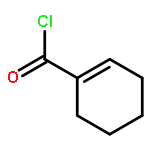
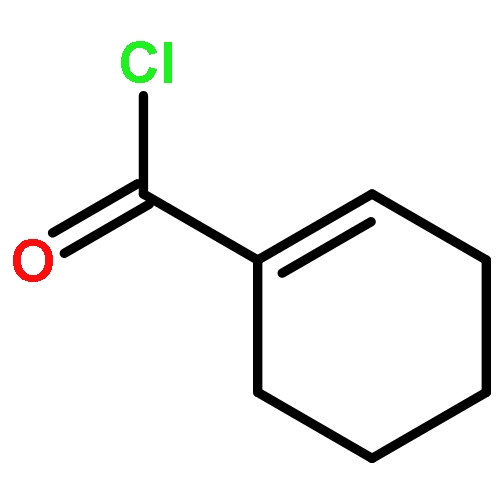
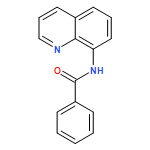
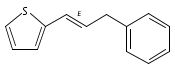
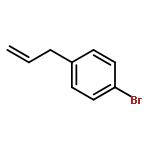
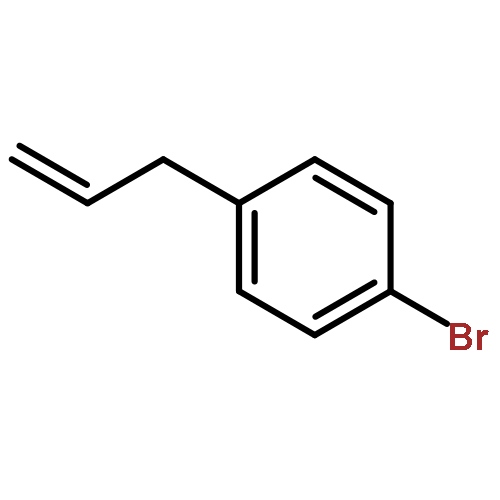
Co-reporter:Takenari Sato, Takumi Yoshida, Hamad H. Al Mamari, Laurean Ilies, and Eiichi Nakamura
Organic Letters October 6, 2017 Volume 19(Issue 19) pp:
Publication Date(Web):September 14, 2017
DOI:10.1021/acs.orglett.7b02778
We report here a manganese-catalyzed C–H methylation reaction of considerable substrate scope, using MeMgBr, a catalytic amount of MnCl2·2LiCl, and an organic dihalide oxidant. The reaction features ambient temperature, low catalyst loading, typically 1%, high catalytic turnover reaching 5.9 × 103, and no need for an extraneous ligand and illustrates a unique catalytic use of simple manganese salts for C–H activation, which so far has relied on catalysis by manganese carbonyls.
Co-reporter:Rui Shang, Laurean Ilies, and Eiichi Nakamura
Chemical Reviews July 12, 2017 Volume 117(Issue 13) pp:9086-9086
Publication Date(Web):April 5, 2017
DOI:10.1021/acs.chemrev.6b00772
Catalytic C–H bond activation, which was an elusive subject of chemical research until the 1990s, has now become a standard synthetic method for the formation of new C–C and C–heteroatom bonds. The synthetic potential of C–H activation was first described for ruthenium catalysis and is now widely exploited by the use of various precious metals. Driven by the increasing interest in chemical utilization of ubiquitous metals that are abundant and nontoxic, iron catalysis has become a rapidly growing area of research, and iron-catalyzed C–H activation has been most actively explored in recent years. In this review, we summarize the development of stoichiometric C–H activation, which has a long history, and catalytic C–H functionalization, which emerged about 10 years ago. We focus in this review on reactions that take place via reactive organoiron intermediates, and we excluded those that use iron as a Lewis acid or radical initiator. The contents of this review are categorized by the type of C–H bond cleaved and the type of bond formed thereafter, and it covers the reactions of simple substrates and substrates possessing a directing group that anchors the catalyst to the substrate, providing an overview of iron-mediated and iron-catalyzed C–H activation reported in the literature by October 2016.
Co-reporter:Laurean Ilies, Yuki Itabashi, Rui Shang, and Eiichi Nakamura
ACS Catalysis January 6, 2017 Volume 7(Issue 1) pp:89-89
Publication Date(Web):November 22, 2016
DOI:10.1021/acscatal.6b02927
An iron(III) salt, (Z)-1,2-bis(diphenylphosphino)ethene or its electron-rich congener, (Z)-1,2-bis[bis(4-methoxyphenyl)phosphine]ethene, and a zinc(II) salt catalyze the arylation, heteroarylation, and alkenylation of propionamides possessing an 8-quinolylamide group with organoborate reagents in the presence of 1,2-dichlorobutane as oxidant at 70 °C. Stoichiometric experiments provided evidence for the involvement of an organoiron(III) species as a key intermediate for C–H activation and C–C bond formation.Keywords: alkenylation; amide; boron compound; C(sp3)−H activation; iron catalysis; zinc catalysis;
