Co-reporter:Kenan Tokmic, Bailey J. Jackson, Andrea Salazar, Toby J. Woods, and Alison R. Fout
Journal of the American Chemical Society September 27, 2017 Volume 139(Issue 38) pp:13554-13554
Publication Date(Web):September 14, 2017
DOI:10.1021/jacs.7b07368
The selective hydrogenation of nitriles to primary amines using a bench-stable cobalt precatalyst under 4 atm of H2 is reported herein. The catalyst precursor was reduced in situ using NaHBEt3, and the resulting Lewis acid formed, BEt3, was found to be integral to the observed catalysis. Mechanistic insights gleaned from para-hydrogen induced polarization (PHIP) transfer NMR studies revealed that the pairwise hydrogenation of nitriles proceeded through a Co(I/III) redox process.
Co-reporter:Zachary Gordon, Michael J. Drummond, Ellen M. Matson, Justin A. Bogart, Eric J. Schelter, Richard L. Lord, and Alison R. Fout
Inorganic Chemistry May 1, 2017 Volume 56(Issue 9) pp:4852-4852
Publication Date(Web):April 10, 2017
DOI:10.1021/acs.inorgchem.6b03071
The derivatization of the imino-functionalized tris(pyrrolylmethyl)amine ligand framework, N(XpiR)3 (XLR; X = H, Br; R = cyclohexyl (Cy), phenyl (Ph), 2,6- diisopropylphenyl (DIPP)), is reported. Modular ligand synthesis allows for facile modification of both the primary and secondary coordination sphere electronics. The iron(II)–hydroxo complexes, N(XpiR)(XafaR)2Fe(II)OH (XLRFeIIOH), are synthesized to establish the impact of the ligand modifications on the complexes’ electronic properties, including their chemical and electrochemical oxidation. Cyclic voltammetry demonstrates that the Fe(II/III) redox couple spans a 400 mV range across the series. The origin of the shifted potential is explained based on spectroscopic, structural, and theoretical analyses of the iron(II) and iron(III) compounds.
Co-reporter:Joseph W. Nugent, Gabriel Espinosa Martinez, Danielle L. Gray, and Alison R. Fout
Organometallics August 14, 2017 Volume 36(Issue 15) pp:2987-2987
Publication Date(Web):August 2, 2017
DOI:10.1021/acs.organomet.7b00463
A bidentate monoanionic NHC-CAryl ligand framework was synthesized, and a host of Ni(II) complexes were prepared. Addition of isocyanides to these complexes led to the formation of NHC-η2-iminoacyl nickel complexes. These complexes were characterized by a suite of spectroscopic techniques, including X-ray crystallography. The η2-iminoacyl was shown to be displaced from the nickel center with oxidant and could then be reattached with reductant.
Co-reporter:Kenan Tokmic and Alison R. Fout
Journal of the American Chemical Society 2016 Volume 138(Issue 41) pp:13700-13705
Publication Date(Web):October 6, 2016
DOI:10.1021/jacs.6b08128
The reactivity of a CoI–H2 complex was extended toward the semihydrogenation of internal alkynes. Under ambient temperatures and moderate pressures of H2, a broad scope of alkynes were semihydrogenated using a CoI-N2 precatalyst, resulting in the formation of trans-alkene products. Furthermore, mechanistic studies using 1H, 2H, and para-hydrogen induced polarization (PHIP) transfer NMR spectroscopy revealed cis-hydrogenation of the alkyne occurs first. The Co-mediated alkene isomerization afforded the E-selective products from a broad group of alkynes with good yields and E/Z selectivity.
Co-reporter:Kenan Tokmic, Charles R. Markus, Lingyang Zhu, and Alison R. Fout
Journal of the American Chemical Society 2016 Volume 138(Issue 36) pp:11907-11913
Publication Date(Web):August 29, 2016
DOI:10.1021/jacs.6b07066
The synthesis of a cobalt dihydrogen CoI-(H2) complex prepared from a CoI-(N2) precursor supported by a monoanionic pincer bis(carbene) ligand, MesCCC (MesCCC = bis(mesityl-benzimidazol-2-ylidene)phenyl), is described. This species is capable of H2/D2 scrambling and hydrogenating alkenes at room temperature. Stoichiometric addition of HCl to the CoI-(N2) cleanly affords the CoIII hydridochloride complex, which, upon the addition of Cp2ZrHCl, evolves hydrogen gas and regenerates the CoI-(N2) complex. Furthermore, the catalytic olefin hydrogenation activity of the CoI species was studied by using multinuclear and parahydrogen (p-H2) induced polarization (PHIP) transfer NMR studies to elucidate catalytically relevant intermediates, as well as to establish the role of the CoI-(H2) in the CoI/CoIII redox cycle.
