Co-reporter:Pan Luo, Wenjuan Xia, Susan L. Morris-Natschke, Kuo-Hsiung Lee, Yu Zhao, Qiong Gu, and Jun Xu
Journal of Natural Products May 26, 2017 Volume 80(Issue 5) pp:1679-1679
Publication Date(Web):April 28, 2017
DOI:10.1021/acs.jnatprod.6b01195
Vitepyrroloids A–D (1–4), four new 2-cyano-substituted pyrrole-ring-containing labdane diterpenoids, were isolated from the leaves of Vitex trifolia. Their structures were elucidated based on spectroscopic data analysis. The absolute configuration of compound 1 was determined by X-ray diffraction. Compounds 1–4 are unprecedented labdane diterpenoids featuring a 2-cyano-substituted pyrrole ring. Compound 1 showed cytotoxic activity against a human nasopharyngeal carcinoma cell line (CNE1) with an IC50 value of 8.7 μM.
Co-reporter:Yongsheng Lin, Qian Wang, Qiong GuHongao Zhang, Cheng Jiang, Jiayuan Hu, Yan Wang, Yuan Yan, Jun Xu
Journal of Natural Products 2017 Volume 80(Issue 1) pp:
Publication Date(Web):January 17, 2017
DOI:10.1021/acs.jnatprod.6b00415
(+)-Rutamarin inhibits EBV lytic DNA replication with an IC50 of 7.0 μM. (−)-Chalepin, a (−)-rutamarin derivative, was isolated from the whole plant of Ruta graveolens and used as a precursor of (−)-rutamarin. Altogether, 28 (−)-rutamarin derivatives were synthesized starting from (−)-chalepin. Of these, 16 compounds (2a–e, 3b–e, 3g, 4f, 4k, 4m–p) were found to be more potent against EBV lytic DNA replication than (−)-chalepin. Compounds 4m, 4n, and 4p exhibited IC50 values of 1.5, 0.32, and 0.83 μM and showed selectivity index values (SI) of 801, 211, and >120, respectively. Thus, compounds 4m, 4n, and 4p are considered promising leads for further laboratory investigation.
Co-reporter:Pan Luo, Qian Yu, Shao-Nan Liu, Wen-Juan Xia, Yu-Ying Fang, Lin-Kun An, Qiong Gu, Jun Xu
Fitoterapia 2017 Volume 120(Volume 120) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.fitote.2017.06.006
Eleven new compounds, including six labdane (1–6), three halimane (7–9), and two clerodane (10–11) diterpenoids and 16 known analogues (12–27), were isolated from the leaves of Vitex trifolia. The structures of 1–11 were established by extensive 1D- and 2D-NMR and HRMS spectroscopic data. The absolute configurations of compounds 3, 7, and 10 were assigned using X-ray diffraction. Compounds 1–27 were evaluated for DNA topoisomerases I (Top1) inhibitory activity and cytotoxicity against HCT 116 cells. Compounds 8 and 11 exhibited equipotent Top1 inhibitory activity to the positive control, camptothecin (CPT), at 100 μM. Compounds 8, 9, 16, and 27 showed moderate cytotoxicity at low micromolar concentrations.Download high-res image (255KB)Download full-size image
Co-reporter:Shao-Nan Liu;Jiayuan Hu;Shen H. Tan;Qian Wang;Jun Xu;Yan Wang;Yan Yuan
RSC Advances (2011-Present) 2017 vol. 7(Issue 74) pp:46938-46947
Publication Date(Web):2017/10/02
DOI:10.1039/C7RA08877A
The phytochemical investigation on the acetone extract of Euphorbia milii afforded thirteen new ent-rosane diterpenoids (1–13) through bioassay guided fractionation for evaluating its effect on Epstein–Barr virus (EBV) DNA lytic replication. Structures were determined by comprehensive spectroscopic analyses including 1D & 2D NMR techniques, chemical methods, and experimental and calculated electronic circular dichroism (ECD) data. The absolute configuration of euphominoid A (1) was established by single crystal X-ray diffraction analysis of its p-bromobenzoate derivative 1a. Compounds 1–3, and 10 displayed inhibitory activity with EC50 values ranging from 5.4 to 29.1 μM and selective index (SI) values varied from 4.5 to 9.3. Compound 2 showed the most potent inhibitory activity with an EC50 value of 5.4 μM comparing with the positive control (+)-rutamarin (EC50 = 5.4 μM). This is the first report of ent-rosane-type diterpenoids exhibiting significant inhibition of EBV lytic replication.
