Co-reporter:Shaheen Shah, Ce Hao
Journal of Environmental Sciences 2017 Volume 57(Volume 57) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.jes.2016.09.023
Sulfamethoxypyridazine (SMP) is one of the commonly used sulfonamide antibiotics (SAs). SAs are mainly studied to undergo triplet-sensitized photodegradation in water under natural sunlight with other coexisting aquatic environmental organic pollutants. In this work, SMP was selected as a representative of SAs. We studied the mechanisms of triplet-sensitized photodegradation of SMP and the influence of selected dissolved inorganic matter, i.e., anions (Br−, Cl−, and NO3−) and cations ions (Ca2 +, Mg2 +, and Zn2 +) on SMP photodegradation mechanism by quantum chemical methods. In addition, the degradation mechanisms of SMP by hydroxyl radical (OH) were also investigated. The creation of SO2 extrusion product was accessed with two different energy pathways (pathway-1 and pathway-2) by following two steps (step-I and step-II) in the triplet-sensitized photodegradation of SMP. Due to low activation energy, the pathway-1 was considered as the main pathway to obtain SO2 extrusion product. Step-II of pathway-1 was measured to be the rate-limiting step (RLS) of SMP photodegradation mechanism and the effect of the selected anions and cations was estimated for this step. All selected anions and cations promoted photodegradation of SMP by dropping the activation energy of pathway-1. The estimated low activation energies of different degradation pathways of SMP with OH radical indicate that OH radical is a very powerful oxidizing agent for SMP degradation via attack through benzene derivative and pyridazine derivative ring.Download high-res image (185KB)Download full-size image
Co-reporter:Hongyu Jing, Suzhen Ren, Yantao Shi, Xuedan Song, Ying Yang, Yanan Guo, Yonglin An, Ce Hao
Electrochimica Acta 2017 Volume 226(Volume 226) pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.electacta.2016.12.190
This work proposes a mild and environmentally-friendly approach to prepare a highly efficient functional graphene (termed as AGO-hv) using methods of ozone oxidation, solvothermal synthesis, and photoreduction. The use of ozone oxidation in the first step can effectively increase the interlaminar distance between graphite oxide sheets, and create active sites for nucleophilic attack on the epoxy carbon from ammonia. The amino groups were successfully grafted on the surface of graphene as evidenced by the amidation reaction, with a maximum nitrogen content of 10.46 wt% and a C/N molar ratio of 8.46. After further photoreduction of the aminated graphite oxide (AGO), the residual oxygen functionalities, such as C-OH, were effectively removed and the conductivity of the graphene sheet was further recovered. The dye-sensitized solar cell (DSC) exhibited a power conversion efficiency (PCE) of 7.51% based on AGO-hv counter electrode (CE), close to that of Pt counterpart (7.79%). The experimental results indicated that the amidation and photoreduction processes were significantly facilitated by the initial ozonization of graphene oxide, and this process significantly improved the electrochemical activity and the conductivity of graphene oxide. Density functional theory (DFT) calculations revealed that AGO-hv had the lowest ionization energy (a better electron-donating ability) and also suitable binding energy with I atoms as well. The combination of ozonization, amination and photoreduction is an efficient route to obtain electrocatalysts with desired compositional distributions and performance for triiodide reduction reaction in DSCs.
Co-reporter:Guanghao Meng, Yantao Shi, Xuedan Song, Min Ji, ... Ce Hao
Current Applied Physics 2017 Volume 17, Issue 10(Volume 17, Issue 10) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.cap.2017.06.004
•Transport orientations consistency parallel to (010), (101) and (111) crystal planes.•Carrier mobility is the highest in (010) crystal plane.•Inconsistency was found that parallel to (100), (110), (011) and (001) crystal planes.All-solid-state organic-inorganic halide perovskite solar cells (PSCs) have attracted wide attention due to the rapid progress of power conversion efficiency in recent years. Hole transport material (HTM) in PSCs plays the role of extracting and transporting photo-excited holes. Anisotropy of carrier mobility is one important property for semiconductors, however, which still remains unclear for the dominant HTM spiro-OMeTAD used in PSCs. Based on Density Functional Theory (DFT) and Marcus theory, we for the first time conducted investigations on the anisotropy of carrier mobility along representative crystal planes of spiro-OMeTAD by recombination energy λ and electronic coupling integral V. Results indicate that the holes and electrons show transport orientations consistency parallel to the (010), (101) and (111) crystal planes while inconsistency was found parallel to (100), (110), (011) and (001) crystal planes (with an angle ranged from 40° to 70° between the hole and electron transport directions). Our work embodies the theoretical significance of controllable and oriented fabrication of HTM in PSCs.Download high-res image (235KB)Download full-size image
Co-reporter:Suxia Liang, Botao Qiao, Xuedan Song, Ce Hao, Aiqin Wang, Tao Zhang, Yantao Shi
Nano Energy 2017 Volume 39(Volume 39) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.nanoen.2017.06.036
