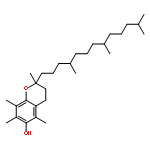
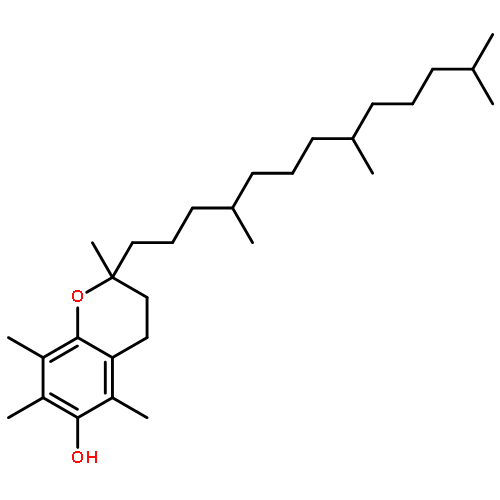
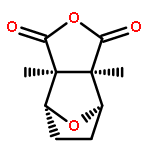
Co-reporter:Yanxiao Xiang;Xinbing Wei;Lin Chen;Huiqing Liu
Journal of Molecular Neuroscience 2014 Volume 52( Issue 4) pp:577-585
Publication Date(Web):2014 April
DOI:10.1007/s12031-013-0113-6
Astrocytes activation has been implicated in the inflammatory responses underlying brain injury and neurodegenerative diseases including bacterial infections, cerebral ischemia, and Parkinson's diseases. Acetylpuerarin is a newly modified isoflavone based on puerarin that has neuroprotective and antioxidant effects. In this study, we investigated the anti-inflammatory action of acetylpuerarin in regulating the eicosanoids generation and its underlying molecular mechanisms in lipopolysaccharide (LPS)-induced production of arachidonic acid (AA) metabolites in primary rat astrocytes. The results showed that acetylpuerarin concentration dependently inhibited the LPS-induced production of AA metabolites such as prostaglandin E2 (PGE2) and leukotriene C4 (LTC4), and acetylpuerarin significantly attenuated the expression and immunoreactivity of group V secretory phospholipase A2 (sPLA2) protein induced by LPS in astrocytes. Furthermore, in astrocytes pretreated with acetylpuerarin, the time course of phosphorylation of extracellular signal-regulated kinase (ERK)1/2 and of cytosolic PLA2 alpha (cPLA2α) and expression of transcription factors, nuclear factor kappa B (NF-κB), was markedly truncated. Acetylpuerarin concentration dependently abolished the LPS-induced expressions of AA-metabolizing enzymes including cyclooxygenase-2 (COX-2) and lipooxygenase-5 (LOX-5). This study indicates that acetylpuerarin inhibited LPS-induced AA-metabolizing enzymes and AA metabolites in astrocytes via downregulation expression of group V sPLA2 and phosphorylation of ERK1/2, cPLA2α, and NF-κB. These findings reveal, in part, the molecular basis underlying the anti-inflammatory properties of acetylpuerarin.
Co-reporter:Yunxue Zhao, Min Liu, Vincent Chagnault, Juying Wang, Xiumei Zhang, Paul V. Murphy
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 2) pp:824-828
Publication Date(Web):15 January 2011
DOI:10.1016/j.bmcl.2010.11.088
Previously the synthesis of novel somatostatin mimetic from 1-deoxynojirimycin (DNJ) led to identification of a compound with affinity for human somatostatin receptor subtypes 4 and 5 (hSSTR4 and hSSTR5). Here we examined the properties of this peptidomimetic in a human umbilical vein endothelial cell (HUVEC) based assays. The peptidomimetic prevented capillary tube formation based on HUVECs. It also inhibited HUVEC proliferation by inducing G1 phase cell cycle arrest and apoptosis. Stress fiber assembly and cell migration in HUVECs was markedly suppressed by the somatostatin receptor ligand.