Co-reporter:Shao-Nan Liu, Dane Huang, Susan L. Morris-Natschke, Hang Ma, Zhi-hong Liu, Navindra P. Seeram, Jun Xu, Kuo-Hsiung Lee, and Qiong Gu
Organic Letters 2016 Volume 18(Issue 23) pp:6132-6135
Publication Date(Web):November 22, 2016
DOI:10.1021/acs.orglett.6b03142
Three highly modified ent-rosane diterpenoids, euphomilones A (1) and B (2) and euphomianol A (3), were isolated from the aerial parts of Euphorbia milii. The structures were elucidated from physical, spectroscopic, and X-ray diffraction data, as well as experimental and calculated electronic circular dichroism (ECD) analysis. Plausible biogenetic pathways to 1–3 are proposed. Also, compound 1 exhibited inhibition of receptor activator of nuclear factor-kappaB ligand (RANKL)-induced osteoclast formation (IC50 = 12.6 μM).
Co-reporter:Xiangkun Luo, Chanjuan Li, Pan Luo, Xin Lin, Hang Ma, Navindra P. Seeram, Ching Song, Jun XuQiong Gu
Journal of Natural Products 2016 Volume 79(Issue 12) pp:3014-3021
Publication Date(Web):November 30, 2016
DOI:10.1021/acs.jnatprod.6b00558
Four new pterosin sesquiterpenoids (1–4), a new ent-kaurane diterpenoid (17), and 18 known compounds were isolated from the aerial parts of Pteris cretica L. The structures of the isolates were elucidated based on spectroscopic data analysis, and their absolute configurations were determined by comparison of experimental and calculated electronic circular dichroism spectra. The compounds were evaluated for lipid-lowering effects in 3T3-L1 adipocytes. Compounds 4, 8, 17, and 22 were more potent than the positive control, berberine, in decreasing triglycerides activity, with compound 4 exerting the most potent activity. Compound 4 activated LXRα/β in a HEK 293T cell-based reporter gene assay. Molecular dynamic simulations revealed that compound 4 activates liver X receptors (LXRs) through hydrogen bonding with the residues of LXRα/β, suggesting that compound 4 reduces total triglycerides through the regulation of LXRα/β.
Co-reporter:Cheng Jiang; Pan Luo; Yu Zhao; Jialing Hong; Susan L. Morris-Natschke; Jun Xu; Chin-Ho Chen; Kuo-Hsiung Lee
Journal of Natural Products 2016 Volume 79(Issue 3) pp:578-583
Publication Date(Web):January 12, 2016
DOI:10.1021/acs.jnatprod.5b01012
Seven new carolignans, including two pairs of enantiomers (±)-erythro-7′-methylcarolignan E (1a/1b) and (±)-threo-7′-methylcarolignan E (2a/2b), (+)-threo-carolignan E (3a), (+)-erythro-carolignan E (4a), and (−)-erythro-carolignan Z (5), together with four known lignans (3b, 4b, 6, and 7) and six polyphenols (8–13) were isolated from the aerial parts of Euphorbia sikkimensis. The structures of the new compounds were elucidated by spectroscopic analysis, and their absolute configurations were determined by electronic circular dichroism calculations. Seven of the isolates were examined for anti-HIV effects, and compounds 1a and 1b showed moderate anti-HIV activity with EC50 values of 6.3 and 5.3 μM.
Co-reporter:Sai Fang, Lei Chen, Miao Yu, Bao Cheng, Yongsheng Lin, Susan L. Morris-Natschke, Kuo-Hsiung Lee, Qiong Gu and Jun Xu
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 16) pp:4714-4726
Publication Date(Web):10 Mar 2015
DOI:10.1039/C5OB00007F
Based on the scaffolds of caffeic acid phenethyl ester (CAPE) as well as bioactive lactone-containing compounds, 6-acrylic phenethyl ester-2-pyranone derivatives were synthesized and evaluated against five tumor cell lines (HeLa, C6, MCF-7, A549, and HSC-2). Most of the new derivatives exhibited moderate to potent cytotoxic activity. Moreover, HeLa cell lines showed higher sensitivity to these compounds. In particular, compound 5o showed potent cytotoxic activity (IC50 = 0.50–3.45 μM) against the five cell lines. Further investigation on the mechanism of action showed that 5o induced apoptosis, arrested the cell cycle at G2/M phases in HeLa cells, and inhibited migration through disruption of the actin cytoskeleton. In addition, ADMET properties were also calculated in silico, and compound 5o showed good ADMET properties with good absorption, low hepatotoxicity, and good solubility, and thus, could easily be bound to carrier proteins, without inhibition of CYP2D6. A structure–activity relationship (SAR) analysis indicated that compounds with ortho-substitution on the benzene ring exhibited obviously increased cytotoxic potency. This study indicated that compound 5o is a promising compound as an antitumor agent.