•The role of Pt single-atom well illustrated both experimentally and theoretically.•A small amount of Pt raised the power conversion efficiency of DSCs immensely.•Single atom Pt could enhance the donating ability of catalysts.•Single atom Pt 5d orbital was accounted for the good catalytic activity.Although single-atom catalysts (SACs) that bridge homogeneous and heterogeneous catalysis exhibit excellent performance in various reactions, only a few examples have reported the use of SACs in electrocatalysis, especially in new types of photovoltaics. This work focused on the association between SAC Pt1/FeOx and the electrocatalysis in hybrid photovoltaics, with the role of single-Pt atom in facilitating triiodide (I3-) catalytic reduction and enhancing the conversion efficiency of dye-sensitized solar cells. Even with an extremely low dispersion density of one Pt atom per 100 nm2 (the atomic ratio between Pt and Fe is 1:12214), the conversion efficiency could be enhanced by 69.3% compared to bare FeOx. DFT calculation indicated that ionization potential (IP), which was responsible for the rate-determining step, decreased with the anchor of single-Pt atoms on an oxygen-terminated Fe2O3(001) slab, thereby the electron-donating ability of catalysts was enhanced. The interaction between I- and O3- terminated Pt1/Fe2O3(001) showed that charge transfer occurred mainly between I and Pt atoms. Single atom Pt played a powerful role in triiodide (I3-) catalytic reduction, since its 5d orbital interacted with the support Fe2O3, accompanied with much more concentrated electronic states and higher density of the occupied states of Pt1/Fe2O3(001) around the Fermi energy.The association between SACs Pt1/FeOx and electrocatalysis in hybrid photovoltaics, together with the powerful role of Pt single-atom in facilitating triiodide (I3-) catalytic reduction and enhancing the conversion efficiency are well illustrated both experimentally and theoretically.Download high-res image (338KB)Download full-size image
Co-reporter:Xiangbin Cao, Jianhui Liu, Pan Hong, Guanglan Li, Ce Hao
Journal of Photochemistry and Photobiology A: Chemistry 2017 Volume 346(Volume 346) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.jphotochem.2017.05.035
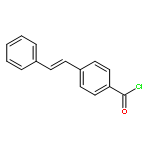
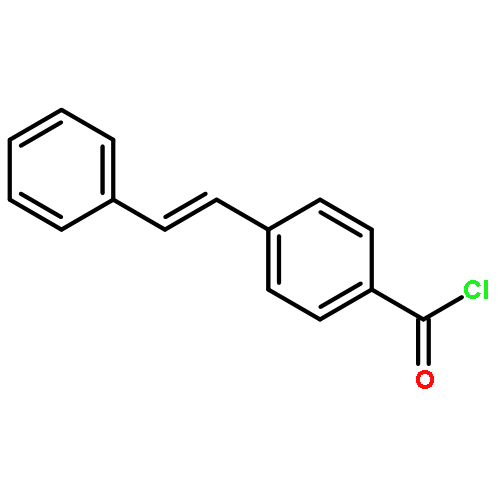
•Two novel styrylcyanine dyes were developed based on ICT.•The probes exhibited red-emission and large Stokes shift.•The probes displayed high sensitivity to viscosity.•The fluorescence intensity was affected by intramolecular rotation.Based on the mechanism of intramolecular charge transfer (ICT), two new styrylcyanine dyes, DPA-1 and DPA-2, composed of an electron-rich N-phenylaniline and a cationic benzothiazene connected with ethylene(s) bridge, were designed and synthesized. These two dyes exhibited red-emission (653/607 nm) and simultaneously impressive large Stokes shift (111/92 nm) due to intramolecular charge transfer effect, twisted geometry and extended conjugation system. When their solution viscosity increased from 1.01 cP to 234 cP in the water-glycerol system, the fluorescence intensity of the synthetic dyes was enhanced by 81-fold and 64-fold, respectively. Additionally, a favorable linear relationship between the fluorescence intensity and the environmental viscosity was observed within the above viscosity range for the obtained samples (R2 > 0.99), which led to the establishment of a method for the quantitative determination of the solution viscosity. Such dyes with improved photophysical properties, including emission wavelength and Stokes shift, could be used as promising candidates for intracellular viscosity detection. Moreover, the mechanism of fluorescence emission of the resultant products toward viscosity was further investigated. Of the two probes studies, DPA-1 is better than DPA-2 in terms of emission wavelength, Stokes shift and the sensitivity of the fluorescence intensity to viscosity.Download high-res image (121KB)Download full-size image
Co-reporter:Hongyu Jing, Xuedan Song, Suzhen Ren, Yantao Shi, Yonglin An, Ying Yang, Mingqiao Feng, Shaobo Ma, Ce Hao
Electrochimica Acta 2016 Volume 213() pp:252-259
Publication Date(Web):20 September 2016
DOI:10.1016/j.electacta.2016.07.129
In this work, a facile one-step approach is reported for using ZIF-67 as a sacrificial template in the synthesis of a counter electrode (CE) catalyst for dye-sensitized solar cells (DSCs). Porous nanocomposites of Co, CoO and N-doped graphitic carbon were synthesized by controlling the carbonization temperature of the templates in a N2 atmosphere. The characterization of the structure of the products indicated that cobalt nanoparticles were embedded in an N-doped graphitic carbon matrix, (a core-shell structure termed Co@NGC) while cobalt and cobalt oxide nanoparticles were exposed on the external surface of the carbon (termed Co/CoO). In particular, the chemical stability of the nanostructure of the Co@NGC was superior to Co/CoO with respect to etching by strong acids such as hydrochloric acid (HCl, 0.1 M). The DSC performance of ZIF-67-850 (pyrolyzed at 850 °C) employed as a CE resulted in a photoelectric conversion efficiency (PCE) of 7.92%, which was close to a Pt CE (8.18%) in the liquid I3−/I− redox couple electrolyte. The excellent performance of ZIF-67-850 can be attributed to the synergetic effects between the Co and CoO coupled with the nitrogen doped graphitic carbon. The cost-effective porous Co/CoO and Co@NGC nanocomposites exhibit great potential for application as high performance CE in solar cells.
Co-reporter:Guoqing Yang, Juanyuan Hao, Jie Cheng, Ning Zhang, Gaohong He, Fengxiang Zhang, Ce Hao
International Journal of Hydrogen Energy 2016 Volume 41(Issue 16) pp:6877-6884
Publication Date(Web):4 May 2016
DOI:10.1016/j.ijhydene.2016.03.067
•The OH− transport mechanism in quaternary ammonium functionalized polystyrene was studied by DFT.•There were two steps in the OH− transport process in anion exchange membrane.•The role of hydrogen bonding was investigated by DFT.•The rotation about ionic groups was studied in detail during OH− transport.A theoretical investigation of hydroxide ion transport mechanism in quaternary ammonium functionalized polystyrene (QAPS) anion exchange membrane (AEM) was studied by density functional theory (DFT). The results showed that there were two steps for OH− transferring through QAPS-AEM. The first step was the movement of OH− in water channel, which was induced by frequently forming and breaking of hydrogen bonds (H-bonds) between H2O and OH−. The second step was that OH− transferred across the quaternary ammonium (QA) groups by following the rotation about CC single bond, which was the rate-determining step for OH− transferring in QAPS-AEM. We presented that the ionic groups on the side chain of polymer with smaller space steric should provide higher ion conductivity due to their lower rotation energy barriers.