Co-reporter:Haiyan Lou, Lei Gao, Xinbing Wei, Zhen Zhang, Dandan Zheng, Dianrui Zhang, Xiumei Zhang, Ying Li, Qiang Zhang
Colloids and Surfaces B: Biointerfaces 2011 Volume 87(Issue 2) pp:319-325
Publication Date(Web):15 October 2011
DOI:10.1016/j.colsurfb.2011.05.037
PurposeThe aim of the present study was to evaluate both the in vitro and in vivo antitumor activity of an oridonin nanosuspension (ORI-N) relative to efficacy of bulk oridonin delivery.MethodsORI-N with a particle size of 897.2 ± 14.2 nm and a zeta potential of −21.8 ± 0.8 mV was prepared by the high-pressure homogenization (HPH) technique. The in vitro cytotoxicity of ORI-N against SMMC-7721 cells was evaluated by MTT[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay, the effects of ORI-N on cell cycle and cell apoptosis was analyzed by flow cytometry; the in vivo anti-tumor activity was observed in H22 tumor bearing mice.ResultsORI-N effectively inhibited the proliferation of SMMC-7721 cells. Flow cytometric analysis demonstrated that ORI-N arrested SMMC-7721 cells in the G2/M cycle, and furthermore, that ORI-N induced a higher apoptotic rate than the bulk ORI solution. In vivo studies ORI-N also showed higher antitumor efficacy as measured by reduced tumor volume and tumor weight, as well as lower toxicity in H22 solid tumor bearing mice compared to free ORI at the same concentration.ConclusionsThese results suggest that the delivery of ORI-N as a nanosuspension is a promising approach for treating tumors.ORI nanosuspensions showed a higher antitumor efficiency as measured by reduced tumor volume in H22 solid tumor bearing mice compared to free ORI solution.
Co-reporter:Li Chao-Wu;Zhang Shuo;Gao Hai-Qing
European Journal of Drug Metabolism and Pharmacokinetics 2011 Volume 36( Issue 4) pp:257-262
Publication Date(Web):2011 December
DOI:10.1007/s13318-011-0045-x
The objective of the study was to establish an HPLC method for the determination of l-tetrahydropalmatine in human plasma, and to investigate the pharmacokinetics after oral administration of l-tetrahydropalmatine disintegrating tablets in healthy Chinese. l-tetrahydropalmatine in human plasma was separated on a Phenomenex luna C18 column (250 mm × 4.6 mm, 5 μm), eluted using methanol–water (75:25, v/v) as mobile phase, and detected by photodiode array detector at a wavelength of 281 nm. A single 60 mg of l-tetrahydropalmatine orally disintegrating tablets were orally given to 12 healthy male volunteers after fasting overnight. Before and after administration 4 mL of blood samples was collected at the scheduled time. The plasma concentration of l-tetrahydropalmatine was determined by the established HPLC method after disposition and its pharmacokinetic parameters were analyzed and evaluated by both compartmental and noncompartmental models using Drug and Statistic (version 2.0). The disintegrating time and the sense of mouth were observed and recorded. The lowest limit of quantification (LLOQ) for l-tetrahydropalmatine in plasma was 0.01 μg mL−1, and a linearity was obtained in the range of 0.01–1 μg mL−1 (r = 0.9998). The disposal procedure of l-tetrahydropalmatine in human was fitted using the DAS program, following a double-compartment open model system (w = 1). l-tetrahydropalmatine was absorbed quickly with t1/2ka of 0.5 ± 0.054 h, distributed fast with t1/2α of 0.74 ± 0.088 h, and eliminated slowly with t1/2β of 11.42 ± 2.43 h. l-tetrahydropalmatine was distributed mainly in the periphery compartment with the V1/F of 133.30 ± 30.78 L. l-tetrahydropalmatine orally disintegrating tablets with good taste were disintegrated in the mouth within 16 s. The established HPLC method was sensitive, rapid, and suitable for both l-tetrahydropalmatine pharmacokinetic studies and its content assay in traditional Chinese medicine (TCM). The procedure of l-tetrahydropalmatine in human was fit to double-compartmental model (w = 1). l-tetrahydropalmatine orally disintegrating tablets were palatable, well-tolerated, disintegrated and absorbed quickly.