Co-reporter:Gabriel Espinosa Martinez; Cristian Ocampo; Yun Ji Park
Journal of the American Chemical Society 2016 Volume 138(Issue 13) pp:4290-4293
Publication Date(Web):March 25, 2016
DOI:10.1021/jacs.5b12827
This communication describes the two-electron oxidation of (DIPPCCC)NiX (DIPPCCC = bis(diisopropylphenyl-benzimidazol-2-ylidene)phenyl); X = Cl or Br) with halogen and halogen surrogates to form (DIPPCCC)NiX3. These complexes represent a rare oxidation state of nickel, as well as an unprecedented reaction pathway to access these species through Br2 and halogen surrogate (benzyltrimethylammonium tribromide). The NiIV complexes have been characterized by a suite of spectroscopic techniques and can readily reduce to the NiII counterpart, allowing for cycling between the NiII/NiIV oxidation states.
Co-reporter:Abdulrahman D. Ibrahim, Steven W. Entsminger, Lingyang Zhu, and Alison R. Fout
ACS Catalysis 2016 Volume 6(Issue 6) pp:3589
Publication Date(Web):May 5, 2016
DOI:10.1021/acscatal.6b01091
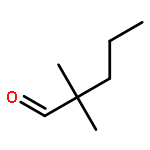
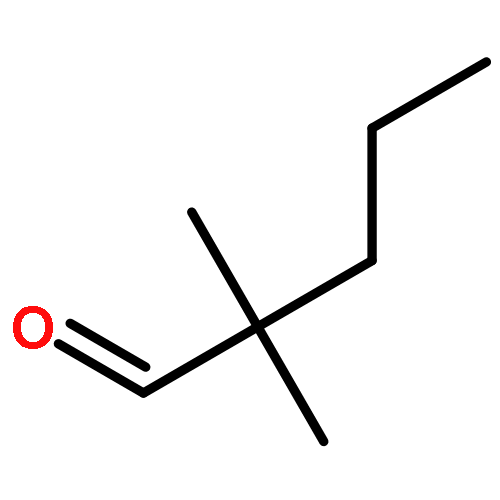
The hydrosilylation of alkene substrates bearing additional functionalities is difficult to achieve using earth-abundant catalysts and has not been extensively realized with both earth-abundant transition metals and tertiary silanes or hydrosiloxanes. Reported herein is a well-defined bis(carbene) cobalt(I)-dinitrogen complex for the efficient, catalytic anti-Markovnikov hydrosilylation of terminal alkenes, featuring a broad substrate scope. Alkenes containing hydroxyl, amino, ester, epoxide, ketone, formyl, and nitrile groups are selectively hydrosilylated in this reaction sequence. Multinuclear NMR studies of reactive intermediates gave insights into the mechanism.Keywords: alkenes; chemoselective; cobalt; hydrosiloxanes; hydrosilylation; tertiary silanes
Co-reporter:Abdulrahman D. Ibrahim, Kenan Tokmic, Marshall R. Brennan, Dongyoung Kim, Ellen M. Matson, Mark J. Nilges, Jeffery A. Bertke and Alison R. Fout
Dalton Transactions 2016 vol. 45(Issue 24) pp:9805-9811
Publication Date(Web):18 Jan 2016
DOI:10.1039/C5DT04723D
The synthesis and characterization of a series of cobalt complexes featuring a pincer bis(carbene) ligand of the meta-phenylene-bridged bis-N-heterocyclic carbene (ArCCC, Ar = 2,6-diispropylphenyl or mesityl) are reported. Cleavage of the aryl C–H bond of the ligand was achieved in a one-pot metalation procedure using Co(N(SiMe3)2)2(py)2, an equivalent of exogenous base, and trityl chloride to form the (DIPPCCC)CoCl2py complex. This species could be reduced to the Co(II) and Co(I)–N2 molecules with the appropriate equivalents of reductant. Subsequent generation of (MesCCC)CoI–III derivatives with the mesityl ligand proceeded in good yields. A suite of characterization techniques and the interconversion between all three oxidation states of the cobalt complexes is described.