Co-reporter:Rui Shang, Laurean Ilies, and Eiichi Nakamura
Journal of the American Chemical Society 2016 Volume 138(Issue 32) pp:10132-10135
Publication Date(Web):August 3, 2016
DOI:10.1021/jacs.6b06908
Iron-catalyzed C–H functionalization of aromatics has attracted widespread attention from chemists in recent years, while the requirement of an elaborate directing group on the substrate has so far hampered the use of simple aromatic carbonyl compounds such as benzoic acid and ketones, much reducing its synthetic utility. We describe here a combination of a mildly reactive methylaluminum reagent and a new tridentate phosphine ligand for metal catalysis, 4-(bis(2-(diphenylphosphanyl)phenyl)phosphanyl)-N,N-dimethylaniline (Me2N-TP), that allows us to convert an ortho C–H bond to a C–CH3 bond in aromatics and heteroaromatics bearing simple carbonyl groups under mild oxidative conditions. The reaction is powerful enough to methylate all four ortho C–H bonds in benzophenone. The reaction tolerates a variety of functional groups, such as boronic ester, halide, sulfide, heterocycles, and enolizable ketones.
Co-reporter:Laurean Ilies, Mayuko Isomura, Shin-ichi Yamauchi, Tomoya Nakamura, and Eiichi Nakamura
Journal of the American Chemical Society 2016 Volume 139(Issue 1) pp:23-26
Publication Date(Web):December 22, 2016
DOI:10.1021/jacs.6b10061
Upon zincation of two acidic protons attached to the nitrogen and the sp-carbon atoms, a N-protected 2-ethynylaniline cyclizes to a 2,3-dizincioindole at 120 °C. Driven by the energy gain due to formation of two C–Zn bonds, this reaction occurs smoothly without side reactions, although this transformation is intrinsically endothermic in its bare anionic form. The resulting dizinc intermediate can be functionalized with one or two different electrophiles either inter- or intramolecularly on either C2 or C3 selectively, depending on the choice of catalyst and the electrophiles. This conversion of 2-ethynylaniline to 2,3-dimetalloindole can be applied to an expeditious synthesis of indenoindolone and benzodipyrrole derivatives, which are compounds of interest for medicinal chemistry and materials science, respectively.
Co-reporter:Tatsuaki Matsubara;Dr. Laurean Ilies;Dr. Eiichi Nakamura
Chemistry – An Asian Journal 2016 Volume 11( Issue 3) pp:380-384
Publication Date(Web):
DOI:10.1002/asia.201501095
Abstract
An iron catalyst combined with a mild organic oxidant promotes both C−H bond cleavage and C−N bond formation, and forms 2-pyridones and isoquinolones from an alkene- or arylamide and an internal alkyne, respectively. An unsymmetrical alkyne gives the pyridone derivative with high regioselectivity, this could be due to the sensitivity of the reaction to steric effects because of the compact size of iron.
Co-reporter:Rui Shang; Laurean Ilies;Eiichi Nakamura
Journal of the American Chemical Society 2015 Volume 137(Issue 24) pp:7660-7663
Publication Date(Web):June 10, 2015
DOI:10.1021/jacs.5b04818
