Co-reporter:Xiaotian Sun;Yunxia Liu;Zhigang Song;Yongdan Li;Weizhou Wang;Haiping Lin;Youyong Li
Journal of Materials Chemistry C 2017 vol. 5(Issue 17) pp:4159-4166
Publication Date(Web):2017/05/04
DOI:10.1039/C7TC00306D
Defects are unavoidable during the synthesis of materials, especially for two-dimensional (2D) nanomaterials. They are usually seen as detrimental to device properties, but sometimes bring about new beneficial effects. In order to clarify the influence of defects on the structural and electronic properties, we have performed first-principles calculations to systematically investigate the structural stability, mobility and electronic properties of typical point defects in 2D arsenene (h-As), antimonene (h-Sb) and antimony arsenide (h-AsSb), including the Stone–Wales defects, single vacancies (SVs), double vacancies (DVs) and adatoms. To provide visual guidance for experimental observations, scanning tunnelling microscopy (STM) images are simulated. Compared to defects in graphene and silicene, these defects form more easily with lower formation energies, and SVs can diffuse very quickly to the edges with a lower diffusion barrier of less than 1 eV. Monolayer arsenene, antimonene and antimony arsenide are indirect band gap semiconductors, and the defective structures significantly reduce the band gaps. Most of the SV and adatom defects carry magnetic moments due to the dangling bonds resulting from the absent or extra atom. Our present results have demonstrated that the point defects induce significant effects on the electronic properties of pristine arsenene, antimonene and the antimony arsenide alloy, which should be considered in their future applications.
Co-reporter:Xiao Yuan;Mingye Yang;Youyong Li
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 21) pp:13846-13854
Publication Date(Web):2017/05/31
DOI:10.1039/C7CP01727H
Based on the first-principles calculations, we systematically studied the structures and electronic properties of two-dimensional (2D) transition metal dichalcogenide (TMD) alloys with half-to-half mixing of S and Se. Using the chemical potentials of S and Se, the energetic phase diagrams for both single phases and mixed phases of TMD were constructed. A new heterolayer structure (for Sc and Ti) and alternating structure (for Cr, Mn, Fe, Zr, Mo, and W) were proposed for the first time, which were thermodynamically stable for MSSe alloys under the S-poor (relatively low chemical potential of S) and Se-rich (relatively high chemical potential of Se) conditions, and further compared with the disordered structures. Moreover, band gaps, carrier effective mass, and work functions were calculated for these stable mixed phases. Compared to the single phases of MS2 and MSe2, MSSe alloys showed superior electronic properties including tunable band gaps and work functions. Importantly, the significantly reduced effective mass of the carriers in the MSSe alloys may induce higher carrier mobility, providing better performance of TMD materials in electronic devices.
Co-reporter:Yujin Ji;Mingye Yang;Huilong Dong;Tingjun Hou;Youyong Li
Nanoscale (2009-Present) 2017 vol. 9(Issue 25) pp:8608-8615
Publication Date(Web):2017/06/29
DOI:10.1039/C7NR00688H
Full utilization of solar energy to split water into hydrogen and oxygen is an efficient way to solve the current energy problem. Graphene-like germanium monochalcogenides (GeS or GeSe) are proposed here as efficient photocatalysts for water splitting under a broad range from ultraviolet, visible to near-infrared light dependent on their thickness. Compared to traditional photocatalysts, GeS and GeSe possess a large intrinsic dipole and introduce an internal electric field directing from Ge surface to S/Se surface, which causes notable band bending. The band bending means they possess favorable band positions located outside the reduction potential and oxidation potential of water, overcoming the restriction of their band gaps. Moreover, multilayer GeS and GeSe further provide a good separation of electrons and holes, which effectively reduces the probability of their recombination and ensures photocatalytic activity with high efficiency.
Co-reporter:Mingye Yang;Tingjun Hou;Youyong Li
Journal of Materials Chemistry C 2017 vol. 5(Issue 1) pp:201-207
Publication Date(Web):2016/12/22
DOI:10.1039/C6TC04487E
Two-dimensional nanomaterials have attracted extensive interest and their heterostructures possess excellent electronic and optical properties suitable for various applications. Based on first-principles calculations, we investigated the structural stability and electronic properties of WS2 and graphene oxide (GO) heterostructures. We considered three types of GO, including epoxy only, hydroxyl only and both epoxy and hydroxyl on the GO surface. Our results show that the interlayer binding energy per WS2 unit increases from 0.117 eV to 0.214 eV as the surface oxygen concentration of GO increases. The band gap of the WS2/GO heterostructures can be efficiently tuned in a wide range from 0.13 eV to 1.91 eV by changing the oxygen functionalities and the concentration. The spatial separation of the conduction band minimum and valence band maximum is observed, which are distributed in different layers. In addition, the work function of WS2 can also be modulated by GO in the range of 4.09 eV to 6.34 eV, which potentially increases the carrier concentration and broadens the applications of WS2 and other transition metal dichalcogenide materials in optoelectronic devices.