Co-reporter:Courtney L. Ford;Yun Ji Park;Ellen M. Matson;Zachary Gordon
Science 2016 Vol 354(6313) pp:741-743
Publication Date(Web):11 Nov 2016
DOI:10.1126/science.aah6886
Biological inspiration for reduction
Microorganisms have evolved sophisticated enzymatic machinery to reduce perchlorate and nitrate ions. Although the energetics of the pathways are different, the heme-containing active sites of the corresponding reductase enzymes are remarkably similar. Ford et al. constructed an inorganic catalyst to mediate these reactions based on these active sites, using a nonheme iron complex. A secondary coordination sphere near the iron center aligned the nitrate or perchlorate oxyanions and formed an iron-oxo complex. Regenerating the catalyst in the presence of protons and electrons released water—a potentially much more sustainable process than reduction strategies that require the use of harsh reagents.
Science, this issue p. 741
Co-reporter:Yun Ji Park, Ellen M. Matson, Mark J. Nilges and Alison R. Fout
Chemical Communications 2015 vol. 51(Issue 25) pp:5310-5313
Publication Date(Web):06 Mar 2015
DOI:10.1039/C4CC08603A
Complexes containing manganese–oxygen bonds have been implicated in a variety of biological and synthetic processes. Herein, we describe the synthesis of a family of stable, high-spin trigonal bipyramidal manganese complexes of the electronically flexible ligand tris(5-cyclohexylimino-pyrrol-2-ylmethyl)amine [H3N(piCy)3] featuring apical water, hydroxyl, and oxo ligands. Terminal MnIII–O complexes are rare and the formation of this species was achieved from a variety of reagents including O2, PhIO and NO2−. Described herein is the preparation, structural and electronic properties of these manganese complexes.
Co-reporter:Ellen M. Matson, Yun Ji Park, Jeffery A. Bertke and Alison R. Fout
Dalton Transactions 2015 vol. 44(Issue 22) pp:10377-10384
Publication Date(Web):13 May 2015
DOI:10.1039/C5DT00985E
The syntheses of M(II) (M = Mn, Fe, Co) complexes bearing the tris(5-cycloaminoazafulvene-2-methyl)amine (H3N(afaCy)3) ligand in its datively coordinated, tautomeric form is reported. The metal–azafulvene complexes [N(afaCy)3M](OTf)2 are generated in high yields, featuring a secondary coordination sphere composed of amino moieties from the ligand platform. To investigate the ability of the hydrogen bonding network to support hydrogen-bond accepting, coordinating anions, pseudohalide derivatives, [N(afaCy)3MX](OTf) (X = NCS−, NCO−, N3−) were synthesized by exposure of [N(afaCy)3M](OTf)2 to an equivalent of the corresponding salt, [(nBu)4N](X). Structural characterization of the products reveals two isomorphs of the desired species. One complex features a single hydrogen bonding interaction with the pseudohalide, while the second compound has two H-bonds from the secondary coordination sphere to the coordinated anion. These complexes showcase the structural and electron flexibility of the ligand platform, presenting a scaffold capable of accessing a different number of hydrogen bonds for stabilizing a given moiety.
Co-reporter:Ellen M. Matson, Gabriel Espinosa Martinez, Abdulrahman D. Ibrahim, Bailey J. Jackson, Jeffrey A. Bertke, and Alison R. Fout
Organometallics 2015 Volume 34(Issue 2) pp:399-407
Publication Date(Web):September 10, 2014
DOI:10.1021/om5007177
The synthesis and characterization of a series of nickel(II) pincer complexes of the meta-phenylene-bridged bis-N-heterocyclic DIPPCCC ligand framework are reported. Characterization of the Ni(II)Cl complex revealed a square planar species with Cl– and the anionic carbon trans to one another. Formation of Ni(II) alkyl complexes derived from complex 1 was accomplished by addition of LiR [R = CH3 (2); CH2SiMe3 (3)]. Furthermore, we report a synthetic pathway to access the catalytically relevant Ni(II)H species (DIPPCCC)NiH (4), by direct oxidative addition of an aryl C–H bond across a Ni(0) center. Complexes 1–4 have been characterized by 1H and 13C NMR and electronic absorption spectroscopies as well as X-ray crystallography.