Conversion of a C(sp2)–H or C(sp3)–H bond to the corresponding C–Me bond can be achieved by using AlMe3 or its air-stable diamine complex in the presence of catalytic amounts of an inorganic iron(III) salt and a diphosphine along with 2,3-dichlorobutane as a stoichiometric oxidant. The reaction is applicable to a variety of amide substrates bearing a picolinoyl or 8-aminoquinolyl directing group, enabling methylation of a variety of (hetero)aryl, alkenyl, and alkyl amides. The use of the mild aluminum reagent prevents undesired reduction of iron and allows the reaction to proceed with catalyst turnover numbers as high as 6500.
Co-reporter:Laurean Ilies ; Tatsuaki Matsubara ; Saki Ichikawa ; Sobi Asako ;Eiichi Nakamura
Journal of the American Chemical Society 2014 Volume 136(Issue 38) pp:13126-13129
Publication Date(Web):July 17, 2014
DOI:10.1021/ja5066015
Alkenes, arenes, and heteroarenes possessing an 8-quinolylamide group as the directing group are alkylated with primary and secondary alkyl tosylates, mesylate, and halides in the presence of Fe(acac)3/diphosphine as a catalyst and ArZnBr as a base. The reaction proceeds stereospecifically for alkene substrates and takes place without loss of regiochemical integrity of the starting secondary tosylate, but with loss of the stereochemistry of the chiral center.
Co-reporter:Rui Shang ; Laurean Ilies ; Sobi Asako ;Eiichi Nakamura
Journal of the American Chemical Society 2014 Volume 136(Issue 41) pp:14349-14352
Publication Date(Web):September 30, 2014
DOI:10.1021/ja5070763
We report here that an iron-catalyzed directed C–H functionalization reaction allows the coupling of a variety of aromatic, heteroaromatic, and olefinic substrates with alkenyl and aryl boron compounds under mild oxidative conditions. We rationalize these results by the involvement of an organoiron(III) reactive intermediate that is responsible for the C–H bond-activation process. A zinc salt is crucial to promote the transfer of the organic group from the boron atom to the iron(III) atom.
Co-reporter:Sobi Asako;Jakob Norinder;Naohiko Yoshikai;Eiichi Nakamura
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 7) pp:1481-1485
Publication Date(Web):
DOI:10.1002/adsc.201400063
Co-reporter:Sobi Asako ; Laurean Ilies ;Eiichi Nakamura
Journal of the American Chemical Society 2013 Volume 135(Issue 47) pp:17755-17757
Publication Date(Web):November 11, 2013
DOI:10.1021/ja4106368
Arenes possessing an N-(quinolin-8-yl)amide directing group are ortho-allylated with allyl phenyl ether in the presence of an iron/diphosphine catalyst and an organometallic base at 50–70 °C. The reaction proceeds via fast iron-catalyzed C–H activation, followed by reaction of the resulting iron intermediate with the allyl ether in γ-selective fashion.
Co-reporter:Tatsuaki Matsubara ; Sobi Asako ; Laurean Ilies ;Eiichi Nakamura
Journal of the American Chemical Society 2013 Volume 136(Issue 2) pp:646-649
Publication Date(Web):December 31, 2013
DOI:10.1021/ja412521k
Arenes possessing an 8-quinolinylamide group as a directing group are ortho aminated with N-chloroamines and N-benzoyloxyamines in the presence of an iron/diphosphine catalyst and an organometallic base to produce anthranilic acid derivatives in high yield. The reaction proceeds via iron-catalyzed C–H activation, followed by the reaction of the resulting iron intermediate with N-chloroamine. The choice of the directing group and diphosphine ligand is crucial for obtaining the anthranilic acid derivative with high yield and product selectivity.