Co-reporter:Taizong Wu, Qian Wang, Cheng Jiang, Susan L. Morris-Natschke, Hui Cui, Yan Wang, Yuan Yan, Jun Xu, Kuo-Hsiung Lee, and Qiong Gu
Journal of Natural Products 2015 Volume 78(Issue 3) pp:500-509
Publication Date(Web):February 3, 2015
DOI:10.1021/np500988m
Bioassay-guided fractionation was conducted on an EtOAc-soluble extract of the whole plants of Scutellaria barbata, monitored by inhibition of Epstein–Barr virus (EBV) lytic replication. Twenty-six neo-clerodane diterpenoids were isolated, of which 13 are new (1–13, scutolides A–L) and 13 previously known (14–26). The structures of 1–13 were elucidated by analysis of their NMR and MS spectroscopic data. Furthermore, the configurations of the new compounds 1 and 11 were confirmed by single-crystal X-ray diffraction. All of the isolated compounds were evaluated for inhibitory effects against EBV lytic replication. Eleven compounds (3, 4, 6, 11, 12, 15, 16, 17, 20, 22, and 24) exhibited moderate to potent inhibition, with EC50 values from 3.2 to 23.6 μM and selective index (SI) values from 2.1 to 109.2. More specifically, the new compound 4 showed the most potent activity, with EC50 and SI values of 3.2 μM and 46.1, respectively, while compound 24 (EC50 = 16.4 μM) exhibited the highest SI of 109.2. This study is the first to report that neo-clerodane diterpenoids demonstrate significant inhibition against EBV lytic replication.
Co-reporter:Taizong Wu; Cheng Jiang; Ling Wang; Susan L. Morris-Natschke; Hui Miao; Lianquan Gu; Jun Xu; Kuo-Hsiung Lee
Journal of Natural Products 2015 Volume 78(Issue 7) pp:1593-1599
Publication Date(Web):June 23, 2015
DOI:10.1021/acs.jnatprod.5b00156
Four new 3,5-diarylpyrazole analogues (1–4) were isolated from an extract of the flowers of Chrysanthemun indicum using a combination of ammonolysis of the total flavonoid extract and an Aβ aggregation inhibitory activity guided purification procedure. All four compounds (1–4) showed moderate to potent activity against Aβ aggregation with EC50 values of 4.3, 15.8, 1.3, and 2.9 μM, respectively. Moreover, compound 3 showed low cytotoxicity and significant neuroprotective activity against Aβ-induced cytotoxicity in the SH-SY5Y cell line. This report is the first to show that 3,5-diarylpyrazole analogues can inhibit Aβ aggregation and exhibit neuroprotective activity with potential for the treatment of Alzheimer’s disease. Taken together, the method presented here offers an alternative approach to yield bioactive compounds.
Co-reporter:Xiu Le, Qiong Gu and Jun Xu
RSC Advances 2015 vol. 5(Issue 51) pp:40536-40545
Publication Date(Web):28 Apr 2015
DOI:10.1039/C5RA03079J
The glutamate racemase (MurI) is essential for Helicobacter pylori (H. pylori) cell wall biosynthesis. In this work, we report a new method that correlates decomposed binding free energies with MurI inhibition based upon the data from pyrazolopyrimidinedione series MurI uncompetitive inhibitors. With the molecular mechanics/generalized Born surface areas (MM/GBSA) approach, we were able to decompose the binding interaction into van der Waals, electrostatic, and polar solvation surfaces. The decomposed binding energies were correlated with MurI inhibitory activity with partial least squares regression (PLSR). Hence, the method is termed MM/GBSA-PLSR. The non-cross-validation (R2) and leave-one-out cross-validation (LOOCV) (Q2) correlation coefficients of the 3D-QSAR model are 0.962 and 0.822, respectively. The external testing yields a predicted correlation coefficient (Rpred2) of 0.817. This study demonstrated that the activity-contribution fractions from the three types of ligand–receptor interactions are 29.5% from van der Waal interactions, 38.2% from electrostatic interactions, and 32.3% from polar solvation interactions. Comparing with molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA), we find that the CoMFA/CoMSIA steric interaction fields can be interpreted as the MM/GBSA-PLSR van der Waals interactions; CoMFA/CoMSIA electrostatic and H-bond acceptor/donor interaction fields can be interpreted as the MM/GBSA-PLSR electrostatic interactions. However, there is no explicit association between MM/GBSA-PLSR solvation interactions (polar or non-polar) and CoMFA/CoMSIA fields. It is worth noting that the solvation interaction is important for ligand design. Moreover, MM/GBSA-PLSR maps the decomposed binding interactions on to pharmacophore surfaces (van der Waals, electrostatic, and polar solvation surfaces) to aid drug design.