Co-reporter:Min Li, Lu Wang, Ningning Yu, Xiaotian Sun, Tingjun Hou and Youyong Li
Journal of Materials Chemistry A 2015 vol. 3(Issue 15) pp:3645-3649
Publication Date(Web):25 Feb 2015
DOI:10.1039/C5TC00209E
Based on density functional theory (DFT) calculations, we have constructed and investigated different types of single-side hydrogenated graphene (SSHG) structures from their structural motifs. The structural stability and electronic properties of these SSHG structures are extensively analyzed and compared with the reported structures. The single-side hydrogenation causes a severe bending in graphene at high H coverage, which leads to a greater formation energy with increasing H coverage. Among the SSHG structures that we have considered, the configurations with H attached along the armchair direction show the lowest formation energies due to a relatively small buckling compared to other configurations. Moreover, only the armchair hydrogenated graphene opens a band gap near the Fermi level, and the band gap can be modulated from zero to 1.44 eV by varying the H coverage from zero to 50%. Our results suggest an efficient way to prepare graphene-based materials and devices with suitable band gaps.
Co-reporter:Ningning Yu, Lu Wang, Min Li, Xiaotian Sun, Tingjun Hou and Youyong Li
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 17) pp:11700-11704
Publication Date(Web):25 Mar 2015
DOI:10.1039/C5CP00161G
Molybdenum disulfide (MoS2), a kind of graphene-like, two-dimensional material, has attracted great interest because of its unique properties and potential applications in electronics and sensors. In this paper, first-principle calculations and grand canonical Monte Carlo (GCMC) simulations are performed and used to show that the MoS2 layer is efficient at absorbing non-polar gases. Compared with the popular gas sorbents (metal organic frameworks and carbon-based materials), MoS2 has additional advantages, including large surface to volume ratio and tunable properties. The non-polar gas [carbon dioxide (CO2) and methane (CH4)] adsorption on the MoS2 layer with and without vacancies has been systematically studied. The perfect MoS2 shows little or no adsorption for CO2 and CH4 molecules, but the MoS2 with a single S vacancy and double S vacancies exhibits an excellent adsorption ability for CO2 and CH4 gases. The adsorption energies were 65 kJ mol−1 for CO2 and 47 kJ mol−1 for CH4 (van der Waals-D2), respectively. An orbital coupling between the p orbital of the CO2 (or CH4) molecule and the d orbital of the Mo atom was observed. GCMC simulation results show that MoS2 with a single S vacancy could absorb 42.1 wt% of CO2 and 37.6 wt% of CH4 under a pressure of 80 bar at room temperature. The results given in this paper indicate that monolayer MoS2 with defects is a highly efficient absorbent for non-polar gases.
Co-reporter:Shuo Deng
The Journal of Physical Chemistry C 2015 119(52) pp: 28783-28788
Publication Date(Web):December 2, 2015
DOI:10.1021/acs.jpcc.5b10354
On the basis of first-principles calculations, we have systematically studied the adsorption and diffusion of Li ions on monolayer MnO2 and compared with other transition metal dichalcogenides (TMDs) and transition metal dioxides (TMOs). Monolayer MnO2 shows a relatively high Li adsorption energy of 4.37 eV and low Li diffusion barrier of 0.148 eV. The electronic analysis indicates that the electron transferred from Li to the empty orbital of the O atom and there is some orbital coupling between the s orbital of the Li atom and the pz orbital of the O atom in MnO2. Due to Li adsorption on both sides of the MnO2 layer, the theoretical Li storage capacity reaches as high as 616 mAh/g. Our results demonstrated that, compared to other two-dimensional (2D) nanomaterials, monolayer or few-layer MnO2 exhibits excellent performance on Li storage capacity and diffusion rate and is believed to be a promising electrode material for high-capacity Li ion batteries.