Co-reporter:Ellen M. Matson ; Yun Ji Park
Journal of the American Chemical Society 2014 Volume 136(Issue 50) pp:17398-17401
Publication Date(Web):December 3, 2014
DOI:10.1021/ja510615p
Reaction of tetrabutylammonium nitrite with [N(afaCy)3Fe(OTf)](OTf) cleanly resulted in the formation of an iron(III)-oxo species, [N(afaCy)3Fe(O)](OTf), and NO(g). Formation of NO(g) as a byproduct was confirmed by reaction of the iron(II) starting material with half an equivalent of nitrite, resulting in a mixture of two products, the iron-oxo and an iron-NO species, [N(afaCy)3Fe(NO)](OTf)2. Formation of the latter was confirmed through independent synthesis. The results of this study provide insight into the role of hydrogen bonding in the mechanism of nitrite reduction and the binding mode of nitrite in biological heme systems.
Co-reporter:Marshall R. Brennan, Dongyoung Kim and Alison R. Fout
Chemical Science 2014 vol. 5(Issue 12) pp:4831-4839
Publication Date(Web):26 Aug 2014
DOI:10.1039/C4SC01257G
Employing first-row transition metals in catalytic two-electron transformations remains a synthetic challenge. In order to overcome the common and often deleterious single-electron reactivity, an electron rich ligand was targeted on cobalt. Herein, we report the Co(I) catalyzed amination of aryl halides with lithium hexamethyldisilazide. This transformation features (PPh3)3CoCl (1) as the catalyst and affords structurally diverse and electronically varied primary arylamines in good chemical yields, with the scope of the reaction featuring arylamines that cannot be synthesized via traditional metal-catalyzed amination routes, including 4-aminophenylboronic acid pinacol ester. Stoichiometric reactivity revealed that (PPh3)2CoN(SiMe3)2 (2) is likely generated within the catalytic cycle and could be independently synthesized from the reaction of (PPh3)3CoCl with LiN(SiMe3)2. Catalytic reactivity featuring the Co–amide complex, (PPh3)2CoN(SiMe3)2, showed that it is a competent catalyst, implying that the (PPh3)3CoCl may be serving as a pre-catalyst in the reaction. Both stoichiometric and kinetic studies support the catalytic cycle involving a Co(I) complex. Catalytic reactions featuring Co(II) complexes resulted in undesired biaryl formation, a product that is not observed under standard catalytic conditions and any productive catalytic reactivity likely arises from an in situ reduction of Co(II) to Co(I). A Hammett study was carried out to differentiate between a closed-shell or radical mechanism, the results of which are consistent with the proposed closed-shell mechanism. Initial studies indicate that this reactivity may be expanded to other bulky nucleophiles.
Co-reporter:Ellen M. Matson, Jeffrey A. Bertke, and Alison R. Fout
Inorganic Chemistry 2014 Volume 53(Issue 9) pp:4450-4458
Publication Date(Web):April 23, 2014
DOI:10.1021/ic500102c
A tripodal ligand platform, tris(5-cycloiminopyrrol-2-ylmethyl)amine (H3[N(piCy)3]), that features a hydrogen bond-accepting secondary coordination sphere when bound anionically to an iron center is reported. Neutral coordination to iron affords ligand tautomerization, resulting in a hydrogen bond-donating secondary coordination sphere, and formation of the tris(5-cyclohexyl-amineazafulvene-2-methyl)amine, H3[N(afaCy)3], scaffold. Both binding motifs result in formation of stable, high-spin iron(II) complexes featuring ancillary water, triflate, or hydroxo ligands. Structural analysis reveals that these complexes exhibit distorted trigonal-bipyramidal geometries with coordination of the apical nitrogen to iron as well as three equatorial amine or imine nitrogens, depending on the axial ancillary ligand. Formation of the aqua complex K[(N(piCy)3)Fe(OH2)] (3) illustrated the propensity of the ligand to be hydrogen bond-accepting, whereas the iron triflate species [N(afaCy)3Fe](OTf)2 (4) features a hydrogen bond-donating secondary coordination sphere. The ability of each of the three arms of the ligand to tautomerize independently was observed during the formation of the iron–hydroxyl species [N(afaCy)2(piCy)]FeOH (5) and characterized by X-ray crystallography and IR spectroscopy. The combined data for the iron complexes established that each arm of the tripodal ligand can tautomerize independently and is likely dependent on the electronic needs of the iron center when binding various substrates.
