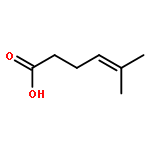
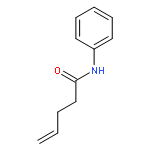
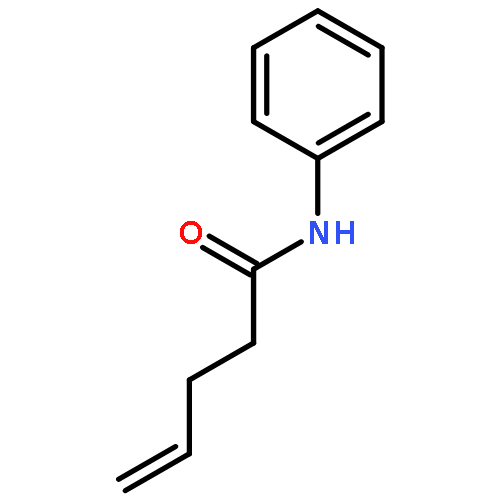
Co-reporter:Andrew J. Musacchio;Saeed G. Naguib;Brendan C. Lainhart;Xin Zhang;Trevor C. Sherwood
Science 2017 Volume 355(Issue 6326) pp:727-730
Publication Date(Web):17 Feb 2017
DOI:10.1126/science.aal3010
Hydroamination gets a light push uphill
Hydroamination of olefins is a broadly useful method for making carbon-nitrogen bonds. However, when both the amine and the olefin have multiple alkyl substituents, the reaction can become energetically unfavorable. Musacchio et al. used the energy in blue light to surmount this obstacle (see the Perspective by Buchanan and Hull). A photo-excited iridium complex oxidized the amine, which in turn bonded efficiently to the olefin, after which a thiophenol cocatalyst shuttled the electron back. The reaction could operate across a wide range of amine and olefin partners.
Science, this issue p. 727; see also p. 690
Co-reporter:Emily C. Gentry and Robert R. Knowles
Accounts of Chemical Research 2016 Volume 49(Issue 8) pp:1546
Publication Date(Web):July 29, 2016
DOI:10.1021/acs.accounts.6b00272
ConspectusRedox events in which an electron and proton are exchanged in a concerted elementary step are commonly referred to as proton-coupled electron transfers (PCETs). PCETs are known to operate in numerous important biological redox processes, as well as recent inorganic technologies for small molecule activation. These studies suggest that PCET catalysis might also function as a general mode of substrate activation in organic synthesis. Over the past three years, our group has worked to advance this hypothesis and to demonstrate the synthetic utility of PCET through the development of novel catalytic radical chemistries. The central aim of these efforts has been to demonstrate the ability of PCET to homolytically activate a wide variety of common organic functional groups that are energetically inaccessible using known molecular H atom transfer catalysts.To do so, we made use of a simple formalism first introduced by Mayer and co-workers that allowed us to predict the thermodynamic capacity of any oxidant/base or reductant/acid pair to formally add or remove H· from a given substrate. With this insight, we were able to rationally select catalyst combinations thermodynamically competent to homolyze the extraordinarily strong E–H σ-bonds found in many common protic functional groups (BDFEs > 100 kcal/mol) or to form unusually weak bonds to hydrogen via the reductive action of common organic π-systems (BDFEs < 35 kcal/mol). These ideas were reduced to practice through the development of new catalyst systems for reductive PCET activations of ketones and oxidative PCET activation of amide N–H bonds to directly furnish reactive ketyl and amidyl radicals, respectively. In both systems, the reaction outcomes were found to be successfully predicted using the effective bond strength formalism, suggesting that these simple thermochemical considerations can provide useful and actionable insights into PCET reaction design.The ability of PCET catalysis to control enantioselectivity in free radical processes has also been established. Specifically, multisite PCET requires the formation of a pre-equilibrium hydrogen bond between the substrate and a proton donor/acceptor prior to charge transfer. We recognized that these H-bond interfaces persist following the PCET event, resulting in the formation of noncovalent complexes of the nascent radical intermediates. When chiral proton donors/acceptors are employed, this association can provide a basis for asymmetric induction in subsequent bond-forming steps. We discuss our efforts to capitalize on this understanding via the development of a catalytic protocol for enantioselective aza-pinacol cyclizations.Lastly, we highlight an alternative PCET mechanism that exploits the ability of redox-active metals to homolytically weaken the bonds in coordinated ligands, enabling nominally strong bonds (BDFEs ∼ 100 kcal) to be abstracted by weak H atom acceptors with concomitant oxidation of the metal center. This “soft homolysis” mechanism enables the generation of metalated intermediates from protic substrates under completely neutral conditions. The first example of this form of catalysis is presented in the context of a catalytic C–N bond forming reaction jointly mediated by bulky titanocene complexes and the stable nitroxyl radical TEMPO.
Co-reporter:Hatice G. Yayla, Huaiju Wang, Kyle T. Tarantino, Hudson S. Orbe, and Robert R. Knowles
Journal of the American Chemical Society 2016 Volume 138(Issue 34) pp:10794-10797
Publication Date(Web):August 12, 2016
DOI:10.1021/jacs.6b06517
We report a new photocatalytic protocol for the redox-neutral isomerization of cyclic alcohols to linear ketones via C–C bond scission. Mechanistic studies demonstrate that key alkoxy radical intermediates in this reaction are generated via the direct homolytic activation of alcohol O–H bonds in an unusual intramolecular PCET process, wherein the electron travels to a proximal radical cation in concert with proton transfer to a weak Brønsted base. Effective bond strength considerations are shown to accurately forecast the feasibility of alkoxy radical generation with a given oxidant/base pair.