Co-reporter:Long Li, Xiu Le, Ling Wang, Qiong Gu, Huihao Zhou and Jun Xu
RSC Advances 2015 vol. 5(Issue 128) pp:105600-105608
Publication Date(Web):07 Dec 2015
DOI:10.1039/C5RA22568J
Bacterial DNA gyrase is not expressed in eukaryotes. It is a promising target for broad-spectrum antibiotics. This paper reports new DNA gyrase inhibitors as broad-spectrum antibacterial agents discovered by means of target-based in silico and in vitro models. Two machine learning methods (naïve Bayesian and recursive partitioning) were employed to build in silico models based on physicochemical descriptors and structural fingerprints. For both training and testing sets, the overall predictive accuracies of the best in silico models were greater than 80%. The best 11 models were used to virtually screen a molecular database to identify DNA gyrase inhibitors. The in vitro models were used to verify the virtual hits activities against Escherichia coli, methicillin-resistant Staphylococcus aureus and other bacteria, and DNA gyrase. The MIC values of the confirmed DNA gyrase inhibitors range between 1 and 32 μg mL−1 and, the relative inhibition rates of the inhibitors range between 42% to 75% at 1 μM. Cell-based cytotoxicity assays demonstrated that the confirmed DNA gyrase inhibitors were not toxic. In silico studies indicated that the new DNA gyrase inhibitors have similar binding modes to the reported inhibitors.
Co-reporter:Qianzhi Ding, Chanjuan Li, Ling Wang, Yali Li, Huihao Zhou, Qiong Gu and Jun Xu
MedChemComm 2015 vol. 6(Issue 7) pp:1393-1403
Publication Date(Web):08 Jun 2015
DOI:10.1039/C5MD00149H
The farnesoid X receptor (FXR), a ligand-modulated transcription factor, is a multiple functional hepatic cell protector. Therefore, FXR agonists present as promising dyslipidemia and anti-diabetes agents. To identify novel FXR agonists, models were created from 144 known FXR agonists by naïve Bayesian (NB) and recursive partitioning (RP) approaches. The predictive and reliable models were selected with Matthews correlation coefficient (MCC) criterion (>0.900 with 117 testing compounds). The top 4 models were validated with the external data (282 compounds having cell-free activities and 500 decoys). Two optimal FXR agonist models (one from the NB method and the other from the RP method) were obtained from the top models by further validation. A virtual screening campaign was conducted against our in-house compound library with the optimal models and produced 15 virtual hits, which were further confirmed with cell-based luciferase assays. Finally, we discovered two new FXR agonists. Molecular docking studies indicated that the two new FXR agonists have similar binding modes to the known FXR agonists. This work demonstrated that a machine learning approach with combined NB and RP methods was able to identify novel FXR agonists and that the approach could be applied in other lead identification processes.
Co-reporter:Hui Cui, Bo Xu, Taizong Wu, Jun Xu, Yan Yuan, and Qiong Gu
Journal of Natural Products 2014 Volume 77(Issue 1) pp:100-110
Publication Date(Web):December 20, 2013
DOI:10.1021/np400757k
Epstein–Barr virus (EBV) is a member of the γ-herpes virus subfamily and has been implicated in the pathogenesis of several human malignancies. Bioassay-guided fractionation was conducted on an EtOAc-soluble extract of the roots of Saururus chinensis and monitored using an EBV lytic replication assay. This led to the isolation of 19 new (1–19) and nine known (20–28) lignans. The absolute configurations of the new lignans were established by Mosher’s ester, ECD, and computational methods. Eight lignans, including three sesquineolignans (19, 23, and 24) and five dineolignans (3, 4, 26, 27, and 28), exhibited inhibitory effects toward EBV lytic replication with EC50 values from 1.09 to 7.55 μM and SI values from 3.3 to 116.4. In particular, manassantin B (27) exhibited the most promising inhibition, with an EC50 of 1.72 μM, low cytotoxicity, CC50 > 200 μM, and SI > 116.4. This is the first study demonstrating that lignans possess anti-EBV lytic replication activity.
Co-reporter:Ling Wang, Xiu Le, Long Li, Yingchen Ju, Zhongxiang Lin, Qiong Gu, and Jun Xu
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 11) pp:3186-3197
Publication Date(Web):November 6, 2014
DOI:10.1021/ci500253q
To discover new agents active against methicillin-resistant Staphylococcus aureus (MRSA), in silico models derived from 5451 cell-based anti-MRSA assay data were developed using four machine learning methods, including naïve Bayesian, support vector machine (SVM), recursive partitioning (RP), and k-nearest neighbors (kNN). A total of 876 models have been constructed based on physicochemical descriptors and fingerprints. The overall predictive accuracies of the best models exceeded 80% for both training and test sets. The best model was employed for the virtual screening of anti-MRSA compounds, which were then validated by a cell-based assay using the broth microdilution method with three types of highly resistant MRSA strains (ST239, ST5, and 252). A total of 12 new anti-MRSA agents were confirmed, which had MIC values ranging from 4 to 64 mg/L. This work proves the capacity of combined multiple ligand-based approaches for the discovery of new agents active against MRSA with cell-based assays. We think this work may inspire other lead identification processes when cell-based assay data are available.