Co-reporter:Min Li, Lu Wang, Ningning Yu, Xiaotian Sun, Tingjun Hou and Youyong Li
Journal of Materials Chemistry A 2015 - vol. 3(Issue 15) pp:NaN3649-3649
Publication Date(Web):2015/02/25
DOI:10.1039/C5TC00209E
Based on density functional theory (DFT) calculations, we have constructed and investigated different types of single-side hydrogenated graphene (SSHG) structures from their structural motifs. The structural stability and electronic properties of these SSHG structures are extensively analyzed and compared with the reported structures. The single-side hydrogenation causes a severe bending in graphene at high H coverage, which leads to a greater formation energy with increasing H coverage. Among the SSHG structures that we have considered, the configurations with H attached along the armchair direction show the lowest formation energies due to a relatively small buckling compared to other configurations. Moreover, only the armchair hydrogenated graphene opens a band gap near the Fermi level, and the band gap can be modulated from zero to 1.44 eV by varying the H coverage from zero to 50%. Our results suggest an efficient way to prepare graphene-based materials and devices with suitable band gaps.
Co-reporter:Xiao Yuan, Mingye Yang, Lu Wang and Youyong Li
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 21) pp:NaN13854-13854
Publication Date(Web):2017/05/02
DOI:10.1039/C7CP01727H
Based on the first-principles calculations, we systematically studied the structures and electronic properties of two-dimensional (2D) transition metal dichalcogenide (TMD) alloys with half-to-half mixing of S and Se. Using the chemical potentials of S and Se, the energetic phase diagrams for both single phases and mixed phases of TMD were constructed. A new heterolayer structure (for Sc and Ti) and alternating structure (for Cr, Mn, Fe, Zr, Mo, and W) were proposed for the first time, which were thermodynamically stable for MSSe alloys under the S-poor (relatively low chemical potential of S) and Se-rich (relatively high chemical potential of Se) conditions, and further compared with the disordered structures. Moreover, band gaps, carrier effective mass, and work functions were calculated for these stable mixed phases. Compared to the single phases of MS2 and MSe2, MSSe alloys showed superior electronic properties including tunable band gaps and work functions. Importantly, the significantly reduced effective mass of the carriers in the MSSe alloys may induce higher carrier mobility, providing better performance of TMD materials in electronic devices.
Co-reporter:Xiaotian Sun, Yunxia Liu, Zhigang Song, Yongdan Li, Weizhou Wang, Haiping Lin, Lu Wang and Youyong Li
Journal of Materials Chemistry A 2017 - vol. 5(Issue 17) pp:NaN4166-4166
Publication Date(Web):2017/03/24
DOI:10.1039/C7TC00306D
Defects are unavoidable during the synthesis of materials, especially for two-dimensional (2D) nanomaterials. They are usually seen as detrimental to device properties, but sometimes bring about new beneficial effects. In order to clarify the influence of defects on the structural and electronic properties, we have performed first-principles calculations to systematically investigate the structural stability, mobility and electronic properties of typical point defects in 2D arsenene (h-As), antimonene (h-Sb) and antimony arsenide (h-AsSb), including the Stone–Wales defects, single vacancies (SVs), double vacancies (DVs) and adatoms. To provide visual guidance for experimental observations, scanning tunnelling microscopy (STM) images are simulated. Compared to defects in graphene and silicene, these defects form more easily with lower formation energies, and SVs can diffuse very quickly to the edges with a lower diffusion barrier of less than 1 eV. Monolayer arsenene, antimonene and antimony arsenide are indirect band gap semiconductors, and the defective structures significantly reduce the band gaps. Most of the SV and adatom defects carry magnetic moments due to the dangling bonds resulting from the absent or extra atom. Our present results have demonstrated that the point defects induce significant effects on the electronic properties of pristine arsenene, antimonene and the antimony arsenide alloy, which should be considered in their future applications.
Co-reporter:Mingye Yang, Lu Wang, Tingjun Hou and Youyong Li
Journal of Materials Chemistry A 2017 - vol. 5(Issue 1) pp:NaN207-207
Publication Date(Web):2016/11/29
DOI:10.1039/C6TC04487E
Two-dimensional nanomaterials have attracted extensive interest and their heterostructures possess excellent electronic and optical properties suitable for various applications. Based on first-principles calculations, we investigated the structural stability and electronic properties of WS2 and graphene oxide (GO) heterostructures. We considered three types of GO, including epoxy only, hydroxyl only and both epoxy and hydroxyl on the GO surface. Our results show that the interlayer binding energy per WS2 unit increases from 0.117 eV to 0.214 eV as the surface oxygen concentration of GO increases. The band gap of the WS2/GO heterostructures can be efficiently tuned in a wide range from 0.13 eV to 1.91 eV by changing the oxygen functionalities and the concentration. The spatial separation of the conduction band minimum and valence band maximum is observed, which are distributed in different layers. In addition, the work function of WS2 can also be modulated by GO in the range of 4.09 eV to 6.34 eV, which potentially increases the carrier concentration and broadens the applications of WS2 and other transition metal dichalcogenide materials in optoelectronic devices.