Co-reporter:Ellen M. Matson, Zachary Gordon, Benjamin Lin, Mark J. Nilges and Alison R. Fout
Dalton Transactions 2014 vol. 43(Issue 45) pp:16992-16995
Publication Date(Web):17 Oct 2014
DOI:10.1039/C4DT02327G
The synthesis of a novel dipodal ligand framework, H2[MeN(piCy)2], is summarized. Upon metalation with MCl2 salts (M = Fe, Cu), the ligand undergoes a conformational change, resulting in the formation of a trigonal bipyramidal metal center with a pseudoplanar, meridionally-bound ligand framework. This tautomerization positions pendant amines in the metal's secondary coordination sphere. Metalation with M(OTf)2 in coordinating solvent yields octahedral metal complexes, where two solvent molecules bind in the apical positions with one outer sphere counter ion. Reactivity of these complexes, (MeN(afaCy)2)M(X)2 (X = Cl, OTf), with 2,2′-bypyridine results in ligand reorganization, yielding a facial coordination geometry of the dipodal framework. The described complexes have been characterized by 1H NMR, EPR, IR, Mössbauer and electronic absorption spectroscopies as well as X-ray crystallography.
Co-reporter:Ellen M. Matson, Zachary Gordon, Benjamin Lin, Mark J. Nilges and Alison R. Fout
Dalton Transactions 2014 - vol. 43(Issue 45) pp:NaN16995-16995
Publication Date(Web):2014/10/17
DOI:10.1039/C4DT02327G
The synthesis of a novel dipodal ligand framework, H2[MeN(piCy)2], is summarized. Upon metalation with MCl2 salts (M = Fe, Cu), the ligand undergoes a conformational change, resulting in the formation of a trigonal bipyramidal metal center with a pseudoplanar, meridionally-bound ligand framework. This tautomerization positions pendant amines in the metal's secondary coordination sphere. Metalation with M(OTf)2 in coordinating solvent yields octahedral metal complexes, where two solvent molecules bind in the apical positions with one outer sphere counter ion. Reactivity of these complexes, (MeN(afaCy)2)M(X)2 (X = Cl, OTf), with 2,2′-bypyridine results in ligand reorganization, yielding a facial coordination geometry of the dipodal framework. The described complexes have been characterized by 1H NMR, EPR, IR, Mössbauer and electronic absorption spectroscopies as well as X-ray crystallography.
Co-reporter:Yun Ji Park, Ellen M. Matson, Mark J. Nilges and Alison R. Fout
Chemical Communications 2015 - vol. 51(Issue 25) pp:NaN5313-5313
Publication Date(Web):2015/03/06
DOI:10.1039/C4CC08603A
Complexes containing manganese–oxygen bonds have been implicated in a variety of biological and synthetic processes. Herein, we describe the synthesis of a family of stable, high-spin trigonal bipyramidal manganese complexes of the electronically flexible ligand tris(5-cyclohexylimino-pyrrol-2-ylmethyl)amine [H3N(piCy)3] featuring apical water, hydroxyl, and oxo ligands. Terminal MnIII–O complexes are rare and the formation of this species was achieved from a variety of reagents including O2, PhIO and NO2−. Described herein is the preparation, structural and electronic properties of these manganese complexes.
