Co-reporter:Matthew R. Kelley and Jan-Uwe Rohde
Dalton Transactions 2014 vol. 43(Issue 2) pp:527-537
Publication Date(Web):14 Oct 2013
DOI:10.1039/C3DT52283K
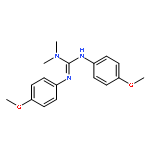
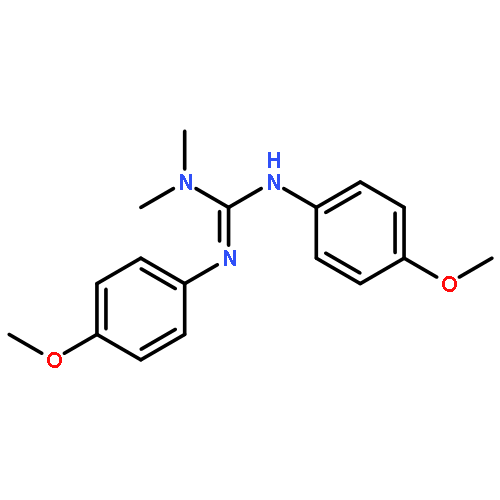
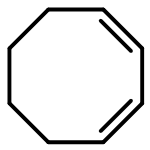
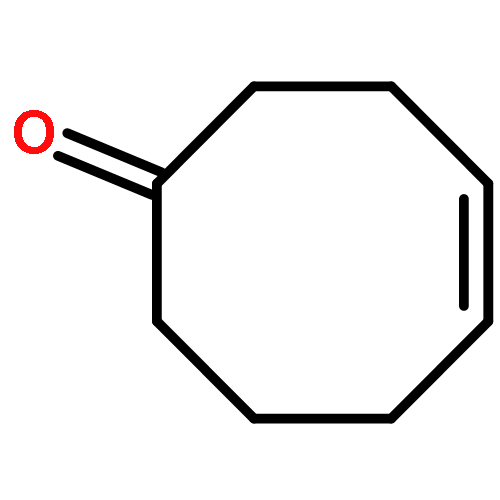
An IrI complex of an acetamidinato ligand was synthesized by reaction of N,N′-diphenylacetamidine, PhNC(Me)NHPh, with either MeLi and [{Ir(cod)}2(μ-Cl)2] or [{Ir(cod)}2(μ-OMe)2] and was characterized by X-ray crystallography as a mononuclear complex, [Ir{PhNC(Me)NPh}(cod)] (1; where cod = 1,5-cyclooctadiene). Reaction of 1 with CO afforded a dinuclear carbonyl complex, [{Ir(CO)2}2{μ-PhNC(Me)NPh-κN:κN′}2] (2), as indicated by EI mass spectrometry and solution- and solid-state IR spectroscopy [νCO (n-pentane) = 2067, 2034 and 1992 cm−1]. Activation of O2 by 1 in solution at 20 °C was irreversible and produced an (alkene)peroxoiridium(III) intermediate, [Ir{PhNC(Me)NPh}(cod)(O2)] (3), which was characterized by one- and two-dimensional NMR techniques and IR spectroscopy (for 3, νOO = 860 cm−1; for 3–18O2, νOO = 807 cm−1). Complex 3 oxidized PPh3 to OPPh3, and its decay in the absence of added substrates followed by reaction with cod yielded 4-cycloocten-1-one and a minor amount of 1. In comparison with the results for the previously reported guanidinato complex [Ir{PhNC(NMe2)NPh}(cod)(O2)] (4), the formation of 3 and its reaction with PPh3 are significantly faster, indicating considerable ligand effects in these reactions.