Co-reporter:Yingying Cao, Ling Wang, Zhongxiang Lin, Fengyin Liang, Zhong Pei, Jun Xu and Qiong Gu
MedChemComm 2014 vol. 5(Issue 11) pp:1736-1743
Publication Date(Web):27 Aug 2014
DOI:10.1039/C4MD00305E
Dehydroabietylamine derivatives have been reported to eliminate the superoxide anion (O2−˙) and 1,1-dipheny-2-picrylhydrazyl radicals. In this work, several dehydroabietylamine derivatives were shown to have potent anti-oxidative activity in SH-SY5Y cells and exhibited significant inhibition of Aβ42 self-mediated aggregation and the disaggregation of Aβ42 aging fibrils. In particular, the compound 12N,18N-bis(caffeoyl)-14-nitrodehydroabietylamine (3b) showed the most potent inhibitory activity against Aβ42 self-mediated aggregation with an IC50 value of 3.96 ± 0.33 μM. Compound 3b decreased the production of Aβ42 in swAPP HEK293 cells and showed neuroprotective activity against Aβ42-induced cytotoxicity. Through reducing the production of the Aβ42 species, compound 3b alleviated Aβ42-induced paralysis in transgenic Caenorhabditis elegans strain CL4176. Considering its multifunctional activity and low cytotoxicity, 3b is considered to be a potential multifunctional agent for the treatment of Alzheimer's disease.
Co-reporter:Minghao Zheng;Zhihong Liu;Xin Yan;Qianzhi Ding;Jun Xu
Molecular Diversity 2014 Volume 18( Issue 4) pp:829-840
Publication Date(Web):2014 November
DOI:10.1007/s11030-014-9545-3
Abundant data on compound bioactivity and publicly accessible chemical databases increase opportunities for ligand-based drug discovery. In order to make full use of the data, an online platform for ligand-based virtual screening (LBVS) using publicly accessible databases has been developed. LBVS adopts Bayesian learning approach to create virtual screening models because of its noise tolerance, speed, and efficiency in extracting knowledge from data. LBVS currently includes data derived from BindingDB and ChEMBL. Three validation approaches have been employed to evaluate the virtual screening models created from LBVS. The tenfold cross validation results of twenty different LBVS models demonstrate that LBVS achieves an average AUC value of 0.86. Our internal and external testing results indicate that LBVS is predictive for lead identifications. LBVS can be publicly accessed at http://rcdd.sysu.edu.cn/lbvs.
Co-reporter:Lei Chen;Ling Wang;Jun Xu
Molecular Diversity 2014 Volume 18( Issue 4) pp:841-852
Publication Date(Web):2014 November
DOI:10.1007/s11030-014-9543-5
The mammalian target of rapamycin (mTOR) is an anti-cancer target. In this study, we propose an in silico protocol for identifying mTOR inhibitors from the ZINC natural product database. First, a three-dimensional quantitative structure–activity relationship pharmacophore model was built based on known mTOR inhibitors. The model was validated with an external test set, Fischer’s randomization method, a decoy set and pharmacophore mapping conformation testing. The results showed that the model can predict the mTOR inhibition activity of the tested compounds. Virtual screening was performed based on the best pharmacophore model, and the results were then filtered using a molecular docking approach. In addition, molecular mechanics/generalized born surface area analysis was used to refine the selected candidates. The top 20 natural products were selected as potential mTOR inhibitors, and their structural scaffolds could serve as building blocks in designing drug-like molecules for mTOR inhibition.
Co-reporter:Ling Wang, Qiong Gu, Xuehua Zheng, Jiming Ye, Zhihong Liu, Jiabo Li, Xiaopeng Hu, Arnold Hagler, and Jun Xu
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 9) pp:2409-2422
Publication Date(Web):July 31, 2013
DOI:10.1021/ci400322j
Aldose reductase reduces glucose to sorbitol. It plays a key role in many of the complications arising from diabetes. Thus, aldose reductase inhibitors (ARI) have been identified as promising therapeutic agents for treating such complications of diabetes, as neuropathy, nephropathy, retinopathy, and cataracts. In this paper, a virtual screening protocol applied to a library of compounds in house has been utilized to discover novel ARIs. IC50’s were determined for 15 hits that inhibited ALR2 to greater than 50% at 50 μM, and ten of these have an IC50 of 10 μM or less, corresponding to a rather substantial hit rate of 14% at this level. The specificity of these compounds relative to their cross-reactivity with human ALR1 was also assessed by inhibition assays. This resulted in identification of novel inhibitors with IC50’s comparable to the commercially available drug, epalrestat, and greater than an order of magnitude better selectivity.
Co-reporter:Qiong Gu, Yaoyao Chen, Hui Cui, Dane Huang, Jingwei Zhou, Taizong Wu, Yiping Chen, Lina Shi and Jun Xu
RSC Advances 2013 vol. 3(Issue 26) pp:10168-10172
Publication Date(Web):13 May 2013
DOI:10.1039/C3RA23172K
Chrysanolide A, a novel guaianolide-type sesquiterpenoid trimer (3), along with its biogenetically related monomer (Chrysanolide B, 1) and dimer (Chrysanolide C, 2), were simultaneously isolated from Chrysanthemum indicum L flowers. Their structures and absolute configurations were elucidated via spectroscopic and computational methods. All isolated compounds were evaluated for their anti-HBV activities.