Co-reporter:Abdulrahman D. Ibrahim, Kenan Tokmic, Marshall R. Brennan, Dongyoung Kim, Ellen M. Matson, Mark J. Nilges, Jeffery A. Bertke and Alison R. Fout
Dalton Transactions 2016 - vol. 45(Issue 24) pp:NaN9811-9811
Publication Date(Web):2016/01/18
DOI:10.1039/C5DT04723D
The synthesis and characterization of a series of cobalt complexes featuring a pincer bis(carbene) ligand of the meta-phenylene-bridged bis-N-heterocyclic carbene (ArCCC, Ar = 2,6-diispropylphenyl or mesityl) are reported. Cleavage of the aryl C–H bond of the ligand was achieved in a one-pot metalation procedure using Co(N(SiMe3)2)2(py)2, an equivalent of exogenous base, and trityl chloride to form the (DIPPCCC)CoCl2py complex. This species could be reduced to the Co(II) and Co(I)–N2 molecules with the appropriate equivalents of reductant. Subsequent generation of (MesCCC)CoI–III derivatives with the mesityl ligand proceeded in good yields. A suite of characterization techniques and the interconversion between all three oxidation states of the cobalt complexes is described.
Co-reporter:Marshall R. Brennan, Dongyoung Kim and Alison R. Fout
Chemical Science (2010-Present) 2014 - vol. 5(Issue 12) pp:NaN4839-4839
Publication Date(Web):2014/08/26
DOI:10.1039/C4SC01257G
Employing first-row transition metals in catalytic two-electron transformations remains a synthetic challenge. In order to overcome the common and often deleterious single-electron reactivity, an electron rich ligand was targeted on cobalt. Herein, we report the Co(I) catalyzed amination of aryl halides with lithium hexamethyldisilazide. This transformation features (PPh3)3CoCl (1) as the catalyst and affords structurally diverse and electronically varied primary arylamines in good chemical yields, with the scope of the reaction featuring arylamines that cannot be synthesized via traditional metal-catalyzed amination routes, including 4-aminophenylboronic acid pinacol ester. Stoichiometric reactivity revealed that (PPh3)2CoN(SiMe3)2 (2) is likely generated within the catalytic cycle and could be independently synthesized from the reaction of (PPh3)3CoCl with LiN(SiMe3)2. Catalytic reactivity featuring the Co–amide complex, (PPh3)2CoN(SiMe3)2, showed that it is a competent catalyst, implying that the (PPh3)3CoCl may be serving as a pre-catalyst in the reaction. Both stoichiometric and kinetic studies support the catalytic cycle involving a Co(I) complex. Catalytic reactions featuring Co(II) complexes resulted in undesired biaryl formation, a product that is not observed under standard catalytic conditions and any productive catalytic reactivity likely arises from an in situ reduction of Co(II) to Co(I). A Hammett study was carried out to differentiate between a closed-shell or radical mechanism, the results of which are consistent with the proposed closed-shell mechanism. Initial studies indicate that this reactivity may be expanded to other bulky nucleophiles.
Co-reporter:Ellen M. Matson, Yun Ji Park, Jeffery A. Bertke and Alison R. Fout
Dalton Transactions 2015 - vol. 44(Issue 22) pp:NaN10384-10384
Publication Date(Web):2015/05/13
DOI:10.1039/C5DT00985E
The syntheses of M(II) (M = Mn, Fe, Co) complexes bearing the tris(5-cycloaminoazafulvene-2-methyl)amine (H3N(afaCy)3) ligand in its datively coordinated, tautomeric form is reported. The metal–azafulvene complexes [N(afaCy)3M](OTf)2 are generated in high yields, featuring a secondary coordination sphere composed of amino moieties from the ligand platform. To investigate the ability of the hydrogen bonding network to support hydrogen-bond accepting, coordinating anions, pseudohalide derivatives, [N(afaCy)3MX](OTf) (X = NCS−, NCO−, N3−) were synthesized by exposure of [N(afaCy)3M](OTf)2 to an equivalent of the corresponding salt, [(nBu)4N](X). Structural characterization of the products reveals two isomorphs of the desired species. One complex features a single hydrogen bonding interaction with the pseudohalide, while the second compound has two H-bonds from the secondary coordination sphere to the coordinated anion. These complexes showcase the structural and electron flexibility of the ligand platform, presenting a scaffold capable of accessing a different number of hydrogen bonds for stabilizing a given moiety.