Co-reporter:Peipei Wang, Xuedan Song, Zhengyan Zhao, Lei Liu, Wensheng Mu, Ce Hao
Chemical Physics Letters 2016 Volume 661() pp:257-262
Publication Date(Web):16 September 2016
DOI:10.1016/j.cplett.2016.06.085
Highlights
- •
Luminescent mechanism of LMOF1 is the electron transfer from ligand to ZnO quantum dot.
- •
Intermolecular electron transfer occurred when LMOF1 interacts with the electron-withdrawing nitro-explosives.
- •
There was no significant intermolecular electron transfer when LMOF1 interacts with the electron-withdrawing nitro-explosives.
- •
In the excited state, the intermolecular hydrogen bondings of LMOF1-NB are strengthened, while those of LMOF1-TO are weakened.
Co-reporter:Hong-Yu Cao, Duan-Hui Si, Qian Tang, Xue-Fang Zheng, Ce Hao
Computational and Theoretical Chemistry 2016 Volume 1081() pp:18-24
Publication Date(Web):1 April 2016
DOI:10.1016/j.comptc.2016.01.012
•Introducing the carbon–nitrogen-swap strategy onto the porphyrin skeleton effectively varied the multi-band photon absorption.•The unsymmetrical neo-confused porphyrin (NeoCP) and Ni coordinated NeoCP showed broader and better visible light absorption capacity.•The light absorption performance of NeoCP derivatives varied in different polarity solvents.Unsymmetrical porphyrin derivatives and metal-coordinated porphyrin have received great attention in potential application for their intriguing photo-physical and photo-chemical properties. Density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations were applied to simulate the molecular and electronic structures with the electronic absorption spectra of the unsymmetrical neo-confused porphyrin (NeoCP) derivatives. Molecular structure and fragment charge distribution results revealed that the carbon–nitrogen-swap structure lead to the redistribution of charges and the differentiation of inner hydrogen in the Neo-confused Porphyrin ring. Introducing the unsymmetrical strategy onto the porphyrin skeleton effectively varied the energy levels of molecular orbitals, resulting in significant multi-band photon absorption and Soret band splitting for the porphyrin derivatives. Ni coordinated NeoCP (NiNeoCP) showed broader visible light absorption capacity and better light absorption performance due to the contribution of Ni atom. The blue-shifted Q bands illuminate that the light absorption performance of NeoCP and NiNeoCP varies in different polarity solvents based on the integral equation formalism polarizable continuum model (IEF-PCM) results. These theoretical researches would be conducive to the molecular design of novel multi-band photon absorption porphyrin derivatives.
Co-reporter:Lei Liu, Xiaofang Chen, Jieshan Qiu and Ce Hao
Dalton Transactions 2015 vol. 44(Issue 6) pp:2897-2906
Publication Date(Web):07 Jan 2015
DOI:10.1039/C4DT03185G
Luminescent metal–organic frameworks (LMOFs) have emerged as a group of new and very promising optic sensors in the detection of explosives. However, fundamental understanding of the sensing mechanisms on these materials is still immature and detailed investigations are needed. In this contribution, density functional theory (DFT) and time-dependent density functional theory (TD-DFT) are applied to reveal the underlying principles for the sensing mechanism by comprehensively studying the analyte–sensor interactions. Three molecules namely nitrobenzene, benzene and acetone are chosen as analytes while a newly reported explosives-detecting LMOF [Zn2(L)(bipy)(H2O)2]·(H2O)3(DMF)2 is chosen as the sensor. Roles of two fundamental weak interactions namely hydrogen bonding interaction and π–π stacking interaction are clarified for the first time. By studying both the periodic crystal models and cluster models we obtained an in-depth understanding of the detecting mechanism from the view of electronic coupling. We find that intermolecular electron transfer is the inducement for the luminescence quenching detection of explosives. A brand new pathway for this electron transfer process is proposed for the first time. Most significantly, we discover that the hydrogen bond shows multi-functions during the detecting processes which, on the one hand, serves as the electron transfer bridge, and on the other hand, reinforces the π–π stacking. This cooperative effect of the two weak forces inside MOFs is investigated for the first time, which not only provides valuable insights into the understanding of the analyte–sensor interactions inside the sensors but also offers useful guidance in the design of MOF sensors to achieve high sensitivity.
Co-reporter:Zhengyan Zhao, Juanyuan Hao, Xuedan Song, Suzhen Ren and Ce Hao
RSC Advances 2015 vol. 5(Issue 61) pp:49752-49758
Publication Date(Web):29 May 2015
DOI:10.1039/C5RA07373A
Density functional theory and time-dependent density functional theory methods have been used to investigate the hydrogen bonding between the Metal–organic framework [Zn2(H2L)(2,2′-bpy)2(H2O)]n and formaldehyde in the electronically excited state. The calculated geometric configuration, 1H NMR chemical shift and IR spectra of the hydrogen-bonded complex demonstrated that the hydrogen bond was strengthened in the excited state S1. The strengthening of the hydrogen bond in the S1 state would lead to a luminescence decreasing phenomenon of [Zn2(H2L)(2,2′-bpy)2(H2O)]n, and the fluorescent rate constant of [Zn2(H2L)(2,2′-bpy)2(H2O)]n was decreased when encapsulating formaldehyde into it. Taken together, these results indicated that [Zn2(H2L)(2,2′-bpy)2(H2O)]n could be used for the detection of formaldehyde.