Co-reporter:Lucas Q. Nguyen and Robert R. Knowles
ACS Catalysis 2016 Volume 6(Issue 5) pp:2894
Publication Date(Web):March 30, 2016
DOI:10.1021/acscatal.6b00486
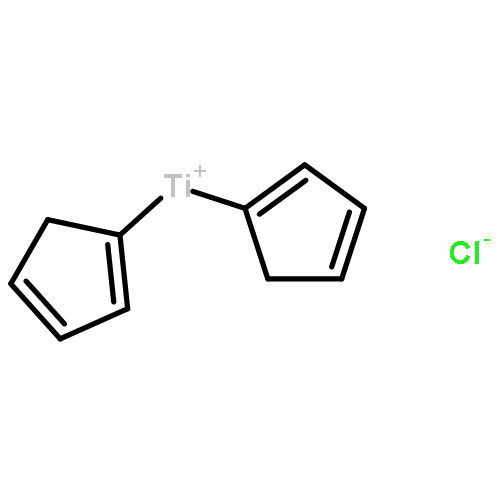
Over the past three years, our group has become interested in the ability of proton-coupled electron transfer (PCET) to facilitate direct homolytic bond activations of common organic functional groups that are challenging substrates for conventional hydrogen atom transfer catalysts. This perspective details our efforts to develop oxidative PCET platforms for activating the strong N–H bonds of amides, providing catalytic access to synthetically useful amidyl radicals. We successfully identified compatible combinations of one-electron oxidants and Brønsted bases that, although unable to activate the amide substrates independently, act concomitantly with the requisite energetics to selectively homolyze the N–H bond via concerted PCET. The resulting amidyls were utilized in the development of new catalytic protocols for alkene carboamination and hydroamidation. We also highlight our efforts to develop a PCET-based bond-weakening protocol for the catalytic conjugate aminations using amide substrates. In this work, coordination to a Ti(III) catalyst significantly decreases the strength of a ligated amide N–H bond, enabling a facile PCET event to occur with the weak H atom acceptor TEMPO. Although this discussion focuses on amide activation, we anticipate that the design parameters presented here are general and should provide a framework for the development of PCET catalyst systems for other challenging homolytic bond activations as well.Keywords: amidyl radicals; bond weakening; C−N bond formation; hydrogen atom transfer; proton-coupled electron transfer
Co-reporter:Hatice G. Yayla, Feng Peng, Ian K. Mangion, Mark McLaughlin, Louis-Charles Campeau, Ian W. Davies, Daniel A. DiRocco and Robert R. Knowles
Chemical Science 2016 vol. 7(Issue 3) pp:2066-2073
Publication Date(Web):2015/12/07
DOI:10.1039/C5SC03350K
Elbasvir is a potent NS5A antagonist for the treatment of chronic hepatitis C. A seemingly trivial indoline oxidation en route to the target compound was complicated by epimerization of a stereogenic hemiaminal center under most standard oxidation conditions. To address this issue, a novel visible light photoredox process for indoline oxidation was developed involving an iridium photosensitizer and environmentally-benign perester oxidant. The reaction was discovered through a high-throughput experimentation campaign and the optimized process was demonstrated on 100 g scale in flow to afford a key intermediate towards the target compound. A battery of kinetic, electrochemical, and spectroscopic studies of this process indicates a radical chain mechanism of dehydrogenation involving selective HAT from the substrate by an alkoxy radicals. Notably, isotope effects were used to validate the chain mechanism when quantum yield data proved ambiguous.
Co-reporter:David C. Miller; Gilbert J. Choi; Hudson S. Orbe
Journal of the American Chemical Society 2015 Volume 137(Issue 42) pp:13492-13495
Publication Date(Web):October 5, 2015
DOI:10.1021/jacs.5b09671
Here we report a ternary catalyst system for the intramolecular hydroamidation of unactivated olefins using simple N-aryl amide derivatives. Amide activation in these reactions occurs via concerted proton-coupled electron transfer (PCET) mediated by an excited state iridium complex and weak phosphate base to furnish a reactive amidyl radical that readily adds to pendant alkenes. A series of H-atom, electron, and proton transfer events with a thiophenol cocatalyst furnish the product and regenerate the active forms of the photocatalyst and base. Mechanistic studies indicate that the amide substrate can be selectively homolyzed via PCET in the presence of the thiophenol, despite a large difference in bond dissociation free energies between these functional groups.
Co-reporter:Kyle T. Tarantino; David C. Miller; Ted A. Callon
Journal of the American Chemical Society 2015 Volume 137(Issue 20) pp:6440-6443
Publication Date(Web):May 6, 2015
DOI:10.1021/jacs.5b03428
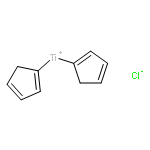
The ability of redox-active metal centers to weaken the bonds in associated ligands is well precedented, but has rarely been utilized as a mechanism of substrate activation in catalysis. Here we describe a catalytic bond-weakening protocol for conjugate amination wherein the strong N–H bonds in N-aryl amides (N–H bond dissociation free energies ∼100 kcal/mol) are destabilized by ∼33 kcal/mol upon by coordination to a reducing titanocene complex, enabling their abstraction by the weak H-atom acceptor TEMPO through a proton-coupled electron transfer process. Significantly, this soft homolysis mechanism provides a method to generate closed-shell, metalated nucleophiles under neutral conditions in the absence of a Brønsted base.
Co-reporter:Gilbert J. Choi
Journal of the American Chemical Society 2015 Volume 137(Issue 29) pp:9226-9229
Publication Date(Web):July 13, 2015
DOI:10.1021/jacs.5b05377