Co-reporter:Xia Sun, Xinbing Wei, Sifeng Qu, Yunxue Zhao, Xiumei Zhang
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 14) pp:4120-4124
Publication Date(Web):15 July 2010
DOI:10.1016/j.bmcl.2010.05.076
Hydroxysafflor Yellow A has been demonstrated to attenuate pressure overloaded hypertrophy in rats and inhibit platelet aggregation. Herein we found that Hydroxysafflor Yellow A prevented cerebral ischemia–reperfusion injury by inhibition of thrombin generation. In addition, treatment with Hydroxysafflor Yellow A significantly inhibited NF-κB p65 nuclear translation and p65 binding activity, both mRNA and protein levels of ICAM-1 and the infiltration of neutrophils. Mean while, Hydroxysafflor Yellow A had the capacity to improve neurological deficit scores, increase the number of the surviving hippocampal CA1 pyramidal cells and decrease the plasma angiotensin II level. These results illustrated that anti-cerebral ischemic mechanism of Hydroxysafflor Yellow A may be due to its suppression of thrombin generation and inhibition of thrombin-induced inflammatory responses by reducing angiotensin II content.
Co-reporter:Haiyan Lou, Xu Jing, Xinbing Wei, Huanying Shi, Dongmei Ren, Xiumei Zhang
Neuropharmacology (April 2014) Volume 79() pp:380-388
Publication Date(Web):1 April 2014
DOI:10.1016/j.neuropharm.2013.11.026
•Naringenin activates the Nrf2/ARE pathway in SH-SY5Y cells.•Naringenin blocks 6-OHDA-induced neurotoxicity in SH-SY5Y cells.•Naringenin up regulates protein levels of Nrf2/ARE genes in vivo.•Naringenin reduces 6-OHDA-induced striatal oxidative stress in mice.•Naringenin protects against DA neurodegeneration in 6-OHDA-leisoned mice.There is increasing evidence that oxidative stress is critically involved in the pathogenesis of Parkinson's disease (PD), suggesting that pharmacological targeting of the antioxidant machinery may have therapeutic value. Naringenin, a natural flavonoid compound, has been reported to possess neuroprotective effect against PD related pathology; however the mechanisms underlying its beneficial effects are poorly defined. Thus, the purpose of the present study was to investigate the potential neuroprotective role of naringenin and to delineate its mechanism of action against 6-hydroxydopamine (6-OHDA)-induced neurotoxicity in models of PD both in vitro and in vivo. Naringenin treatment resulted in an increase in nuclear factor E2-related factor 2 (Nrf2) protein levels and subsequent activation of antioxidant response element (ARE) pathway genes in SH-SY5Y cells and in mice. Exposure of SH-SY5Y cells to naringenin provided protection against 6-OHDA-induced oxidative insults that was dependent on Nrf2, since treatment with Nrf2 siRNA failed to block against 6-OHDA neurotoxicity or induce Nrf2-dependent cytoprotective genes in SH-SY5Y cells. In mice, oral administration of naringenin resulted in significant protection against 6-OHDA-induced nigrostriatal dopaminergic neurodegeneration and oxidative damage. Our results indicate that activation of Nrf2/ARE signaling by naringenin is strongly associated with its neuroprotective effects against 6-OHDA neurotoxicity and suggest that targeting the Nrf2/ARE pathway may be a promising approach for therapeutic intervention in PD.Download full-size image
Co-reporter:Tian Wang, Fenghua Fu, Bing Han, Leiming Zhang, Xiumei Zhang
Journal of Neuroimmunology (April 2012) Volume 245(Issues 1–2) pp:79-86
Publication Date(Web):1 April 2012
DOI:10.1016/j.jneuroim.2012.02.008
Spatial learning and memory are impaired in diabetic animals. The interaction of advanced glycation end products (AGEs) with the receptor of AGEs (RAGE), resulting in the activation of nuclear factor-κB (NF-κB), plays an important role in pathways leading to the cytotoxic effects to neurons. Danshensu, a compound from Salvia miltiorrhiza Bunge, has neuroprotective effects. This study aimed to investigate the role of AGE-mediated neuroinflammation in learning and memory deficits and the effect of Danshensu on the cognitive decline in diabetic mice. C57BL/6 mice were injected intraperitoneally with streptozotocin. Sodium Danshensu (sodium salt of Danshensu) was administered at a dose of 15, 30, or 60 mg/kg for 12 weeks. The results showed that diabetes caused impairment in acquisition and retrieval processes, as demonstrated by performance in the Morris water maze test. Danshensu not only reduced the mean escape latency but also increased the percentage of time spent in the target quadrant. Western blot analysis revealed that there was a significant increase in the expression of RAGE, p-p38, and COX-2, and the NF-κB activation. Danshensu partly blocked the expression of RAGE, p-p38, and COX-2, and NF-κB activation, and inhibited the increase of TNF-α, IL-6, and PGE2. However, Danshensu did not affect body weight and the levels of blood glucose, glycosylated hemoglobin, insulin, and AGEs. These findings demonstrate that AGE-mediated neuroinflammation plays an important role in learning and memory deficits in diabetic mice and that Danshensu may provide a potential alternative for the prevention of cognitive impairment associated with diabetes by attenuating AGE-mediated neuroinflammation.