Co-reporter:Juanyuan Hao, Fengyu Li, Hongjiang Li, Xiaoyu Chen, Yuyan Zhang, Zhongfang Chen and Ce Hao
RSC Advances 2015 vol. 5(Issue 43) pp:34383-34389
Publication Date(Web):08 Apr 2015
DOI:10.1039/C4RA16236F
Relativistic density functional theory (DFT) computations were performed to investigate the dynamic motion of an encapsulated Lu pair inside a C76(Td) cage. The results revealed that the lowest-energy configuration of Lu2@C76(Td) adopts C2 symmetry; four electrons are transferred to the outer carbon cage and the two encapsulated Lu atoms form a metal–metal single bond (with an electronic structure of Lu24+@C764−), and the good electron delocalization in the C764−(Td) cage partially contributes the thermodynamic preference of Lu2@C76(Td). The rather small barrier (3.2 kcal mol−1) for Lu2 atoms to hop from one stable site to another leads to flexible motion of the Lu pair inside the parent fullerene cage, and the Oh symmetrical motion trajectory of two Lu atoms is consistent with the STM image. The computed 13C NMR spectrum with this trajectory also agrees well with the experimental results.
Co-reporter:Xuedan Song, Zhiqiao Liu, Yantao Shi, Juanyuan Hao, Jieshan Qiu, Ce Hao
Chemical Physics 2015 Volume 446() pp:65-69
Publication Date(Web):13 January 2015
DOI:10.1016/j.chemphys.2014.10.014
Highlights
- •
TDDFT method is used to study the excited state hydrogen bonding.
- •
The origin of the luminescence of the MOF is LLCT.
- •
The hydrogen bond between CH3OH and MOF leads to enhanced luminescence.
Co-reporter:Xue-Dan Song, Se Wang, Ce Hao, Jie-Shan Qiu
Inorganic Chemistry Communications 2014 Volume 46() pp:277-281
Publication Date(Web):August 2014
DOI:10.1016/j.inoche.2014.06.003
•Reveal what kinds of MOF materials have good SO2 adsorption capacity.•Find some of MOFs show higher SO2 storage capacity than hydrotalcite-like materials.•ZIF-8 and Cu-BTC as the sulfur-transfer catalysts are expected to apply in industry.Sulfur dioxide (SO2) is responsible for the formation of acid rain and many other undesirable environmental and health hazards. Therefore, the capture or separation of sulfur dioxide is an important gas treatment process in industry. In this study, we predicted the adsorption of SO2 in ten metal–organic frameworks (MOFs), and investigated the effects of heat of adsorption, free volume, and surface area on sulfur dioxide uptake using grand canonical Monte Carlo simulations over a wide range of pressures. The molecular simulations of the SO2 adsorption isotherms in the MOF materials revealed that having high SO2 adsorption capacity and a suitable pore size greater than 0.4 nm is essential. In this pore size range, at low pressures the amount of SO2 adsorbed in these MOF materials is correlated with the heat of adsorption. Furthermore, at moderate pressures the amount of SO2 adsorbed is not well-correlated with the free volume and the accessible surface area, but the free volume and surface area are still clearly important characteristics in evaluating any potential SO2 absorption and storage absorbent. In addition, it was found that MOFs show higher SO2 storage capacity than most hydrotalcite-like materials at low pressure. This result suggests that MOF materials have potential as sulfur-transfer catalysts in industry.We investigated the effects of heat of adsorption, free volume and surface area on adsorption of SO2 in the MOFs using MC simulations. It demonstrates that some of the MOFs show higher SO2 storage capacity than most hydrotalcite-like materials. These MOFs as sulfur-transfer catalysts are expected to be applied in industry.
Co-reporter:Se Wang, Xuedan Song, Ce Hao, Zhanxian Gao, Jingwen Chen, Jieshan Qiu
Computational and Theoretical Chemistry 2014 Volume 1042() pp:49-56
Publication Date(Web):15 August 2014
DOI:10.1016/j.comptc.2014.03.024
•The photoreactivity of dioxins increases with an increase of halogenation degree.•The order of photoreactivity of Cl/Br of O8CDD/BDD follows: 2, 3 > 1, 4 positions.•The order of photoreactivity of Cl/Br of O8CDF/BDF follows: 1 > 2 > 3 > 4 position.•BDEC−Cl/Br of O8-dioxins at 4 and 6 positions were drastically decreased with Mg2+.•The predicted main photodehalogenation product of O8CDD/BDD is changed by Mg2+.Quantum chemical calculations studies were performed to investigate the underlying photodehalogenation mechanisms of polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/Fs) and polybrominated dibenzo-p-dioxins and dibenzofurans (PBDD/Fs), as well as Mg2+ effects. The bond dissociation energies of C−Cl/Br bonds, electronic absorption spectra, vibrational frequencies, and NBO charges of PCDD/Fs and PBDD/Fs were calculated by density functional theory and time-dependent density functional theory. Results show that the photodehalogenation reactivity of PCDD/Fs and PBDD/Fs increases with an increase of halogenation degree. Halogen atoms attached to the benzene ring with more halogens substituted have higher photoreactivity. For PCDD/BDDs, the photoreactivity of halogens at 2 and 3 positions are higher than that of halogens at 1 and 4 positions. For PCDF/BDFs without halogen substitution at 9 position, the order of photoreactivity of halogens follows: 2, 3 > 1, 4 position. However, for PCDFs/BDFs with halogen substitutions at 1 and 9 positions, the order of photoreactivity of Cl/Br atoms follows: 1 > 2 > 3 > 4 position. 1,2,3,4,6,7,9-H7CDD/BDD has been predicted to be the main photodehalogenation products of O8CDD/BDD. However, by the presence of Mg2+, the bond dissociation energies of C−Cl/Br bonds of O8CDD/BDD at 4 and 6 positions are drastically reduced and the main photodehalogenation products are changed to 1,2,3,4,6,7,8-H7CDD/BDD. Furthermore, the presence of Mg2+ significantly reduces the bond dissociation energies of C−Cl/Br bonds of O8CDF/BDF at 4 and 6 positions, but has no effects on the main photodehalogenation products. These findings provide an insight into the evaluation and prediction of photochemical behavior of persistent organic pollutants.Graphical abstract
Co-reporter:Yuhang Yao, Xuedan Song, Jieshan Qiu, and Ce Hao
The Journal of Physical Chemistry A 2014 Volume 118(Issue 32) pp:6191-6196
Publication Date(Web):July 28, 2014
DOI:10.1021/jp503722m
The hydrogen bond between formaldehyde and the luminescent metal–organic framework (MOF) [Zn(NH2bdc)(bix)]n was investigated using density functional theory and time-dependent density functional theory. The frontier molecular orbitals and electronic configuration demonstrate that the origin of the luminescence can be attributed to ligand-to-ligand charge transfer. Examination of the hydrogen bond behavior in the electronic excited state, with comparison of the electronic transition energies, bond distances, binding energy, 1H-NMR chemical shifts, and infrared spectra with those of the ground state, demonstrate that the hydrogen bond is stronger when in the electronic excited state. Strengthening of the hydrogen bond weakens the radioactive transition of [Zn(NH2bdc)(bix)]n, which thus leads to a luminescence decrease or quenching phenomenon, meaning that the luminescent MOF [Zn(NH2bdc)(bix)]n may be applied to the detection of formaldehyde.
