, Xiaohong Zhao, Maodu Chen, Fengcai Ma
Journal of Molecular Structure: THEOCHEM 2008 Volume 867(1–3) pp:10-16
Publication Date(Web):30 October 2008
DOI:10.1016/j.theochem.2008.07.028
Two novel metal-free organic dyes (JK-1, JK-2), composed of bis-dimethylfluoreneaniline moiety as the electron donor and cyanoacrylic acid moiety as the electron acceptor, are theoretically investigated with quantum chemical methods. The energies and densities of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), as well as the binding energies, are compared between the two dyes. The HOMO → LUMO electronic transition describes their lowest singlet excited states. The orientation and strength of transition dipole moment are revealed visually with transition density. We also show the orientation and results of the intramolecular charge transfer with charge difference density. Transition density matrices provide information about the electron–hole coherence and excitation delocalization. Theoretical results reveal that exciton size and transition dipole moment increase with the expansion of the π conjugation and the charge transfer occurs from donor unit across the π bridge to the acceptor unit.
Co-reporter:Shufen Wang, Jiuchuang Yuan, Huixing Li and Maodu Chen
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 30) pp:NaN19880-19880
Publication Date(Web):2017/05/30
DOI:10.1039/C7CP02153D
In order to study the dynamics of the reaction H(2S) + NaH(X1Σ+) → Na(2S) + H2(X1Σg+), a new potential energy surface (PES) for the ground state of the NaH2 system is constructed based on 35730 ab initio energy points. Using basis sets of quadruple zeta quality, multireference configuration interaction calculations with Davidson correction were carried out to obtain the ab initio energy points. The neural network method is used to fit the PES, and the root mean square error is very small (0.00639 eV). The bond lengths, dissociation energies, zero-point energies and spectroscopic constants of H2(X1Σg+) and NaH(X1Σ+) obtained on the new NaH2 PES are in good agreement with the experiment data. On the new PES, the reactant coordinate-based time-dependent wave packet method is applied to study the reaction dynamics of H(2S) + NaH(X1Σ+) → Na(2S) + H2(X1Σg+), and the reaction probabilities, integral cross-sections (ICSs) and differential cross-sections (DCSs) are obtained. There is no threshold in the reaction due to the absence of an energy barrier on the minimum energy path. When the collision energy increases, the ICSs decrease from a high value at low collision energy. The DCS results show that the angular distribution of the product molecules tends to the forward direction. Compared with the LiH2 system, the NaH2 system has a larger mass and the PES has a larger well at the H–NaH configuration, which leads to a higher ICS value in the H(2S) + NaH(X1Σ+) → Na(2S) + H2(X1Σg+) reaction. Because the H(2S) + NaH(X1Σ+) → Na(2S) + H2(X1Σg+) reaction releases more energy, the product molecules can be excited to a higher vibrational state.
Co-reporter:Jiuchuang Yuan, Di He and Maodu Chen
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 17) pp:NaN11739-11739
Publication Date(Web):2015/03/31
DOI:10.1039/C4CP05352D
A new global potential energy surface (PES) is obtained for the ground electronic state of the LiH2 system based on high-level energies. The energy points are calculated at the multireference configuration interaction level with aug-cc-pVXZ (X = Q, 5) basis sets, and these energies are extrapolated to the complete basis set limit. The neural network method and hierarchical construction scheme are applied in the fitting process and the root mean square error of the fitting result is very small (0.004 eV). The dissociation energies and equilibrium distances for LiH(X1Σ+) and H2(X1Σg+) obtained from the new PES are in good agreement with the experimental data. On the new PES, time-dependent wave packet studies for the H(2S) + LiH(X1Σ+) → Li(2S) + H2(X1Σg+) reaction have been carried out. In this reaction, no threshold is found due to the absence of an energy barrier on the minimum energy path. The calculated integral cross sections are high at low collision energy and will decrease with the increase of the collision energy. The product molecule H2 tends to be forward scattering due to direct reactive collisions, which becomes more evident at higher collision energies.