Here we describe a dual catalyst system comprised of an iridium photocatalyst and weak phosphate base that is capable of both selectively homolyzing the N–H bonds of N-arylamides (bond dissociation free energies ∼ 100 kcal/mol) via concerted proton-coupled electron transfer (PCET) and mediating efficient carboamination reactions of the resulting amidyl radicals. This manner of PCET activation, which finds its basis in numerous biological redox processes, enables the formal homolysis of a stronger amide N–H bond in the presence of weaker allylic C–H bonds, a selectivity that is uncommon in conventional molecular H atom acceptors. Moreover, this transformation affords access to a broad range of structurally complex heterocycles from simple amide starting materials. The design, synthetic scope, and mechanistic evaluation of the PCET process are described.
Co-reporter:Robert R. Knowles
ACS Central Science 2015 Volume 1(Issue 5) pp:224
Publication Date(Web):August 6, 2015
DOI:10.1021/acscentsci.5b00257
Co-reporter:Andrew J. Musacchio ; Lucas Q. Nguyen ; G. Hudson Beard
Journal of the American Chemical Society 2014 Volume 136(Issue 35) pp:12217-12220
Publication Date(Web):August 15, 2014
DOI:10.1021/ja5056774
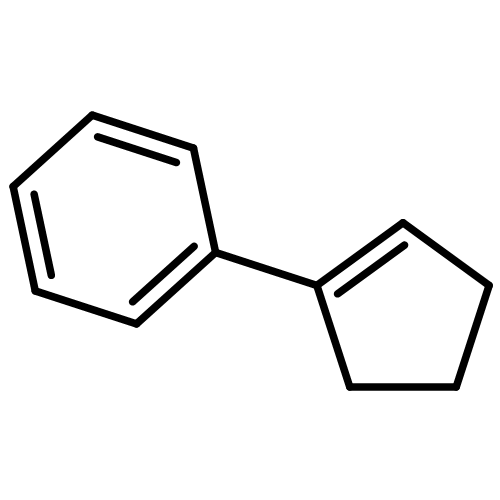
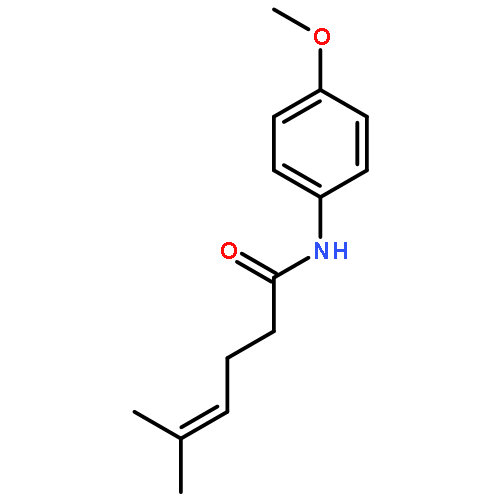
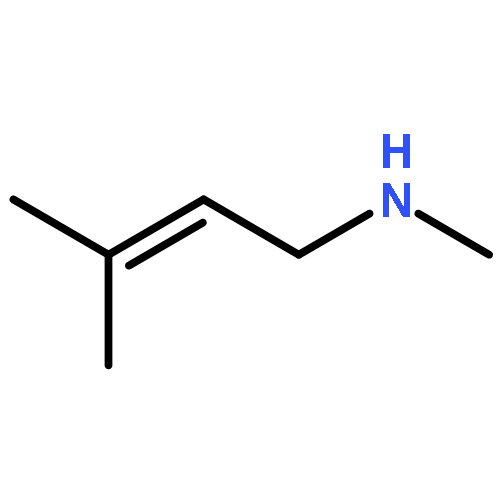
While olefin amination with aminium radical cations is a classical method for C–N bond formation, catalytic variants that utilize simple 2° amine precursors remain largely undeveloped. Herein we report a new visible-light photoredox protocol for the intramolecular anti-Markovnikov hydroamination of aryl olefins that proceeds through catalytically generated aminium radical intermediates. Mechanistic studies are consistent with a process involving amine oxidation via electron transfer, turnover-limiting C–N bond formation, and a second electron transfer step to reduce a carbon-centered radical, rendering the overall process redox-neutral. A range of structurally diverse N-aryl heterocycles can be prepared in good to excellent yields under conditions significantly milder than those required by conventional aminium-based protocols.
Co-reporter:Lydia J. Rono ; Hatice G. Yayla ; David Y. Wang ; Michael F. Armstrong
Journal of the American Chemical Society 2013 Volume 135(Issue 47) pp:17735-17738
Publication Date(Web):November 11, 2013
DOI:10.1021/ja4100595
The first highly enantioselective catalytic protocol for the reductive coupling of ketones and hydrazones is reported. These reactions proceed through neutral ketyl radical intermediates generated via a concerted proton-coupled electron transfer (PCET) event jointly mediated by a chiral phosphoric acid catalyst and the photoredox catalyst Ir(ppy)2(dtbpy)PF6. Remarkably, these neutral ketyl radicals appear to remain H-bonded to the chiral conjugate base of the Brønsted acid during the course of a subsequent C–C bond-forming step, furnishing syn 1,2-amino alcohol derivatives with excellent levels of diastereo- and enantioselectivity. This work provides the first demonstration of the feasibility and potential benefits of concerted PCET activation in asymmetric catalysis.
Co-reporter:Kyle T. Tarantino ; Peng Liu
Journal of the American Chemical Society 2013 Volume 135(Issue 27) pp:10022-10025
Publication Date(Web):June 24, 2013
DOI:10.1021/ja404342j
Concerted proton-coupled electron transfer is a key mechanism of substrate activation in biological redox catalysis. However, its applications in organic synthesis remain largely unexplored. Herein, we report the development of a new catalytic protocol for ketyl-olefin coupling and present evidence to support concerted proton-coupled electron transfer being the operative mechanism of ketyl formation. Notably, reaction outcomes were correctly predicted by a simple thermodynamic formalism relating the oxidation potentials and pKa values of specific Brønsted acid/reductant combinations to their capacity to act jointly as a formal hydrogen atom donor.
Co-reporter:Hatice G. Yayla, Feng Peng, Ian K. Mangion, Mark McLaughlin, Louis-Charles Campeau, Ian W. Davies, Daniel A. DiRocco and Robert R. Knowles
Chemical Science (2010-Present) 2016 - vol. 7(Issue 3) pp:NaN2073-2073
Publication Date(Web):2015/12/07
DOI:10.1039/C5SC03350K
Elbasvir is a potent NS5A antagonist for the treatment of chronic hepatitis C. A seemingly trivial indoline oxidation en route to the target compound was complicated by epimerization of a stereogenic hemiaminal center under most standard oxidation conditions. To address this issue, a novel visible light photoredox process for indoline oxidation was developed involving an iridium photosensitizer and environmentally-benign perester oxidant. The reaction was discovered through a high-throughput experimentation campaign and the optimized process was demonstrated on 100 g scale in flow to afford a key intermediate towards the target compound. A battery of kinetic, electrochemical, and spectroscopic studies of this process indicates a radical chain mechanism of dehydrogenation involving selective HAT from the substrate by an alkoxy radicals. Notably, isotope effects were used to validate the chain mechanism when quantum yield data proved ambiguous.