Co-reporter:Sam P. de Visser, Jan-Uwe Rohde, Yong-Min Lee, Jaeheung Cho, Wonwoo Nam
Coordination Chemistry Reviews 2013 Volume 257(Issue 2) pp:381-393
Publication Date(Web):15 January 2013
DOI:10.1016/j.ccr.2012.06.002
Iron–oxygen species, such as iron(IV)-oxo, iron(III)-superoxo, iron(III)-peroxo, and iron(III)-hydroperoxo complexes, are key intermediates often detected in the catalytic cycles of dioxygen activation by heme and nonheme iron enzymes. Our understanding of the chemistry of these key intermediates has improved greatly by studies of the structural and spectroscopic properties and reactivities of their synthetic analogues. One class of biomimetic coordination complexes that has proven to be particularly versatile in studying dioxygen activation by metal complexes is comprised of FeIVO and FeIIIO2(H) complexes of the macrocyclic tetramethylcyclam ligand (TMC, 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane). Several recent advances have been made in the synthesis and isolation of new iron–oxygen complexes of this ligand, their structural and spectroscopic characterization, and elucidation of their reactivities in various oxidation reactions. In this review, we summarize the chemistry of the first structurally characterized mononuclear nonheme iron(IV)-oxo complex, in which the FeIVO group was stabilized by the TMC ligand. Complexes with different axial ligands, [FeIV(O)(TMC)(X)]n+, and complexes of other cyclam ligands are discussed as well. Very recently, significant progress has also been reported in the area of other iron–oxygen intermediates, such as iron(III)-superoxo, iron(III)-peroxo, and iron(III)-hydroperoxo complexes bearing the TMC ligand. The present results demonstrate how synthetic and mechanistic developments in biomimetic research can advance our understanding of dioxygen activation occurring in mononuclear nonheme iron enzymes.Highlights► FeIVO, FeIII (η2O2), and FeIIIO2H complexes having a common ligand environment. ► Models of intermediates in oxygen activation by mononuclear nonheme iron enzymes. ► Axial ligands influence FeO bonding and spectroscopic properties of FeIVO complexes. ► Generation of FeIVO species by activating O implicates an FeIII-superoxo intermediate. ► Reactivity studies with model substrates establish catalytic potency of short-lived intermediates.
Co-reporter:Matthew R. Kelley and Jan-Uwe Rohde
Inorganic Chemistry 2013 Volume 52(Issue 5) pp:2564-2580
Publication Date(Web):February 19, 2013
DOI:10.1021/ic302570s
A series of seven [Ir{ArNC(NR2)NAr}(cod)] complexes (1a–1g; where R = Me or Et; Ar = Ph, 4-MeC6H4, 4-MeOC6H4, 2,6-Me2C6H3, or 2,6-iPr2C6H3; and cod = 1,5-cyclooctadiene) were synthesized by two different methods from the neutral guanidines, ArN═C(NR2)NHAr, using either MeLi and [{Ir(cod)}2(μ-Cl)2] or [{Ir(cod)}2(μ-OMe)2]. Reaction of 1a–1g with CO produced the corresponding [Ir{ArNC(NR2)NAr}(CO)2] complexes (2a–2g), which were characterized by NMR and solution- and solid-state IR spectroscopy. Complexes 1b (R = Et, Ar = Ph), 1d (R = Et, Ar = 4-MeC6H4), 1f (R = Me, Ar = 2,6-Me2C6H3), and 2b (R = Et, Ar = Ph) were characterized by X-ray crystallography as mononuclear complexes with a guanidinato-κ2N,N′ ligand and a cod or two CO ligands coordinated to the Ir center in a distorted square-planar environment. On the basis of the CO stretching frequencies of 2a–2g [avg. νCO (n-pentane) = 2016–2019 cm–1] and the alkene 13C chemical shifts of 1a–1g [δ(13CC═C) = 58.7–61.0 ppm], the donor strength of the guanidinato ligands was evaluated and compared to that of related monoanionic ligands. Reaction of 1a–1g in solution with O2 at 20 °C afforded (alkene)peroxoiridium(III) intermediates, [Ir{ArNC(NR2)NAr}(cod)(O2)] (3). The steric properties of the supporting ligand play a decisive role in O2 binding in that complexes without ortho substituents react largely irreversibly with O2 (1a–1e; where Ar = Ph, 4-MeC6H4 or 4-MeOC6H4), whereas complexes with ortho substituents exhibit fully reversible O2 binding (1f and 1g; where Ar = 2,6-Me2C6H3 or 2,6-iPr2C6H3). Complexes 3a–3f were characterized by 1H NMR and IR spectroscopy (νOO = 857–872 cm–1). Decay of the new intermediates and subsequent reaction with cod produced 4-cycloocten-1-one and the respective IrI precursor.
Co-reporter:Matthew R. Kelley and Jan-Uwe Rohde
Chemical Communications 2012 vol. 48(Issue 23) pp:2876-2878
Publication Date(Web):07 Feb 2012
DOI:10.1039/C2CC17332H
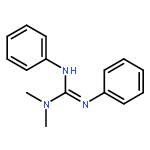
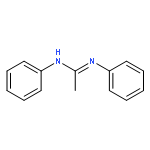
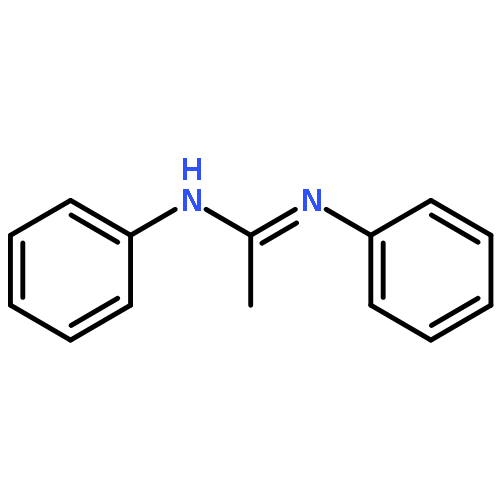
An (alkene)peroxoiridium(III) complex, [Ir(L)(cod)(O2)] [where LH = PhNC(NMe2)NHPh and cod = 1,5-cyclooctadiene], was identified as an intermediate in the reaction of the IrI precursor [Ir(L)(cod)] with O2 and characterized by spectroscopic methods. Decay of the intermediate and further reaction with 1,5-cyclooctadiene produced 4-cycloocten-1-one.