.png)
![2-[4-(4-Methoxy-phenylethynyl)-phenyl]-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane](http://img.cochemist.com/ccimg/1190400/1190376-24-9.png)
![2-[4-(4-Methoxy-phenylethynyl)-phenyl]-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane](http://img.cochemist.com/ccimg/1190400/1190376-24-9_b.png)
![2-HEXANONE, 6-[1,3,3,3-TETRAMETHYL-1-[(TRIMETHYLSILYL)OXY]DISILOXANYL]-](http://img.cochemist.com/ccimg/849900/849830-58-6.png)
![2-HEXANONE, 6-[1,3,3,3-TETRAMETHYL-1-[(TRIMETHYLSILYL)OXY]DISILOXANYL]-](http://img.cochemist.com/ccimg/849900/849830-58-6_b.png)


![Furan,3-[2-(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/465600/465521-19-1.png)
![Furan,3-[2-(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/465600/465521-19-1_b.png)



![Benzenamine, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/111400/111359-74-1.png)
![Benzenamine, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/111400/111359-74-1_b.png)
![(E)-[2-(3-furanyl)ethenyl]trimethyl-Silane](http://img.cochemist.com/ccimg/105300/105281-59-2.png)
![(E)-[2-(3-furanyl)ethenyl]trimethyl-Silane](http://img.cochemist.com/ccimg/105300/105281-59-2_b.png)
![Silane, trimethyl[2-(3-thienyl)ethenyl]-, (E)-](/data/chemimg/1049400/105281-57-0.png)
![Silane, trimethyl[2-(3-thienyl)ethenyl]-, (E)-](/data/chemimg/1049400/105281-57-0_b.png)
![Silane, [2-(2-methoxyphenyl)ethenyl]trimethyl-, (E)-](/data/chemimg/1049900/105281-54-7.png)
![Silane, [2-(2-methoxyphenyl)ethenyl]trimethyl-, (E)-](/data/chemimg/1049900/105281-54-7_b.png)




![Silane, [2-(3-cyclohexen-1-yl)ethyl]dimethylphenyl-](http://img.cochemist.com/ccimg/61600/61518-54-5.png)
![Silane, [2-(3-cyclohexen-1-yl)ethyl]dimethylphenyl-](http://img.cochemist.com/ccimg/61600/61518-54-5_b.png)




![Benzene,[(1E)-2-(trimethylsilyl)ethenyl]-](http://img.cochemist.com/ccimg/19400/19372-00-0.png)
![Benzene,[(1E)-2-(trimethylsilyl)ethenyl]-](http://img.cochemist.com/ccimg/19400/19372-00-0_b.png)
![trimethyl[(Z)-2-phenylethenyl]silane](http://img.cochemist.com/ccimg/19400/19319-11-0.png)
![trimethyl[(Z)-2-phenylethenyl]silane](http://img.cochemist.com/ccimg/19400/19319-11-0_b.png)
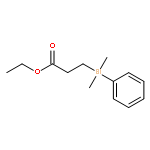
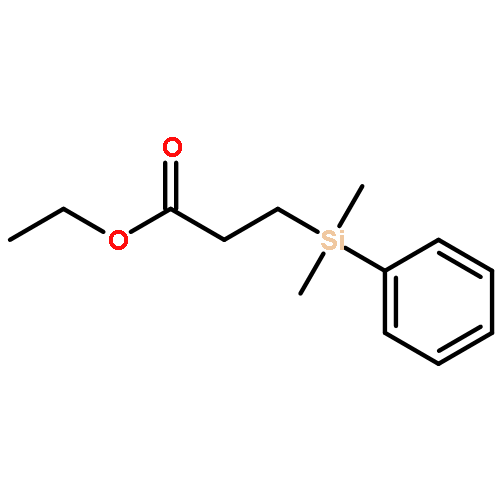
![Bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid,dimethyl ester,(endo,endo)](http://img.cochemist.com/ccimg/17900/17812-27-0.png)
![Bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid,dimethyl ester,(endo,endo)](http://img.cochemist.com/ccimg/17900/17812-27-0_b.png)