![1-BUTANONE, 3-[4-(1,1-DIMETHYLETHYL)PHENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/867200/867178-34-5.png)
![1-BUTANONE, 3-[4-(1,1-DIMETHYLETHYL)PHENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/867200/867178-34-5_b.png)





![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis(triphenylsilyl)-, 4-oxide, (11bR)-](/data/chemimg/116500/791616-55-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis(triphenylsilyl)-, 4-oxide, (11bR)-](/data/chemimg/116500/791616-55-2_b.png)

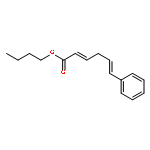
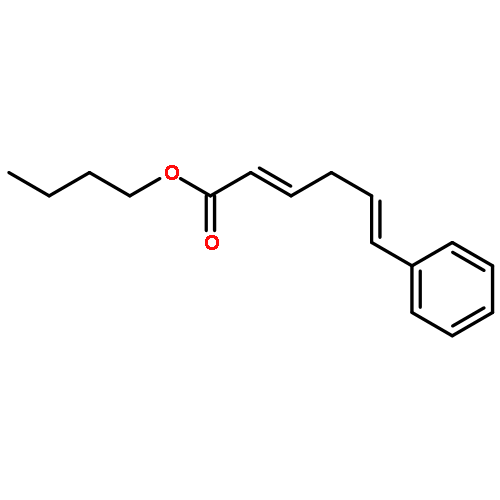
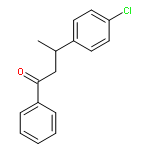
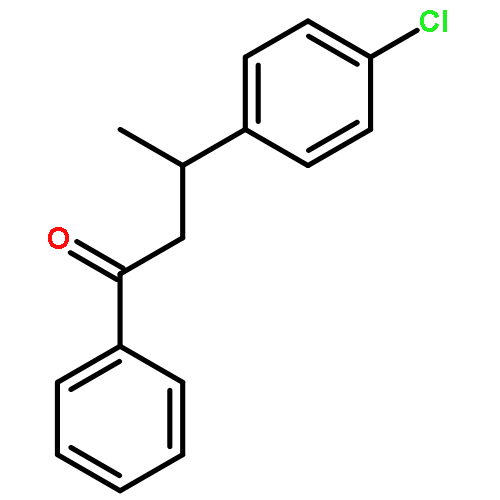
![Ethanone, 1-(4-methoxyphenyl)-2-[(1R,3S)-2,2,3-trimethylcyclopentyl]-](http://img.cochemist.com/ccimg/578100/578006-88-9.png)
![Ethanone, 1-(4-methoxyphenyl)-2-[(1R,3S)-2,2,3-trimethylcyclopentyl]-](http://img.cochemist.com/ccimg/578100/578006-88-9_b.png)
![2,8-diazaspiro[4.5]decan-1-one](http://img.cochemist.com/ccimg/546100/546086-95-7.png)
![2,8-diazaspiro[4.5]decan-1-one](http://img.cochemist.com/ccimg/546100/546086-95-7_b.png)
![Cyclopenta[b]pyrrol-2(1H)-one, hexahydro-1-phenyl-, (3aR,6aR)-rel-](http://img.cochemist.com/ccimg/330000/329950-43-8.png)
![Cyclopenta[b]pyrrol-2(1H)-one, hexahydro-1-phenyl-, (3aR,6aR)-rel-](http://img.cochemist.com/ccimg/330000/329950-43-8_b.png)








![Cyclopropanemethanol, 2-[(phenylmethoxy)methyl]-, trans-](http://img.cochemist.com/ccimg/190100/190004-96-7.png)
![Cyclopropanemethanol, 2-[(phenylmethoxy)methyl]-, trans-](http://img.cochemist.com/ccimg/190100/190004-96-7_b.png)
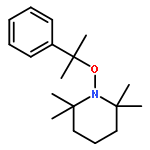
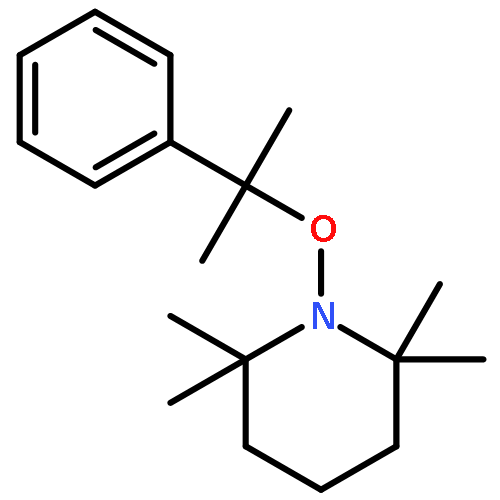
![Piperidine, 1-[1-(4-bromophenyl)ethoxy]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/178700/178625-97-3.png)
![Piperidine, 1-[1-(4-bromophenyl)ethoxy]-2,2,6,6-tetramethyl-](http://img.cochemist.com/ccimg/178700/178625-97-3_b.png)







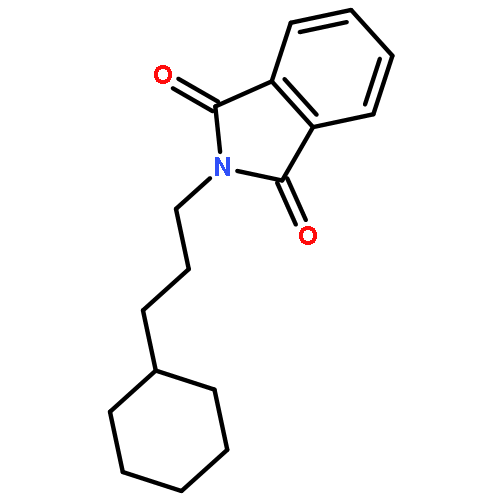
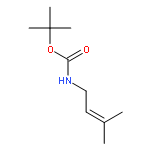
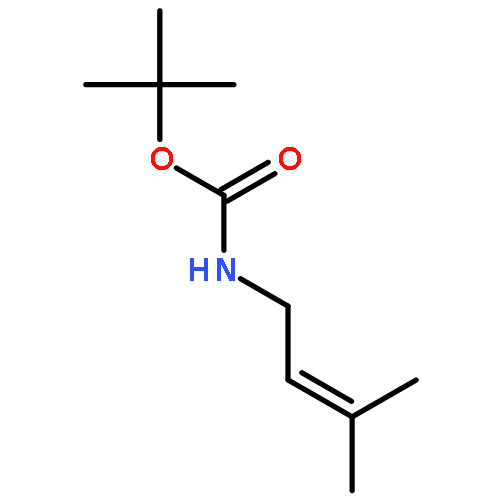


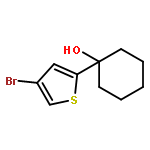



![Benzene, [(1E)-4-bromo-1-methyl-1-butenyl]-](http://img.cochemist.com/ccimg/129000/128902-50-1.png)
![Benzene, [(1E)-4-bromo-1-methyl-1-butenyl]-](http://img.cochemist.com/ccimg/129000/128902-50-1_b.png)
![Benzenemethanamine, N-[2-(2-cyclopenten-1-yl)ethyl]-](http://img.cochemist.com/ccimg/122800/122718-94-9.png)
![Benzenemethanamine, N-[2-(2-cyclopenten-1-yl)ethyl]-](http://img.cochemist.com/ccimg/122800/122718-94-9_b.png)