Co-reporter:Joseph J. Braymer;Kevin P. O'Neill;Dr. Jan-Uwe Rohde;Dr. Mi Hee Lim
Angewandte Chemie 2012 Volume 124( Issue 22) pp:5472-5476
Publication Date(Web):
DOI:10.1002/ange.201200901
Co-reporter:Joseph J. Braymer;Kevin P. O'Neill;Dr. Jan-Uwe Rohde;Dr. Mi Hee Lim
Angewandte Chemie International Edition 2012 Volume 51( Issue 22) pp:5376-5380
Publication Date(Web):
DOI:10.1002/anie.201200901
Co-reporter:Travis M. Owen
Inorganic Chemistry 2011 Volume 50(Issue 11) pp:5283-5289
Publication Date(Web):April 28, 2011
DOI:10.1021/ic2007205
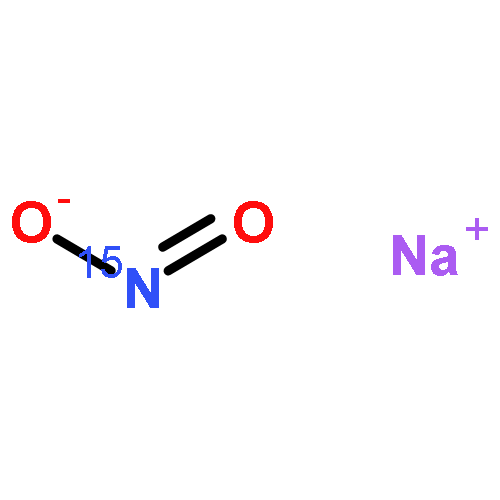
Reaction of [FeO(tmc)(OAc)]+ with the free radical nitrogen monoxide afforded a mixture of two FeII complexes, [Fe(tmc)(OAc)]+ and [Fe(tmc)(ONO)]+ (where tmc = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane and AcO– = acetate anion). The amount of nitrite produced in this reaction (ca. 1 equiv with respect to Fe) was determined by ESI mass spectrometry after addition of 15N-enriched NaNO2. In contrast to oxygen atom transfer to PPh3, the NO reaction of [FeO(tmc)(OAc)]+ proceeds through an FeIII intermediate that was identified by UV–vis–NIR spectroscopy and ESI mass spectrometry and whose decay is dependent on the concentration of methanol. The observations are consistent with a mechanism involving oxide(•1–) ion transfer from [FeO(tmc)(OAc)]+ to NO to form an FeIII complex and NO2–, followed by reduction of the FeIII complex. Competitive binding of AcO– and NO2– to FeII then leads to an equilibrium mixture of two FeII(tmc) complexes. Evidence for the incorporation of oxygen from the oxoiron(IV) complex into NO2– was obtained from an 18O-labeling experiment. The reported reaction serves as a synthetic example of the NO reactivity of biological oxoiron(IV) species, which has been proposed to have physiological functions such as inhibition of oxidative damage, enhancement of peroxidase activity, and NO scavenging.
Co-reporter:Tony D. Manuel
Journal of the American Chemical Society 2009 Volume 131(Issue 43) pp:15582-15583
Publication Date(Web):October 14, 2009
DOI:10.1021/ja9065943
Bis(imino)pyridine complex [Ni{2,6-(ArN═CMe)2C5H3N}Cl] (where Ar = 2,6-iPr2C6H3) was synthesized by reduction of the corresponding dichloride complex and characterized as a ligand-radical complex of NiII. Reaction of this complex with O2 caused intraligand C−C bond cleavage to afford the Ni complex of the new iminoethylpyridylcarboxamidato ligand, which also was isolated as the corresponding carboxamide, 6-(ArN═CMe)C5H3N-2-C(O)NHAr. This reaction serves as an example of small-molecule activation effected directly at the redox-active bis(imino)pyridine ligand without an overall oxidation state change at the Ni center.