Co-reporter:Ji-biao Wu, Ning-ning Song, Xin-bing Wei, Hua-shi Guan, Xiu-mei Zhang
Journal of the Neurological Sciences (15 January 2008) Volume 264(Issues 1–2) pp:50-55
Publication Date(Web):15 January 2008
DOI:10.1016/j.jns.2007.06.057
Mounting evidence has suggested that paeonol possesses plenty of pharmacologic actions. Our research is to determine if paeonol can protect cultured rat hippocampal neurons from oxygen–glucose deprivation(OGD)-induced injury and elucidate the underlying mechanism. We cultivated the rat hippocampal neurons as the object of study and then established the model of oxygen–glucose deprivation. Neuronal viability was measured by the reduction of 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), while intracellular Ca2+ concentration was observed by fluorospectrophotometer. The binding force of N-methyl-d-aspartate (NMDA) receptor was evaluated by liquid scintillation counting.Compared with oxygen–glucose deprivation group, paeonol treatment obviously increased cell survival rate and reduced the activity of the binding force of NMDA receptors, reversing the overload of intracellular Ca2+. These results demonstrate that paeonol protected rat neurons from oxygen–glucose deprivation-induced injury, resulting in alleviating the morphological damage and increasing neuron viability and suggest that paeonol may exhibit its protective effect against oxygen–glucose deprivation-induced injury by targeting on NMDA receptors.

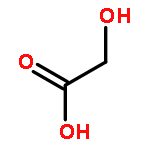
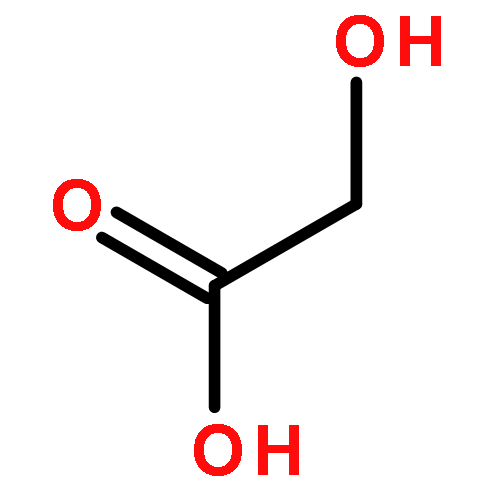


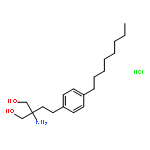
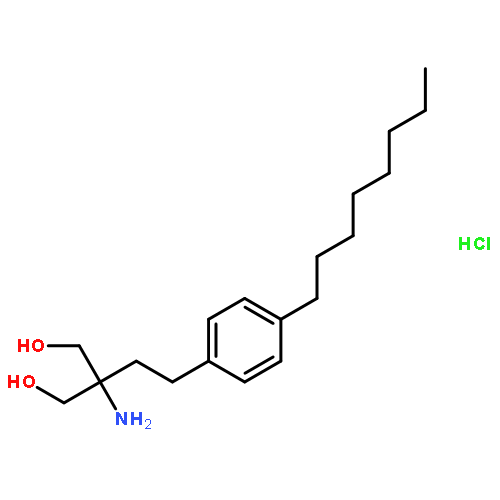






![Pyrazine, [[4-[bis(4-fluorophenyl)methyl]-1-piperazinyl]methyl]trimethyl-](http://img.cochemist.com/ccimg/892400/892396-24-6.png)
![Pyrazine, [[4-[bis(4-fluorophenyl)methyl]-1-piperazinyl]methyl]trimethyl-](http://img.cochemist.com/ccimg/892400/892396-24-6_b.png)