![2-Pyridinecarboxamide, N-[(2-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/867400/867350-26-3.png)
![2-Pyridinecarboxamide, N-[(2-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/867400/867350-26-3_b.png)




![Silane, trimethyl[(3E)-4-phenyl-3-buten-1-ynyl]-](http://img.cochemist.com/ccimg/42200/42134-52-1.png)
![Silane, trimethyl[(3E)-4-phenyl-3-buten-1-ynyl]-](http://img.cochemist.com/ccimg/42200/42134-52-1_b.png)


![Benzene, [(1R,4R)-4-methyl-2-cyclohexen-1-yl]-, rel-](http://img.cochemist.com/ccimg/400100/400091-78-3.png)
![Benzene, [(1R,4R)-4-methyl-2-cyclohexen-1-yl]-, rel-](http://img.cochemist.com/ccimg/400100/400091-78-3_b.png)
![Benzene, 1-fluoro-4-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/485900/485844-19-7.png)
![Benzene, 1-fluoro-4-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/485900/485844-19-7_b.png)


![1H-Indole, 1-[(4-methoxyphenyl)methyl]-2-phenyl-](http://img.cochemist.com/ccimg/665100/665034-12-8.png)
![1H-Indole, 1-[(4-methoxyphenyl)methyl]-2-phenyl-](http://img.cochemist.com/ccimg/665100/665034-12-8_b.png)
![1H-Indole, 1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/22100/22051-21-4.png)
![1H-Indole, 1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/22100/22051-21-4_b.png)

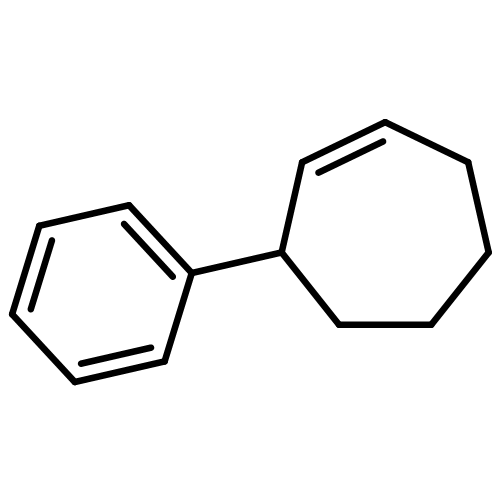
![Benzene, [(1R,4S)-4-methyl-2-cyclohexen-1-yl]-, rel-](http://img.cochemist.com/ccimg/400100/400091-82-9.png)
![Benzene, [(1R,4S)-4-methyl-2-cyclohexen-1-yl]-, rel-](http://img.cochemist.com/ccimg/400100/400091-82-9_b.png)


![PHOSPHINE, BIS[2-(DIPHENYLPHOSPHINO)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/53200/53103-03-0.png)
![PHOSPHINE, BIS[2-(DIPHENYLPHOSPHINO)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/53200/53103-03-0_b.png)




![BENZENEMETHANAMINE, 4-METHOXY-N-[2-[(TRIMETHYLSILYL)ETHYNYL]PHENYL]-](http://img.cochemist.com/ccimg/665100/665033-55-6.png)
![BENZENEMETHANAMINE, 4-METHOXY-N-[2-[(TRIMETHYLSILYL)ETHYNYL]PHENYL]-](http://img.cochemist.com/ccimg/665100/665033-55-6_b.png)


![2-PYRIDINECARBOXAMIDE, N-[(3-METHYLPHENYL)METHYL]-](http://img.cochemist.com/ccimg/867400/867350-27-4.png)
![2-PYRIDINECARBOXAMIDE, N-[(3-METHYLPHENYL)METHYL]-](http://img.cochemist.com/ccimg/867400/867350-27-4_b.png)
![Benzene, 1-bromo-2-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/15900/15861-49-1.png)
![Benzene, 1-bromo-2-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/15900/15861-49-1_b.png)




![Methanone, bis[2-(diphenylphosphino)phenyl]-](/data/chemimg/2291200/845821-92-3.png)
![Methanone, bis[2-(diphenylphosphino)phenyl]-](/data/chemimg/2291200/845821-92-3_b.png)


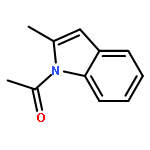
















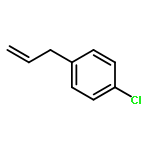
![Benzene, [(1-methyl-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/22600/22509-78-0.png)
![Benzene, [(1-methyl-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/22600/22509-78-0_b.png)
![Benzene, 1-methyl-2-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/19000/18916-11-5.png)
![Benzene, 1-methyl-2-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/19000/18916-11-5_b.png)