Co-reporter:Jan-Uwe Rohde ;Wei-Tsung Lee
Journal of the American Chemical Society 2009 Volume 131(Issue 26) pp:9162-9163
Publication Date(Web):June 10, 2009
DOI:10.1021/ja9033445
Electron-rich tris(guanidinato) complexes of IrIII, [Ir{ArNC(NR2)NAr}3] (where R = Me or Et; Ar = Ph or 4-MeC6H4), were synthesized from the respective [Ir{ArNC(NR2)NAr}(C8H14)2] precursors (C8H14 = cis-cyclooctene), are air-sensitive, and can be electrochemically oxidized in two one-electron transfer steps. The first electron transfer is reversible and occurs at much lower potentials than typical for IrIII. Chemical oxidation by [FeCp2]PF6 afforded isolable, paramagnetic IrIV compounds, [Ir{ArNC(NR2)NAr}3]PF6, which were characterized by analytical and spectroscopic methods and a single-crystal structure determination, demonstrating that IrIV is accessible in a nitrogen-donor ligand environment.
Co-reporter:Jan-Uwe Rohde, Matthew R. Kelley and Wei-Tsung Lee
Inorganic Chemistry 2008 Volume 47(Issue 24) pp:11461-11463
Publication Date(Web):November 14, 2008
DOI:10.1021/ic801867r
Mononuclear [Ir{ArNC(NR2)NAr}(C8H12)] complexes (where R = Me or Et; Ar = Ph, 4-MeC6H4, or 2,6-Me2C6H3; and C8H12 = 1,5-cyclooctadiene) were synthesized from the neutral N,N-dialkyl-N′,N′′-diarylguanidines via deprotonation and transmetalation. As confirmed by single-crystal structure determinations, the guanidinato(1−) ligands coordinate the low-valent d8 IrI center in an N,N′-chelating binding mode, and the 13C NMR chemical shifts of the alkene carbon atoms establish that these ligands function as stronger donors than related monoanionic, bidentate nitrogen-based ligands. In the reactions of the complexes with O2, the observed reactivity trends correlate with the electronic and steric influences of the substituents of the guanidinato ligands.
Co-reporter:Matthew R. Kelley and Jan-Uwe Rohde
Chemical Communications 2012 - vol. 48(Issue 23) pp:NaN2878-2878
Publication Date(Web):2012/02/07
DOI:10.1039/C2CC17332H
An (alkene)peroxoiridium(III) complex, [Ir(L)(cod)(O2)] [where LH = PhNC(NMe2)NHPh and cod = 1,5-cyclooctadiene], was identified as an intermediate in the reaction of the IrI precursor [Ir(L)(cod)] with O2 and characterized by spectroscopic methods. Decay of the intermediate and further reaction with 1,5-cyclooctadiene produced 4-cycloocten-1-one.
Co-reporter:Matthew R. Kelley and Jan-Uwe Rohde
Dalton Transactions 2014 - vol. 43(Issue 2) pp:NaN537-537
Publication Date(Web):2013/10/14
DOI:10.1039/C3DT52283K
An IrI complex of an acetamidinato ligand was synthesized by reaction of N,N′-diphenylacetamidine, PhNC(Me)NHPh, with either MeLi and [{Ir(cod)}2(μ-Cl)2] or [{Ir(cod)}2(μ-OMe)2] and was characterized by X-ray crystallography as a mononuclear complex, [Ir{PhNC(Me)NPh}(cod)] (1; where cod = 1,5-cyclooctadiene). Reaction of 1 with CO afforded a dinuclear carbonyl complex, [{Ir(CO)2}2{μ-PhNC(Me)NPh-κN:κN′}2] (2), as indicated by EI mass spectrometry and solution- and solid-state IR spectroscopy [νCO (n-pentane) = 2067, 2034 and 1992 cm−1]. Activation of O2 by 1 in solution at 20 °C was irreversible and produced an (alkene)peroxoiridium(III) intermediate, [Ir{PhNC(Me)NPh}(cod)(O2)] (3), which was characterized by one- and two-dimensional NMR techniques and IR spectroscopy (for 3, νOO = 860 cm−1; for 3–18O2, νOO = 807 cm−1). Complex 3 oxidized PPh3 to OPPh3, and its decay in the absence of added substrates followed by reaction with cod yielded 4-cycloocten-1-one and a minor amount of 1. In comparison with the results for the previously reported guanidinato complex [Ir{PhNC(NMe2)NPh}(cod)(O2)] (4), the formation of 3 and its reaction with PPh3 are significantly faster, indicating considerable ligand effects in these reactions.
.jpg)