Co-reporter:GuoDong Zhang;Hu Ge;Jun Xu
Science China Chemistry 2013 Volume 56( Issue 10) pp:1402-1412
Publication Date(Web):2013 October
DOI:10.1007/s11426-013-4952-3
Human intestinal carboxyl esterase (hiCE) is a drug target for ameliorating irinotecan-induced diarrhea. By reducing irinotecan-induced diarrhea, hiCE inhibitors can improve the anti-cancer efficacy of irinotecan. To find effective hiCE inhibitors, a new virtual screening protocol that combines pharmacophore models derived from the hiCE structure and its ligands has been proposed. The hiCE structure has been constructed through homology techniques using hCES1’s crystal structure. The hiCE structure was optimized via molecular dynamics simulations with the most known active hiCE inhibitors docked into the structure. An optimized pharmacophore, derived from the receptor, was then generated. A ligand-based pharmacophore was also generated from a larger set of known hiCE inhibitors. The final hiCE inhibitor predictions were based upon the virtual screening hits from both ligand-based and receptor-based pharmacophore models. The hit rates from the ligand-based and receptor-based pharmacophore models are 88% and 86%, respectively. The final hit rate is 94%. The two models are highly consistent with one another (85%). This proves that both models are reliable.
Co-reporter:Ying Ying Cao;Ling Wang;Hu Ge;Xi Lin Lu;Zhong Pei
Molecular Diversity 2013 Volume 17( Issue 3) pp:515-524
Publication Date(Web):2013 August
DOI:10.1007/s11030-013-9452-z
The effects of Salvianolic acid A (Sal A) on the treatment of Alzheimer’s disease (AD) were investigated. Sal A significantly inhibits amyloid beta \((\text{ A }\beta )\) self-aggregation and disaggregates pre-formed \(\text{ A }\beta \) fibrils, reduces metal-induced \(\text{ A }\beta \) aggregation through chelating metal ions, and blocks the formation of reactive oxygen species (ROS) in SH-SY5Y cells. Sal A protects cells against \(\text{ A }\beta _{42}\)-induced toxicity. Furthermore, Sal A, possibly because of the effects of decreasing toxicity effects of \(\text{ A }\beta \) species, alleviates \(\text{ A }\beta \)-induced paralysis in transgenic Caenorhabditis elegans. Circular dichroism (CD) experiments and Molecular dynamic (MD) simulations demonstrate that Sal A inhibits \(\text{ A }\beta \) self-aggregation through binding to the C-terminus of \(\text{ A }\beta \), and therefore stabilizing the \(\alpha \)-helical conformations. Altogether, our data show that Sal A, as the multifunctional agent, is likely to be promising therapeutics for AD.
Co-reporter:Sai Fang, Lei Chen, Miao Yu, Bao Cheng, Yongsheng Lin, Susan L. Morris-Natschke, Kuo-Hsiung Lee, Qiong Gu and Jun Xu
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 16) pp:NaN4726-4726
Publication Date(Web):2015/03/10
DOI:10.1039/C5OB00007F
Based on the scaffolds of caffeic acid phenethyl ester (CAPE) as well as bioactive lactone-containing compounds, 6-acrylic phenethyl ester-2-pyranone derivatives were synthesized and evaluated against five tumor cell lines (HeLa, C6, MCF-7, A549, and HSC-2). Most of the new derivatives exhibited moderate to potent cytotoxic activity. Moreover, HeLa cell lines showed higher sensitivity to these compounds. In particular, compound 5o showed potent cytotoxic activity (IC50 = 0.50–3.45 μM) against the five cell lines. Further investigation on the mechanism of action showed that 5o induced apoptosis, arrested the cell cycle at G2/M phases in HeLa cells, and inhibited migration through disruption of the actin cytoskeleton. In addition, ADMET properties were also calculated in silico, and compound 5o showed good ADMET properties with good absorption, low hepatotoxicity, and good solubility, and thus, could easily be bound to carrier proteins, without inhibition of CYP2D6. A structure–activity relationship (SAR) analysis indicated that compounds with ortho-substitution on the benzene ring exhibited obviously increased cytotoxic potency. This study indicated that compound 5o is a promising compound as an antitumor agent.