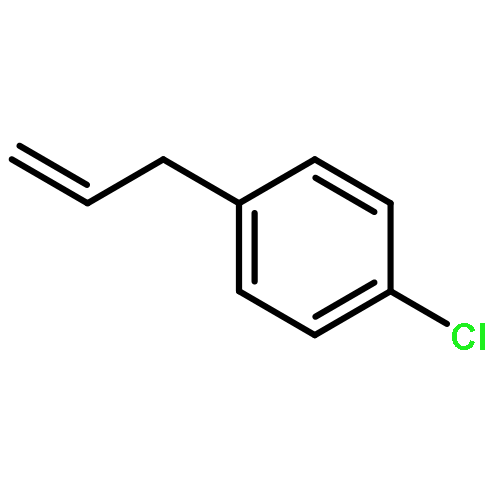

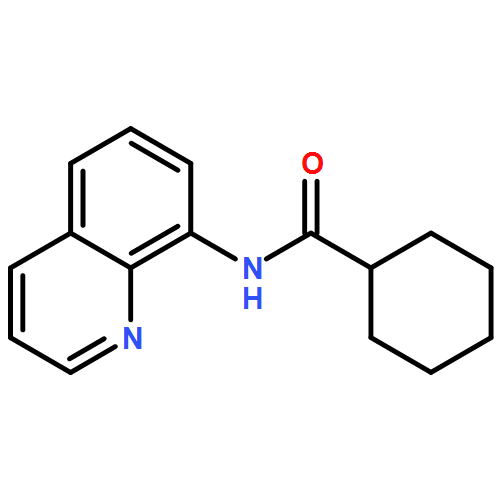


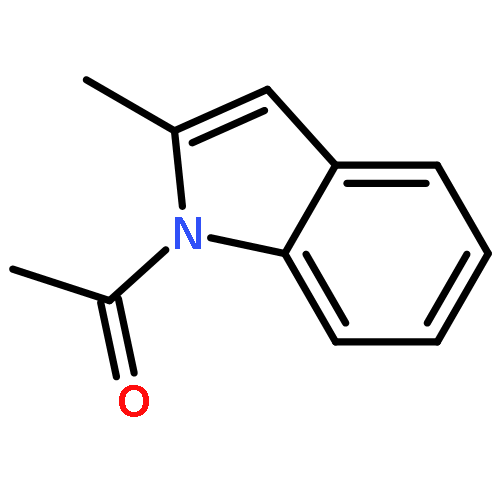
![Ethanone, 1-[2-(diphenylphosphino)phenyl]-](/data/chemimg/2115700/50777-63-4.png)
![Ethanone, 1-[2-(diphenylphosphino)phenyl]-](/data/chemimg/2115700/50777-63-4_b.png)
![Hexanoic acid, 6-[[(4-methylphenyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/122900/122822-50-8.png)
![Hexanoic acid, 6-[[(4-methylphenyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/122900/122822-50-8_b.png)


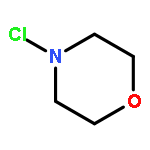
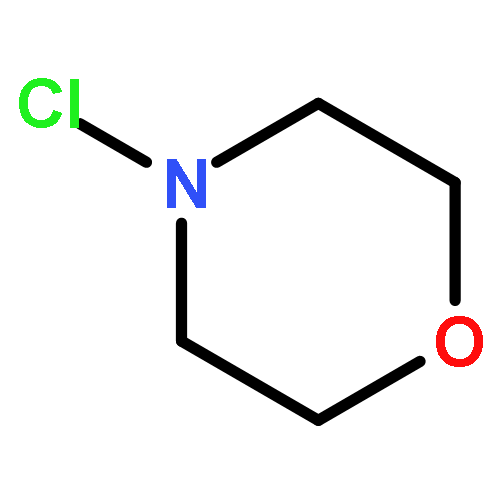
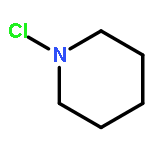