![Propanedioic acid, [(2E)-3-phenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/119800/119793-72-5.png)
![Propanedioic acid, [(2E)-3-phenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/119800/119793-72-5_b.png)
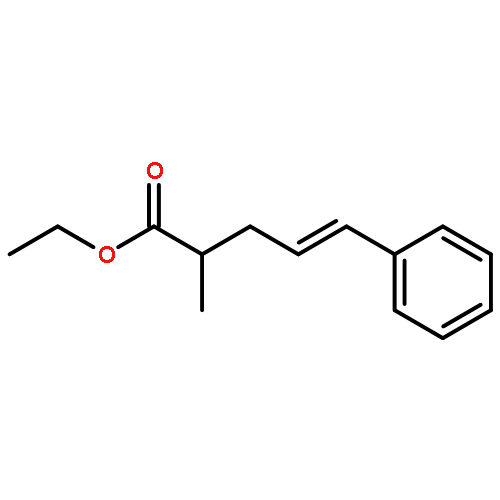
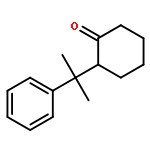
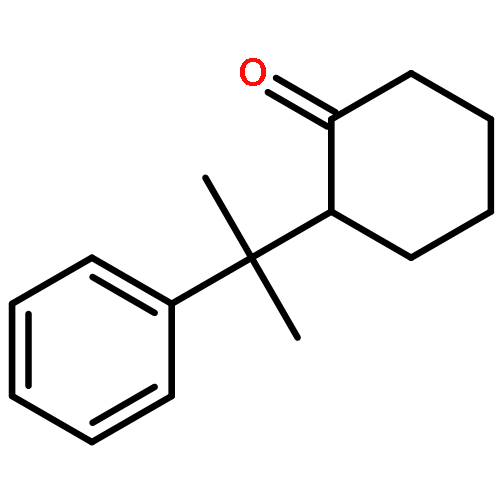
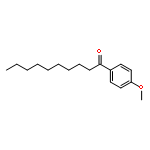
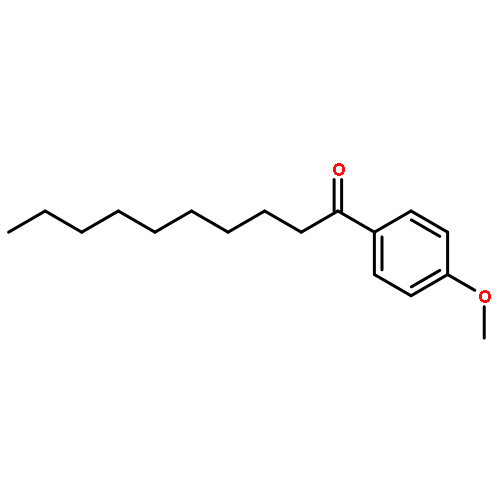




![Ethanol, 2-[[(2E)-3-phenyl-2-propenyl]oxy]-](http://img.cochemist.com/ccimg/99600/99522-43-7.png)
![Ethanol, 2-[[(2E)-3-phenyl-2-propenyl]oxy]-](http://img.cochemist.com/ccimg/99600/99522-43-7_b.png)







![Cyclohexanone, 2-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/84700/84624-35-1.png)
![Cyclohexanone, 2-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/84700/84624-35-1_b.png)
![Piperidine, 1-[2-(4-methoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/83000/82966-18-5.png)
![Piperidine, 1-[2-(4-methoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/83000/82966-18-5_b.png)
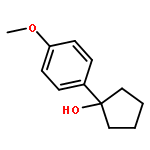
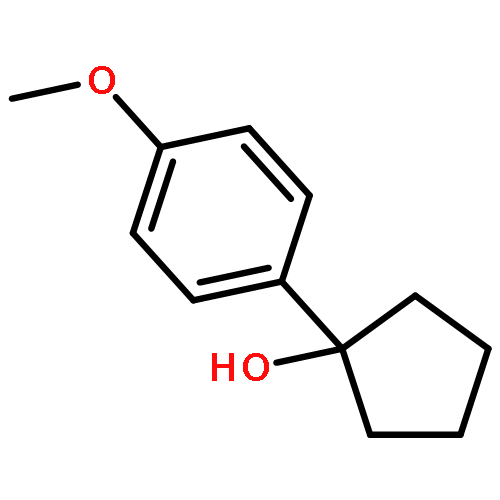
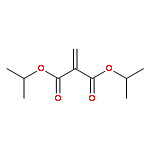
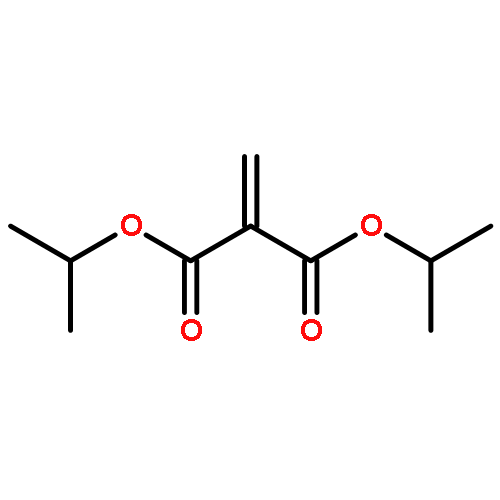


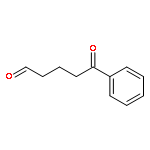
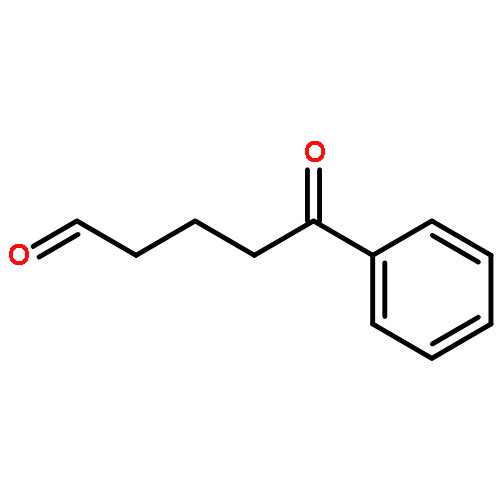
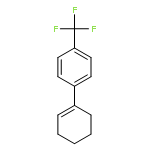
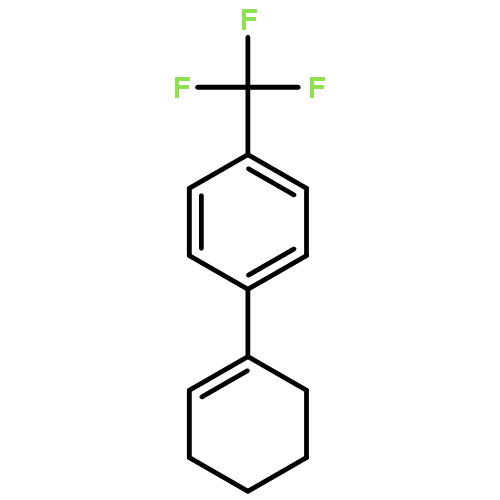





![Acetamide, N-[2-(2-propenyl)phenyl]-](http://img.cochemist.com/ccimg/68300/68267-69-6.png)
![Acetamide, N-[2-(2-propenyl)phenyl]-](http://img.cochemist.com/ccimg/68300/68267-69-6_b.png)


![Ethanone,1-[2-(2-propen-1-yl)phenyl]-](http://img.cochemist.com/ccimg/64700/64664-07-9.png)
![Ethanone,1-[2-(2-propen-1-yl)phenyl]-](http://img.cochemist.com/ccimg/64700/64664-07-9_b.png)