Co-reporter:Xiao Sui, Min Ji, Xin Lan, Weihong Mi, Ce Hao, and Jieshan Qiu
Inorganic Chemistry 2013 Volume 52(Issue 10) pp:5742-5748
Publication Date(Web):May 3, 2013
DOI:10.1021/ic400924n
The electronically excited state and luminescence property of metal–organic framework Zn(3-tzba)(2,2′-bipy)(H2O)·nH2O have been investigated using the density functional theory (DFT) and time-dependent DFT (TDDFT). The calculated geometry and infrared spectra in the ground state are consistent with the experimental results. The frontier molecular orbitals and electronic configuration indicated that the origin of luminescence is attributed to a ligand-to-ligand charge transfer (LLCT). We theoretically demonstrated that the hydrogen bond H47···O5═C is weakened in the excited state S1; the weakening of the excited-state hydrogen bonding should be beneficial to the luminescence. To explore the effect of the water clusters on the luminescence, we studied four complexes Zn(3-tzba)(2,2′-bipy)(H2O)·3H2O, Zn(3-tzba)(2,2′-bipy)(H2O)·2H2O, Zn(3-tzba)(2,2′-bipy)(H2O)·H2O, and Zn(3-tzba)(2,2′-bipy)(H2O). The results reveal that the presence of water should play an important role in the emission characteristics of the MOF. Also, the UV–vis absorption and emission spectra of Zn(3-tzba)(2,2′-bipy)(H2O)·3H2O are in good agreement with the experimental results.
Co-reporter:Min Ji, Ce Hao, Dandan Wang, Hongjiang Li and Jieshan Qiu
Dalton Transactions 2013 vol. 42(Issue 10) pp:3464-3470
Publication Date(Web):21 Nov 2012
DOI:10.1039/C2DT32575F
We have investigated a new silver-based luminescent metal–organic framework (MOF) using density functional theory and time-dependent density functional theory methods. We theoretically demonstrated that the H⋯O hydrogen bond is strengthened and the Ag–O coordination bond is shortened significantly due to strengthening of the hydrogen bond in the S1 state. When the hydrogen bond is formed, the mechanism of luminescence changes from a ligand-to-metal charge transfer (LMCT) coupled with intraligand charge transfer (LLCT) to LMCT, and the luminescence is found to be enhanced.
Co-reporter:Lei Liu, Weihong Mi, Ce Hao, Jieshan Qiu
Inorganic Chemistry Communications 2013 Volume 31() pp:69-73
Publication Date(Web):May 2013
DOI:10.1016/j.inoche.2013.02.023
•We studied the luminescent mechanism of MOF Cu4(L)4•2EtOH using TDDFT method.•The weakening of hydrogen bond in S1 state restrained the non-radiative decay.•We explained how hydrogen and coordination bonds affect MOFs' luminescent behavior.Density functional theory (DFT) and time-dependent density functional theory (TDDFT) were employed to study the solvent dependent luminescence of metal–organic framework (MOF) Cu4(L)4•2EtOH (L = 5-(4-pyridyl) tetrazole). The truncated representative fragment of Cu4(L)4•2EtOH 1 and its solvated form 2 in ethanol were employed for the computation. The structures of 1 and 2 in S0, S1 and T1 states were fully optimized. Based on our frontier molecular orbital and the electronic configuration analysis of the optimized ground state structures of 1 and 2, the ligand-to-metal charge transfer (LMCT) luminescence was confirmed and the charge transfer pathway during the luminescent process was proved to be elongated by the hydrogen bond which led to the luminescence enhancing effect. Besides, the strengthening of hydrogen bond in S1 and T1 states, proved by our TDDFT calculation, enlarged the HOMO-LUMO energy gap which inhibited non-radiative decay thus in favor of luminescence. Particularly, the behaviors of coordination bonds in S1 and T1 states were investigated. The weakening of hydrogen bond greatly strengthened the coordination bond Cu1―N1 in S1 and T1 states. The strengthening of this coordination bond confined the charge transfer from Cu1 to Cu2 which further enhanced the luminescence.The electronic configurations and frontier molecular orbitals of MOF Cu4(L)4•2EtOH were calculated. The results showed that the luminescent of this framework originated from ligand to metal charge transfer (LMCT).
Co-reporter:Dongxu Tian;Suzhen Ren
Journal of Molecular Modeling 2013 Volume 19( Issue 4) pp:1591-1596
Publication Date(Web):2013 April
DOI:10.1007/s00894-012-1703-x
The interaction between lanthanum atom (La) and C74 (D3h) was investigated by all-electron relativistic density function theory (DFT). With the aid of the representative patch of C74 (D3h), we studied the interaction between C74 (D3h) and La and obtained the interaction potential. Optimized structures show that there are three equivalent stable isomers, with La located about 1.7 Å off center. There is one transition state between every two stable isomers. According to the minimum energy pathway, the possible movement trajectory of La atoms in the C74 (D3h) cage was explored. The calculated energy barrier for La atoms moving from the stable isomer to the transition state is 18.4 kcal mol−1. In addition, the dynamic NMR spectra of La@C74 according to the trajectory was calculated.