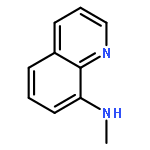


































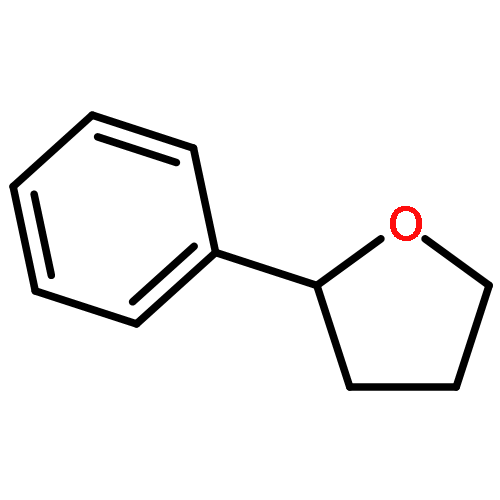




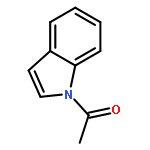
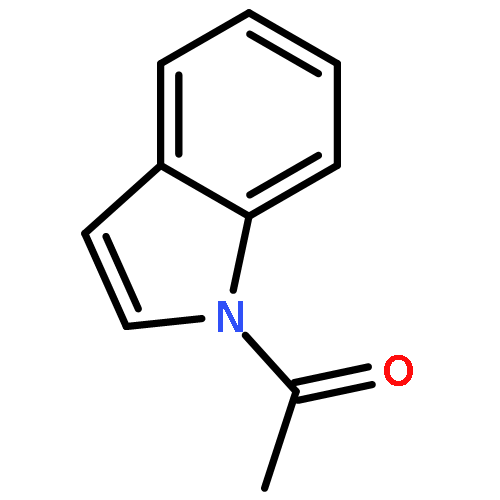
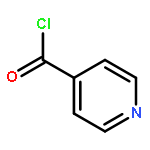
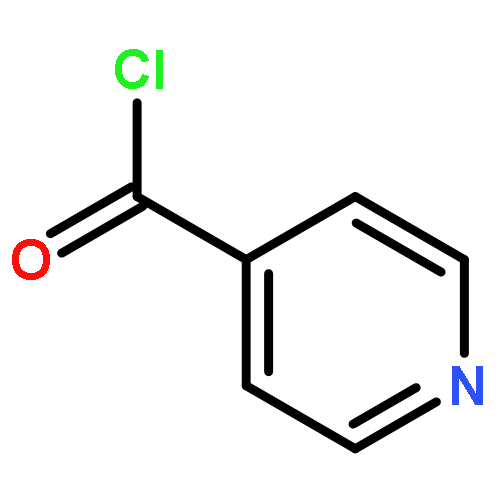






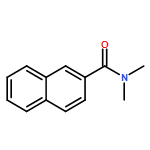













![[1,1'-Biphenyl]-3-carboxylicacid, 4-methyl-](http://img.cochemist.com/ccimg/2900/2840-35-9.png)
![[1,1'-Biphenyl]-3-carboxylicacid, 4-methyl-](http://img.cochemist.com/ccimg/2900/2840-35-9_b.png)






![Benzene, 1-methyl-4-[(1E)-3-phenyl-1-propenyl]-](http://img.cochemist.com/ccimg/134600/134539-87-0.png)
![Benzene, 1-methyl-4-[(1E)-3-phenyl-1-propenyl]-](http://img.cochemist.com/ccimg/134600/134539-87-0_b.png)






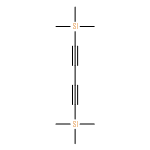
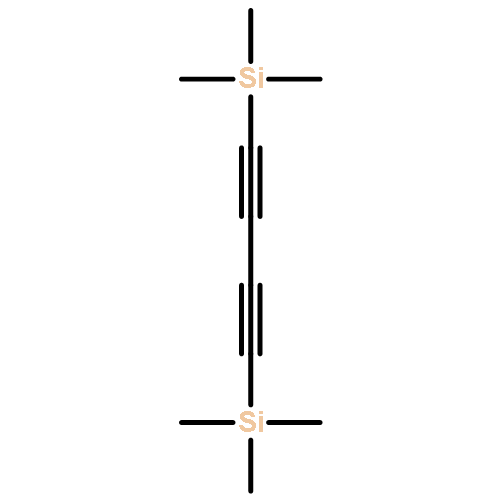
![Pyridine, 2-[1,1'-biphenyl]-2-yl-](/data/chemimg/129800/219843-48-8.png)
![Pyridine, 2-[1,1'-biphenyl]-2-yl-](/data/chemimg/129800/219843-48-8_b.png)