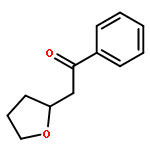
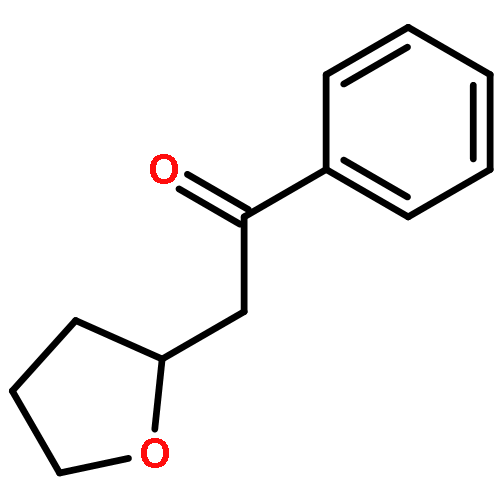
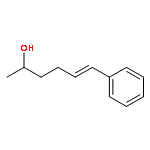
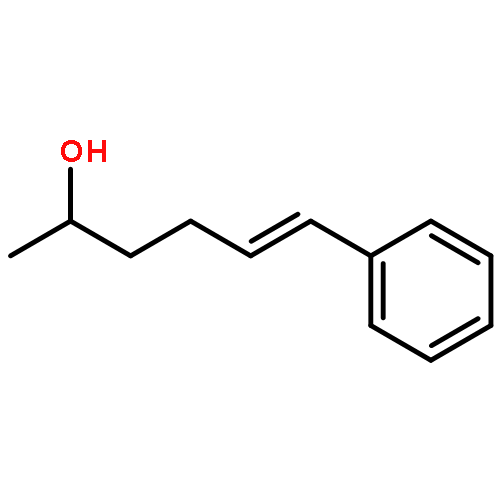

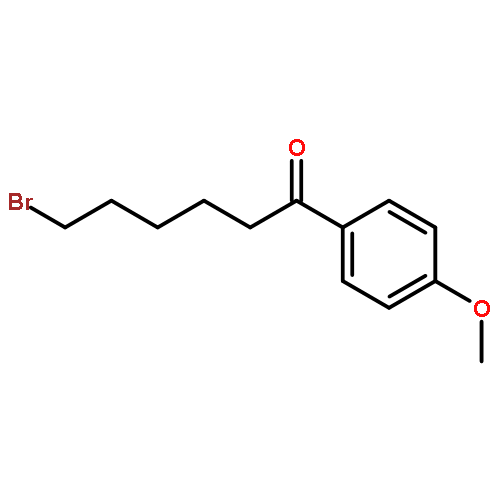
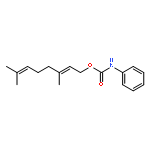

![2-Azatricyclo[3.3.1.1<sup>3,7</sup>]dec-2-yloxidanyl](http://img.cochemist.com/ccimg/57700/57625-08-8.png)
![2-Azatricyclo[3.3.1.1<sup>3,7</sup>]dec-2-yloxidanyl](http://img.cochemist.com/ccimg/57700/57625-08-8_b.png)


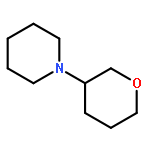

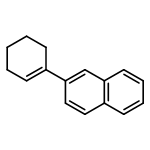
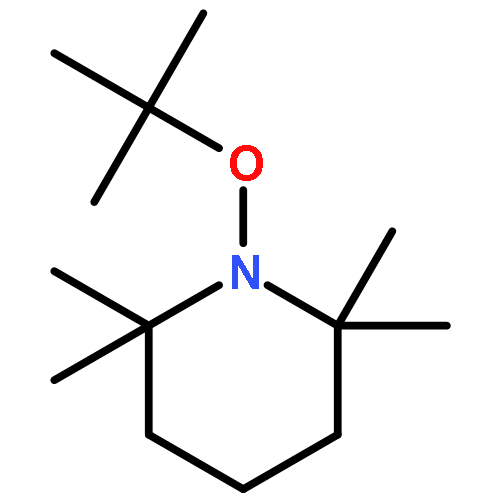

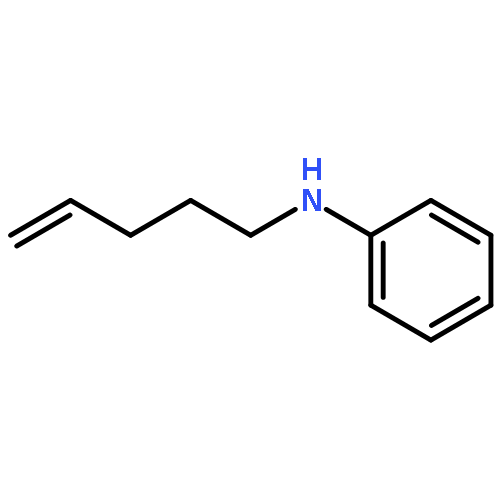
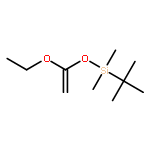
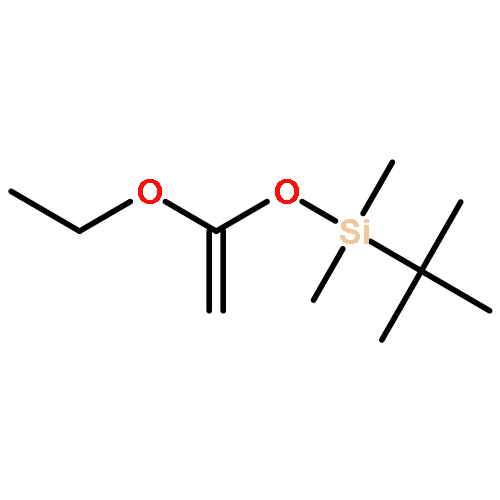
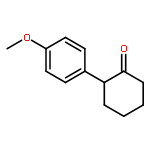