Co-reporter:Yanfang Meng, Chunqing Zhang, Min Ji, Ce Hao, Jieshan Qiu
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2013 Volume 109() pp:14-22
Publication Date(Web):15 May 2013
DOI:10.1016/j.saa.2013.02.010
The luminescent metal organic framework (MOF), Cu2(L)2·MeOH (L = 5-(4-pyridyl)tetrazole), was studied using time-dependent density functional theory (TDDFT). A combination of frontier molecular orbitals and electronic configuration analysis revealed that the emission mechanism was a ligand to metal charge transition (LMCT) rather than a metal to ligand charge transfer (MLCT). Hydrogen bonding significantly changed the nature of the frontier orbital and the luminescence. Electronic transition energies predicted that the hydrogen bonding in excited state would become weaker with an electronic spectral blue-shift. The bond lengths, frequencies, and binding energies indicated weakening of the hydrogen bonding in the excited state, which can affect emissions in two ways, including: (i) a decrease in the electronic coupling between methanol and the motif and suppressing the occurrence of the photo-induced electron transfer (PET); and (ii) increasing the energy gap between S1 and S0, leading to radiative transition. Coordination bonding was also investigated in the excited state through bond lengths, frequencies, and bond orders. Coordination bonds were found to become stronger in the excited state leading to an enhancement of the luminescence.Graphical abstractHighlights► The luminescent metal organic framework was studied by TDDFT method. ► Ligand to metal charge transition mechanism was clarified. ► Excited-state hydrogen bond weakening induces spectral blue-shift. ► Photoinduced electron transfer decreases due to excited-state hydrogen bond weakening.
Co-reporter:Min Ji, Xin Lan, Zhenping Han, Ce Hao, and Jieshan Qiu
Inorganic Chemistry 2012 Volume 51(Issue 22) pp:12389-12394
Publication Date(Web):November 8, 2012
DOI:10.1021/ic301771b
The electronically excited state and luminescence property of metal–organic framework MOF-5 were investigated using relativistic density functional theory (DFT) and time-dependent DFT (TDDFT). The geometry, IR spectra, and UV–vis spectra of MOF-5 in the ground state were calculated using relativistic DFT, leading to good agreement between the experimental and theoretical results. The frontier molecular orbitals and electronic configuration indicated that the luminescence mechanism in MOF-5 follows ligand-to-ligand charge transfer (LLCT), namely, π* → π, rather than emission with the ZnO quantum dot (QD) proposed by Bordiga et al. The geometry and IR spectra of MOF-5 in the electronically excited state have been calculated using the relativistic TDDFT and compared with those for the ground state. The comparison reveals that the Zn4O13 QD is rigid, whereas the ligands BDC2– are nonrigid. In addition, the calculated emission band of MOF-5 is in good agreement with the experimental result and is similar to that of the ligand H2BDC. The combined results confirmed that the luminescence mechanism for MOF-5 should be LLCT with little mixing of the ligand-to-metal charge transfer. The reason for the MOF-5 luminescence is explained by the excellent coplanarity between the six-membered ring consisting of zinc, oxygen, carbon, and the benzene ring.
Co-reporter:Danyang Wu, Weihong Mi, Min Ji, Ce Hao, Jieshan Qiu
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2012 Volume 97() pp:589-593
Publication Date(Web):November 2012
DOI:10.1016/j.saa.2012.07.014
The hydrogen bonding in electronically excited-state of the metal-organic framework [CuCN·EIN] was studied using time-dependent density functional theory (TDDFT). The representative fragment of [CuCN·EIN] was employed for the computation. The geometric structures, binding energies and IR spectra in both ground state and electronically excited state S1 of the complex were computed using DFT and TDDFT methods to investigate excited-state hydrogen-bonding and coordination bonding, respectively. Based on the analysis of the frontier molecular orbitals and the electronic configuration of the complex, the ligand-to-metal charge transfer (LMCT) luminescence was confirmed. Furthermore, furcated hydrogen bonds are both strengthened in the S1 state slightly. And then, the strengthening of the hydrogen bonds in the S1 state goes against the charge transfer from ligand to metal and then should be in favor of the luminescence. In particular, we also discuss strengthening or weakening behavior of the coordination bonds in the S1 state for the first time. Based on the results of the bond lengths and vibration frequency of the coordination bond, we can conclude that the coordination bond Cu7–N8 is strengthened in the S1 state. And the strengthening of the coordination bond Cu7–N8 should also be in favor of the luminescence.Graphical abstractHighlights► The effect of furcated hydrogen bond on luminescent behavior is studied. ► TDDFT is used to study the excited-state hydrogen bonding. ► The effect of coordination bond on luminescent behavior is studied. ► Luminescence can be caused by the ligand-to-metal charge transfer (LMCT) form.
Co-reporter:Ce Hao, Hongjiang Li, Lijuan Guo, Shenmin Li, Jieshan Qiu
Computational and Theoretical Chemistry 2011 Volume 963(2–3) pp:314-318
Publication Date(Web):February 2011
DOI:10.1016/j.comptc.2010.10.033
This study investigates the interaction between C74 (D3h) and fluorine, and the potential energy surface of C74F radical. Our findings show that there are nine distinct isomers of C74F on the surface. The calculations on the structures and energies are further discussed thermodynamically using the density function theory method at the B3LYP/3-21G (d) level. In addition, the transition states, as well as reaction pathways of F transferring between different key points on C74 representative patch, are given to explore the possible reaction mechanism. Finally, the stability of C74F2 is discussed through the density functional-theory.