![5-{2-[(6-Chloro-4-quinolinyl)methyl]-7-(cyclopropylmethyl)-5-meth yl-4,6-dioxo-4,5,6,7-tetrahydro-2H-pyrazolo[3,4-d]pyrimidin-3-yl} -1-methyl-1H-pyrrole-3-carbonitrile](http://img.cochemist.com/ccimg/946500/946411-72-9.png)
![5-{2-[(6-Chloro-4-quinolinyl)methyl]-7-(cyclopropylmethyl)-5-meth yl-4,6-dioxo-4,5,6,7-tetrahydro-2H-pyrazolo[3,4-d]pyrimidin-3-yl} -1-methyl-1H-pyrrole-3-carbonitrile](http://img.cochemist.com/ccimg/946500/946411-72-9_b.png)
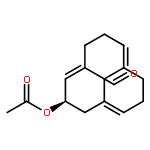
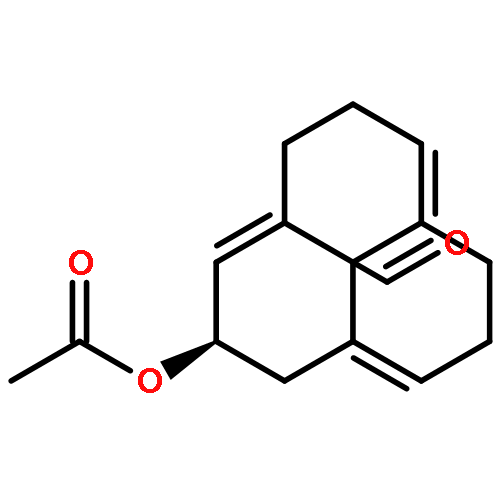





























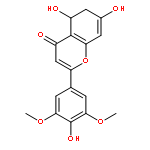
![(1R,2R)-2-[2,6-dimethoxy-4-((E)-prop-1-enyl)phenoxy]-1-(3,4,5-trimethoxyphenyl)propyl acetate](http://img.cochemist.com/ccimg/916300/916264-20-5.png)
![(1R,2R)-2-[2,6-dimethoxy-4-((E)-prop-1-enyl)phenoxy]-1-(3,4,5-trimethoxyphenyl)propyl acetate](http://img.cochemist.com/ccimg/916300/916264-20-5_b.png)
![2-[3-(furan-2-yl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl]pyrazine](http://img.cochemist.com/ccimg/878500/878434-31-2.png)
![2-[3-(furan-2-yl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl]pyrazine](http://img.cochemist.com/ccimg/878500/878434-31-2_b.png)













![Isopropyl 3-(3,4-difluorobenzoyl)-1,1-dimethyl-1,2,3,6-tetrahydroazepino[4,5-b]indole-5-carboxylate](http://img.cochemist.com/ccimg/629700/629664-81-9.png)
![Isopropyl 3-(3,4-difluorobenzoyl)-1,1-dimethyl-1,2,3,6-tetrahydroazepino[4,5-b]indole-5-carboxylate](http://img.cochemist.com/ccimg/629700/629664-81-9_b.png)
![2-Propenoic acid,3-[3-[(cyclohexylcarbonyl)[[4'-(dimethylamino)[1,1'-biphenyl]-4-yl]methyl]amino]phenyl]-,methyl ester](http://img.cochemist.com/ccimg/574100/574013-66-4.png)
![2-Propenoic acid,3-[3-[(cyclohexylcarbonyl)[[4'-(dimethylamino)[1,1'-biphenyl]-4-yl]methyl]amino]phenyl]-,methyl ester](http://img.cochemist.com/ccimg/574100/574013-66-4_b.png)


![(4R)-4-[(3R,5S,6R,7R,8S,9S,10S,13R,14S,17R)-6-ETHYL-3,7-DIHYDROXY-10,13-DIMETHYL-2,3,4,5,6,7,8,9,11,12,14,15,16,17-TETRADECAHYDRO-1H-CYCLOPENTA[A]PHENANTHREN-17-YL]PENTANOIC ACID](http://img.cochemist.com/ccimg/459800/459789-99-2.png)
![(4R)-4-[(3R,5S,6R,7R,8S,9S,10S,13R,14S,17R)-6-ETHYL-3,7-DIHYDROXY-10,13-DIMETHYL-2,3,4,5,6,7,8,9,11,12,14,15,16,17-TETRADECAHYDRO-1H-CYCLOPENTA[A]PHENANTHREN-17-YL]PENTANOIC ACID](http://img.cochemist.com/ccimg/459800/459789-99-2_b.png)






![<br>5-Methyl-4-morpholin-4-yl-2-phenyl-thieno[2,3-d]pyrimidine-6-carboxylic aci d methyl ester](http://img.cochemist.com/ccimg/351500/351437-14-4.png)
![<br>5-Methyl-4-morpholin-4-yl-2-phenyl-thieno[2,3-d]pyrimidine-6-carboxylic aci d methyl ester](http://img.cochemist.com/ccimg/351500/351437-14-4_b.png)






![Benzoic acid,3-[2-(4-morpholinyl)ethoxy]-](http://img.cochemist.com/ccimg/220000/219935-32-7.png)