Co-reporter:Ningning Yu, Lu Wang, Min Li, Xiaotian Sun, Tingjun Hou and Youyong Li
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 17) pp:NaN11704-11704
Publication Date(Web):2015/03/25
DOI:10.1039/C5CP00161G
Molybdenum disulfide (MoS2), a kind of graphene-like, two-dimensional material, has attracted great interest because of its unique properties and potential applications in electronics and sensors. In this paper, first-principle calculations and grand canonical Monte Carlo (GCMC) simulations are performed and used to show that the MoS2 layer is efficient at absorbing non-polar gases. Compared with the popular gas sorbents (metal organic frameworks and carbon-based materials), MoS2 has additional advantages, including large surface to volume ratio and tunable properties. The non-polar gas [carbon dioxide (CO2) and methane (CH4)] adsorption on the MoS2 layer with and without vacancies has been systematically studied. The perfect MoS2 shows little or no adsorption for CO2 and CH4 molecules, but the MoS2 with a single S vacancy and double S vacancies exhibits an excellent adsorption ability for CO2 and CH4 gases. The adsorption energies were 65 kJ mol−1 for CO2 and 47 kJ mol−1 for CH4 (van der Waals-D2), respectively. An orbital coupling between the p orbital of the CO2 (or CH4) molecule and the d orbital of the Mo atom was observed. GCMC simulation results show that MoS2 with a single S vacancy could absorb 42.1 wt% of CO2 and 37.6 wt% of CH4 under a pressure of 80 bar at room temperature. The results given in this paper indicate that monolayer MoS2 with defects is a highly efficient absorbent for non-polar gases.



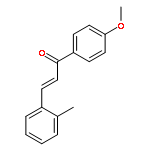
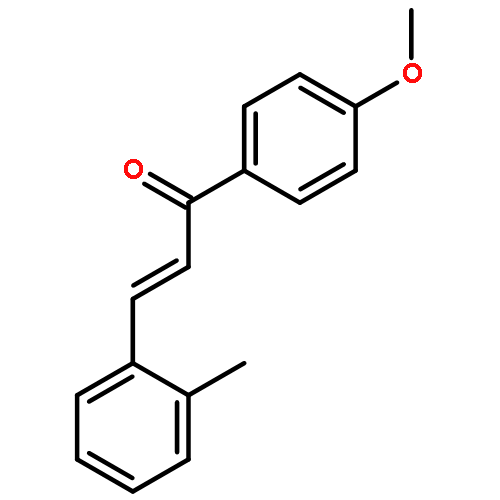












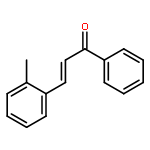
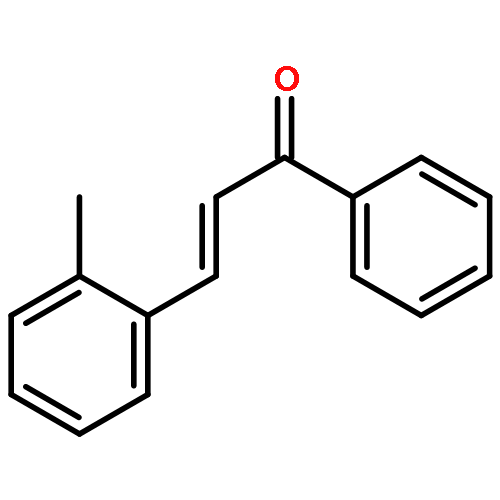















![[4-(2-HYDROXY-2-METHYLPROPYL)PHENYL]ACETIC ACID](/data/chemimg/99000/572-59-8.png)
![[4-(2-HYDROXY-2-METHYLPROPYL)PHENYL]ACETIC ACID](/data/chemimg/99000/572-59-8_b.png)