Co-reporter:Ningning Wei, Peng Li, Ce Hao, Rui Wang, Zhilong Xiu, Jingwen Chen, Peng Song
Journal of Photochemistry and Photobiology A: Chemistry 2010 Volume 210(Issue 1) pp:77-81
Publication Date(Web):5 February 2010
DOI:10.1016/j.jphotochem.2009.12.001
Intermolecular dihydrogen bonding in the electronically excited states of a phenol–diethylmethylsilane (DEMS) complex was studied theoretically using the time-dependent density functional theory (TDDFT) method. Analysis of the frontier molecular orbitals revealed a locally excited S1 state for the dihydrogen-bonded phenol–DEMS complex in which only the phenol moiety is electronically excited. The calculated infrared spectrum of the phenol–DEMS complex is quite different from that of previously studied S1 state of a dihydrogen-bonded phenol–borane-trimethylamine complex. The O–H and Si–H stretching vibrational modes appear as intense, sharp peaks for the S1 state which are slightly red-shifted compared with those predicted for the ground state. Upon electronic excitation to the S1 state, the O–H and Si–H bonds involved in the dihydrogen bond O–H⋯H–Si lengthen slightly, while the C–O bond shortens. The calculated H⋯H distance is significantly shorter in the S1 state than in the ground state. Thus, the intermolecular dihydrogen bond of the phenol–DEMS complex is stronger in the electronically excited state than in the ground state.
Co-reporter:Shuping Liu, Ce Hao, Shenmin Li, Zexin Wang
Applied Surface Science 2009 Volume 255(Issue 7) pp:4232-4238
Publication Date(Web):15 January 2009
DOI:10.1016/j.apsusc.2008.11.031
Co-reporter:Peng Jin, Ce Hao, Zhanxian Gao, Shengbai B. Zhang and Zhongfang Chen
The Journal of Physical Chemistry A 2009 Volume 113(Issue 43) pp:11613-11618
Publication Date(Web):July 16, 2009
DOI:10.1021/jp9019848
The geometries, electronic and spectroscopic properties of two representative endohedral derivatives of B80 fullerene, namely, La2@B80 and Sc3N@B80, and the possibility for their production were investigated by means of density functional computations. The very favorable binding energies suggest a considerable possibility to experimentally realize these novel endohedral metalloborofullerenes. Infrared absorption spectra and 11B nuclear magnetic resonance spectra were also computed to assist future experimental characterization.
Co-reporter:Shaopeng Sun, Jinlan Sun, Ce Hao, Shenmin Li
Journal of Molecular Structure: THEOCHEM 2009 Volume 901(1–3) pp:66-71
Publication Date(Web):15 May 2009
DOI:10.1016/j.theochem.2009.01.003
This study investigates the interaction between C70 and X (X = H and F), and the potential energy surface of C70X radical. Our findings show that there are five distinct isomers of C70X on the surface. The calculations on the structures and energies are further discussed thermodynamically using the density function theory method at the B3LYP/6-31G (d) level. In addition, the transition states, as well as reaction pathways of X (X = H and F) transferring between different key points on C70 representative patch, are given to explore the possible reaction mechanism. Finally, the stability of C70X2 is discussed through the density functional theory.
Co-reporter:Chen Liang, Jin Yang, Ce Hao, Shenmin Li, Yang Li, Yingfu Jin
Journal of Molecular Structure: THEOCHEM 2008 Volume 851(1–3) pp:342-347
Publication Date(Web):28 February 2008
DOI:10.1016/j.theochem.2007.11.035
The interaction between oxygen atom and C36 (D6h) has been investigated by the density function theory method at B3LYP/6-31G level. The calculated potential energy surface of C36O shows that there are eight possible stable isomers, of which the structures and energies are further discussed thermodynamically and kinetically. In addition, the transition states, as well as reaction pathways of oxygen transfer between different key points on C36 (D6h) representative patch are given to explore possible reaction mechanism.
Co-reporter:Yiyi Jia, Yantao Shi, Jieshan Qiu, Ce Hao
Journal of Energy Chemistry (September 2016) Volume 25(Issue 5) pp:861-867
Publication Date(Web):1 September 2016
DOI:10.1016/j.jechem.2016.07.002
For some specific catalytic reaction, how to construct active sites on two dimensional materials is of great scientific significance. Dye-sensitized solar cells (DSCs) can be viewed as one representative photovoltaics because in which liquid electrolyte with triiodide/iodide (I3−/I−) as redox couples are involved. In this study, amino-functionalized graphene (AFG) has been designed according to theoretically analyzing iodine reduction reaction (IRR) processes and rationally screening the volcanic plot. Then, such AFG has been successfully synthesized by a simple hydrothermal method and shows high electrocatalytic activity towards IRR when serving as counter electrode in DSCs. Finally, a high conversion efficiency of 7.39% by AFG-based DSCs was obtained, which is close to that using Pt as counter electrode.Amino-functionalized graphene (AFG) designed through rationally screening the volcanic plot demonstrates high electrocatalytic activity when serving as counter electrode in dye-sensitized solar cells.Download high-res image (157KB)Download full-size image
Co-reporter:Se Wang, Xuedan Song, Ce Hao, Zhanxian Gao, Jingwen Chen, Jieshan Qiu
Chemosphere (March 2015) Volume 122() pp:62-69
Publication Date(Web):1 March 2015
DOI:10.1016/j.chemosphere.2014.11.007
•Triplet-sensitized photolysis mechanism of SDZ was electron transfer or H transfer.•SDZ0 and SDZ− showed different triplet-sensitized photolysis routs.•Presence of Mg2+, Ca2+, or Zn2+ promoted triplet-sensitized photolysis of SDZ0.•Presence of Mg2+, Ca2+, or Zn2+ inhibited triplet-sensitized photolysis of SDZ−.Sulfadiazine (SDZ) mainly proceeds triplet-sensitized photolysis with dissolved organic matter (DOM) in the aquatic environment. However, the mechanisms underlying the triplet-sensitized photolysis of SDZ with DOM have not been fully worked out. In this study, we investigated the mechanisms of triplet-sensitized photolysis of SDZ0 (neutral form) and SDZ− (anionic form) with four DOM analogues, i.e., fluorenone (FL), thioxanthone (TX), 2-acetonaphthone (2-AN), and 4-benzoylbenzoic acid (CBBP), and three metal ions (i.e., Mg2+, Ca2+, and Zn2+) effects using quantum chemical calculations. Results indicated that the triplet-sensitized photolysis mechanism of SDZ0 with FL, TX, and 2-AN was hydrogen transfer, and with CBBP was electron transfer along with proton transfer (for complex SDZ0-CBBP2) and hydrogen transfer (for complex SDZ0-CBBP1). The triplet-sensitized photolysis mechanisms of SDZ− with FL, TX, and CBBP was electron transfer along with proton transfer, and with 2-AN was hydrogen transfer. The triplet-sensitized photolysis product of both SDZ0 and SDZ− was a sulfur dioxide extrusion product (4-(2-iminopyrimidine-1(2H)-yl)aniline), but the formation routs of the products for SDZ0 and SDZ− were different. In addition, effects of the metal ions on the triplet-sensitized photolysis of SDZ0 and SDZ− were different. The metal ions promoted the triplet-sensitized photolysis of SDZ0, but inhibited the triplet-sensitized photolysis of SDZ−.