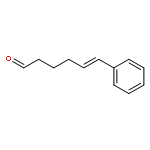
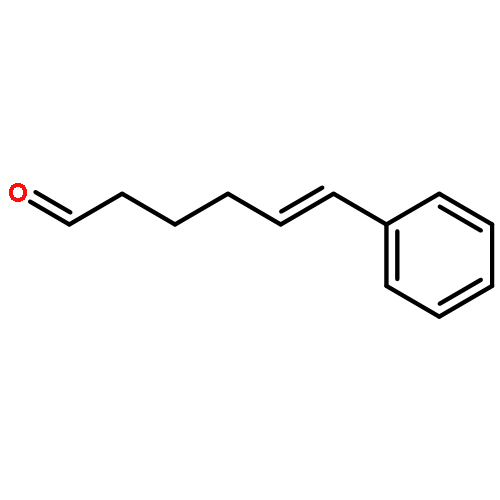
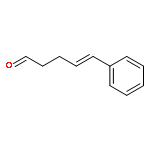
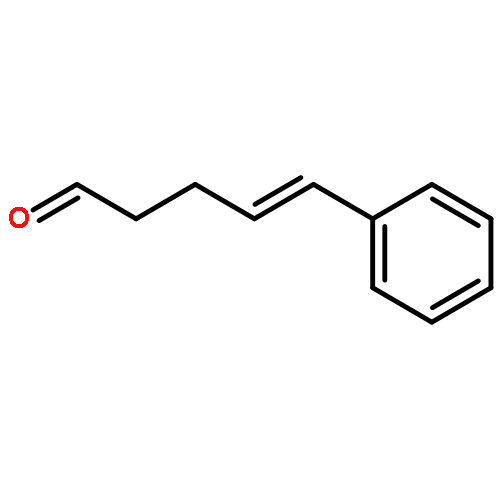
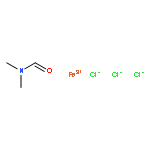
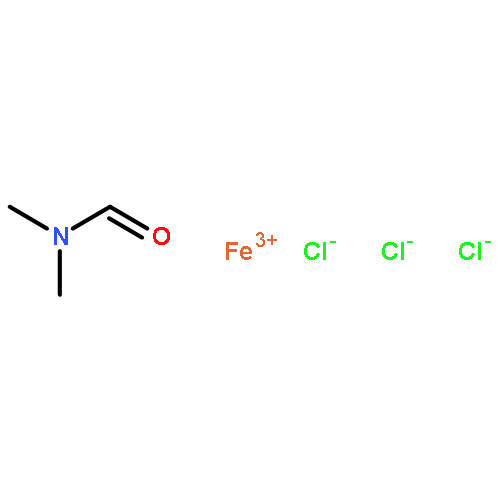








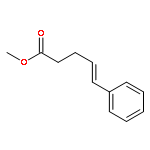






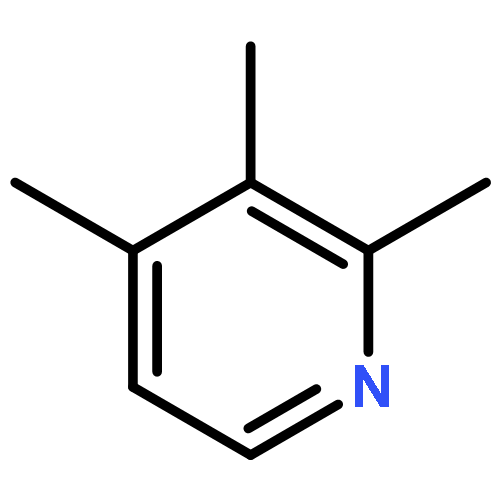
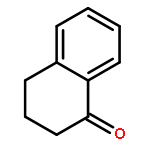
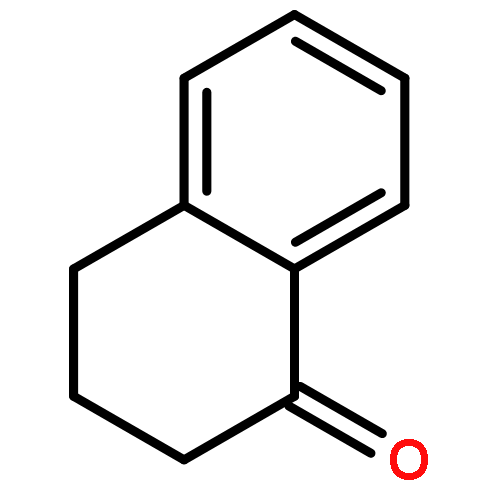
![Ethanone,1-phenyl-2-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/27700/27648-26-6.png)
![Ethanone,1-phenyl-2-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/27700/27648-26-6_b.png)


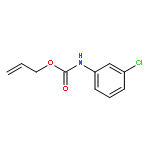
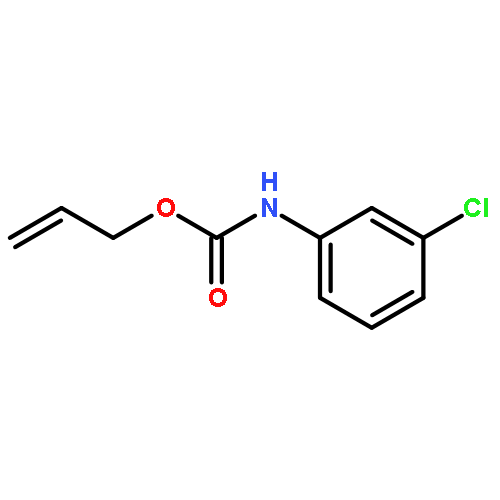
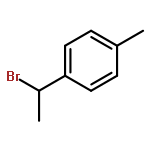



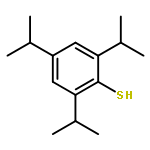

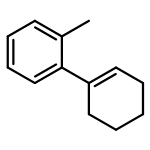



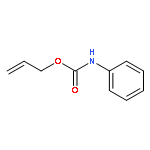

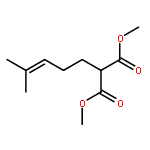
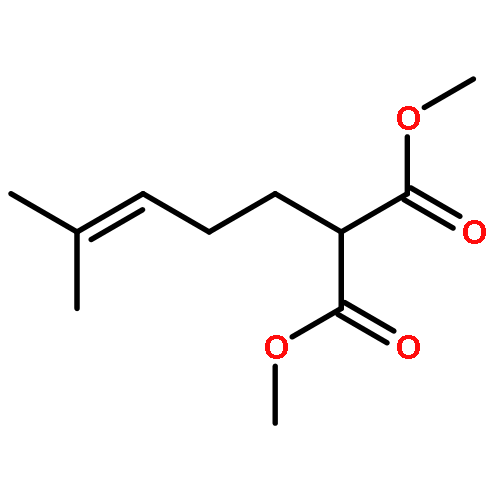
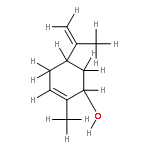




















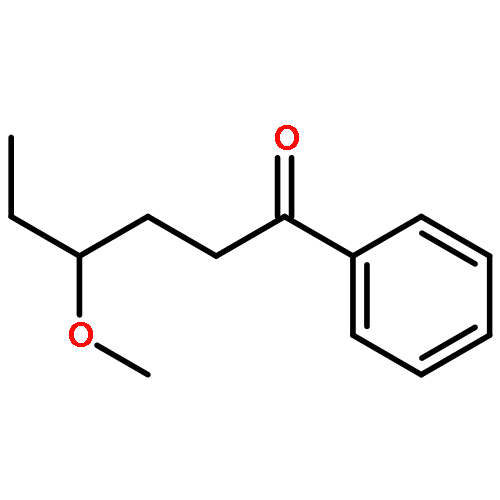




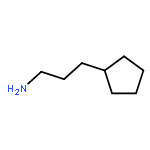
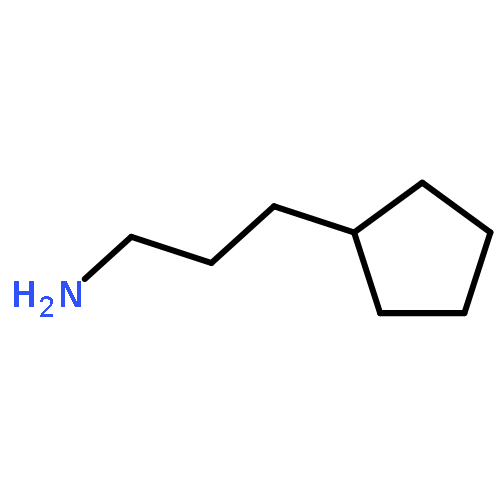

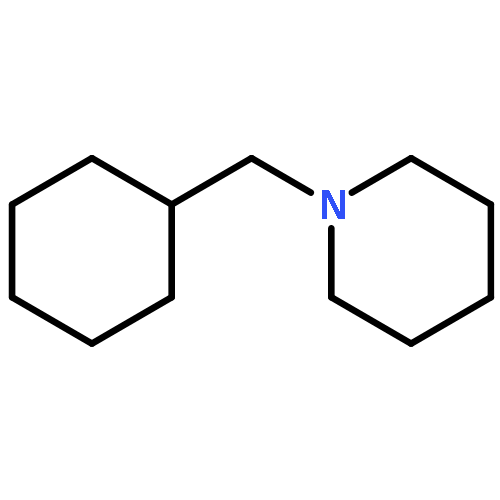
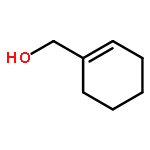
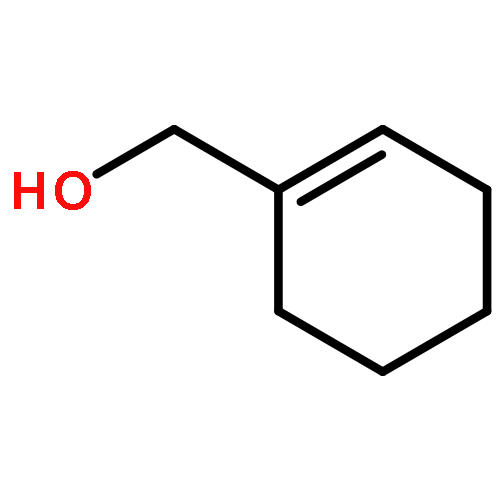
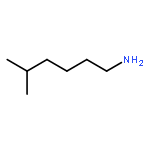



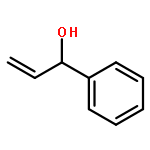
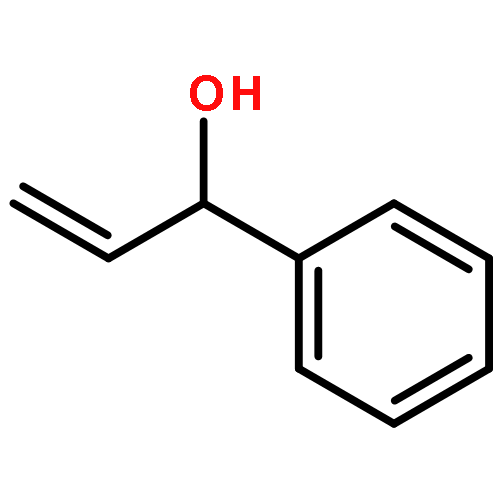




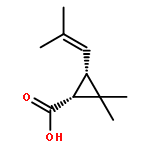
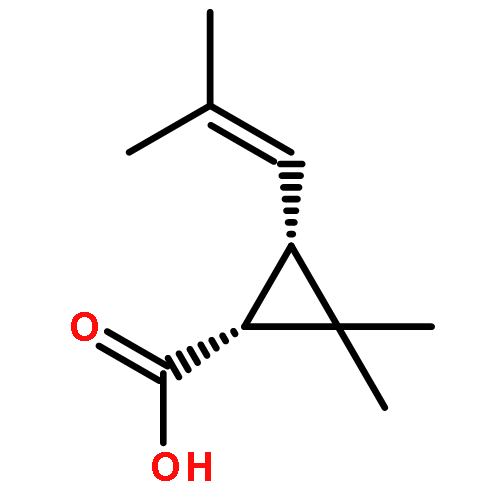



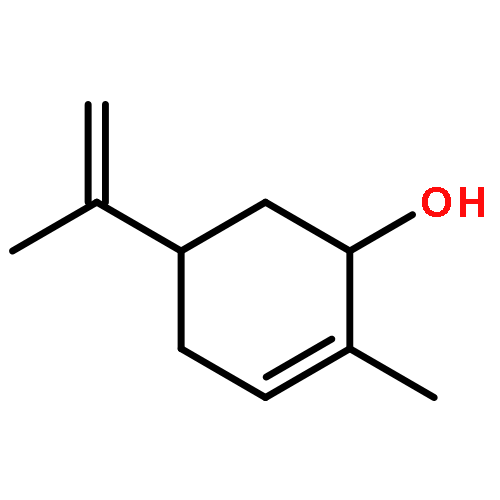



















![1-[(3s,8s,9s,10r,13s,14s,17s)-3-hydroxy-10,13-dimethyl-2,3,6,7,8, 9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-17-yl ]ethanone](http://img.cochemist.com/ccimg/600/566-66-5.png)
![1-[(3s,8s,9s,10r,13s,14s,17s)-3-hydroxy-10,13-dimethyl-2,3,6,7,8, 9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-17-yl ]ethanone](http://img.cochemist.com/ccimg/600/566-66-5_b.png)
![2-hydroxy-7-methylpyrano[3,2-c]pyran-4,5-dione](http://img.cochemist.com/ccimg/500/495-10-3.png)
![2-hydroxy-7-methylpyrano[3,2-c]pyran-4,5-dione](http://img.cochemist.com/ccimg/500/495-10-3_b.png)
















![(1R,2R,4R)-Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/1200/1195-12-6.png)
![(1R,2R,4R)-Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/1200/1195-12-6_b.png)