Co-reporter:Shehnaz, Xuedan Song, Suzhen Ren, Ying Yang, ... Ce Hao
Journal of Energy Chemistry (January 2017) Volume 26(Issue 1) pp:182-192
Publication Date(Web):1 January 2017
DOI:10.1016/j.jechem.2016.11.013
One of the major challenges associated with fuel cells is the design of highly efficient electrocatalysts to reduce the high overpotential of the oxygen reduction reaction (ORR). Here we report Polyaniline (PANI) based micro/nanomaterials with or without transition metals, prepared by the emulsion polymerization and subsequent heat treatment. PANI microspheres with the diameter of about 0.7 µm have been prepared in basic (NH3 solution) condition, using two different types of surfactant (CTAB, SDS) as the stabilizer, ammonium persulphate (APS) as oxidant with aniline/surfactants molar ratio at 1/1 under the hydrothermal treatment. PANI nanorods, FePANI, and FeCoPANI have been synthesized in acidic (HCl) medium with aniline/surfactants molar ratio at 1/2 and polymerization carried out without stirring for 24 h. Products mainly FeCoPANI have shown high current density with increasing sweep rate and excellent specific capacitance 1753 F/g at the scan rate of 1 mV/s. Additionally, it has shown high thermal stability by thermogravimetric analysis (TGA). FePANI has been investigated for excellent performance toward ORR with four electron selectivity in the basic electrolyte. The PANI-based catalysts from emulsion polymerization demonstrate that the method is valuable for making non-precious metal heterogeneous electrocatalysts for ORR or energy storage and conversion technology.Polyaniline-based microspheres, nanorods with or without Fe, Fe–Co have been prepared through emulsion polymerization, and showed good interfacial contact and demonstrated high supercapacity and electrocatalytic activity especially for Fe–Co–PANI sample.Download high-res image (179KB)Download full-size image
Co-reporter:Min Ji, Ce Hao, Dandan Wang, Hongjiang Li and Jieshan Qiu
Dalton Transactions 2013 - vol. 42(Issue 10) pp:NaN3470-3470
Publication Date(Web):2012/11/21
DOI:10.1039/C2DT32575F
We have investigated a new silver-based luminescent metal–organic framework (MOF) using density functional theory and time-dependent density functional theory methods. We theoretically demonstrated that the H⋯O hydrogen bond is strengthened and the Ag–O coordination bond is shortened significantly due to strengthening of the hydrogen bond in the S1 state. When the hydrogen bond is formed, the mechanism of luminescence changes from a ligand-to-metal charge transfer (LMCT) coupled with intraligand charge transfer (LLCT) to LMCT, and the luminescence is found to be enhanced.
Co-reporter:Lei Liu, Xiaofang Chen, Jieshan Qiu and Ce Hao
Dalton Transactions 2015 - vol. 44(Issue 6) pp:NaN2906-2906
Publication Date(Web):2015/01/07
DOI:10.1039/C4DT03185G
Luminescent metal–organic frameworks (LMOFs) have emerged as a group of new and very promising optic sensors in the detection of explosives. However, fundamental understanding of the sensing mechanisms on these materials is still immature and detailed investigations are needed. In this contribution, density functional theory (DFT) and time-dependent density functional theory (TD-DFT) are applied to reveal the underlying principles for the sensing mechanism by comprehensively studying the analyte–sensor interactions. Three molecules namely nitrobenzene, benzene and acetone are chosen as analytes while a newly reported explosives-detecting LMOF [Zn2(L)(bipy)(H2O)2]·(H2O)3(DMF)2 is chosen as the sensor. Roles of two fundamental weak interactions namely hydrogen bonding interaction and π–π stacking interaction are clarified for the first time. By studying both the periodic crystal models and cluster models we obtained an in-depth understanding of the detecting mechanism from the view of electronic coupling. We find that intermolecular electron transfer is the inducement for the luminescence quenching detection of explosives. A brand new pathway for this electron transfer process is proposed for the first time. Most significantly, we discover that the hydrogen bond shows multi-functions during the detecting processes which, on the one hand, serves as the electron transfer bridge, and on the other hand, reinforces the π–π stacking. This cooperative effect of the two weak forces inside MOFs is investigated for the first time, which not only provides valuable insights into the understanding of the analyte–sensor interactions inside the sensors but also offers useful guidance in the design of MOF sensors to achieve high sensitivity.










![Benzenesulfonic acid,4-[2-[4-(dimethylamino)phenyl]diazenyl]-](http://img.cochemist.com/ccimg/600/502-02-3.png)
![Benzenesulfonic acid,4-[2-[4-(dimethylamino)phenyl]diazenyl]-](http://img.cochemist.com/ccimg/600/502-02-3_b.png)