![Benzoic acid,3-[2-(4-morpholinyl)ethoxy]-](http://img.cochemist.com/ccimg/220000/219935-32-7_b.png)




![(2E)-3-[(2S,3R)-3-(Acetoxymethyl)-2-(4-hydroxy-3-methoxyphenyl)-7 -methoxy-2,3-dihydro-1-benzofuran-5-yl]-2-propen-1-yl acetate](http://img.cochemist.com/ccimg/184100/184046-40-0.png)
![(2E)-3-[(2S,3R)-3-(Acetoxymethyl)-2-(4-hydroxy-3-methoxyphenyl)-7 -methoxy-2,3-dihydro-1-benzofuran-5-yl]-2-propen-1-yl acetate](http://img.cochemist.com/ccimg/184100/184046-40-0_b.png)
![(1R,2S,3R,4R,4aS,8aR)-3-Hydroxy-3,4,8,8a-tetramethyl-4-[(E)-2-(5- oxo-2,5-dihydro-3-furanyl)vinyl]-1,2,3,4,4a,5,6,8a-octahydronapht halene-1,2-diyl dinicotinate](http://img.cochemist.com/ccimg/176600/176520-13-1.png)
![(1R,2S,3R,4R,4aS,8aR)-3-Hydroxy-3,4,8,8a-tetramethyl-4-[(E)-2-(5- oxo-2,5-dihydro-3-furanyl)vinyl]-1,2,3,4,4a,5,6,8a-octahydronapht halene-1,2-diyl dinicotinate](http://img.cochemist.com/ccimg/176600/176520-13-1_b.png)




![12-Oxabicyclo[9.2.1]tetradeca-1(14),4,8-trien-13-one,5,9-dimethyl-, (4E,8E,11S)- (9CI)](http://img.cochemist.com/ccimg/128300/128232-71-3.png)
![12-Oxabicyclo[9.2.1]tetradeca-1(14),4,8-trien-13-one,5,9-dimethyl-, (4E,8E,11S)- (9CI)](http://img.cochemist.com/ccimg/128300/128232-71-3_b.png)






![1H-3,9a-Methanocyclopent[c]oxocin-1-one,octahydro-7,10-dihydroxy-6a-methyl-4-(1-methylethenyl)-,(3S,4R,6aS,7S,9aR,10R)- (9CI)](http://img.cochemist.com/ccimg/104000/103963-36-6.png)
![1H-3,9a-Methanocyclopent[c]oxocin-1-one,octahydro-7,10-dihydroxy-6a-methyl-4-(1-methylethenyl)-,(3S,4R,6aS,7S,9aR,10R)- (9CI)](http://img.cochemist.com/ccimg/104000/103963-36-6_b.png)





![3-[2,3-Dihydro-2-[4-[2-hydroxy-2-(4-hydroxy-3-methoxyphenyl)-1-(hydroxymethyl)ethoxy]-3,5-dimethoxyphenyl]-3-hydroxymethyl-7-methoxybenzofuran-5-yl]propenal](http://img.cochemist.com/ccimg/97400/97399-78-5.png)
![3-[2,3-Dihydro-2-[4-[2-hydroxy-2-(4-hydroxy-3-methoxyphenyl)-1-(hydroxymethyl)ethoxy]-3,5-dimethoxyphenyl]-3-hydroxymethyl-7-methoxybenzofuran-5-yl]propenal](http://img.cochemist.com/ccimg/97400/97399-78-5_b.png)




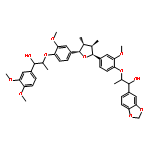

![Benzenemethanol, a,a'-[[(2S,3R,4R,5S)-tetrahydro-3,4-dimethyl-2,5-furandiyl]bis[(2-methoxy-4,1-phenylene)oxy-(1R)-ethylidene]]bis[3,4-dimethoxy-,(aR,a'R)-](http://img.cochemist.com/ccimg/88500/88497-87-4.png)
![Benzenemethanol, a,a'-[[(2S,3R,4R,5S)-tetrahydro-3,4-dimethyl-2,5-furandiyl]bis[(2-methoxy-4,1-phenylene)oxy-(1R)-ethylidene]]bis[3,4-dimethoxy-,(aR,a'R)-](http://img.cochemist.com/ccimg/88500/88497-87-4_b.png)








![[4-(4-AMINO-6,7-DIMETHOXY-QUINAZOLIN-2- YL)PIPERAZIN-1-YL]-(2,5-DIOXABI CYCLO[4.4.0]DECA-6,8,10-TRIEN-4-YL)METHANONE](http://img.cochemist.com/ccimg/74200/74191-85-8.png)
![[4-(4-AMINO-6,7-DIMETHOXY-QUINAZOLIN-2- YL)PIPERAZIN-1-YL]-(2,5-DIOXABI CYCLO[4.4.0]DECA-6,8,10-TRIEN-4-YL)METHANONE](http://img.cochemist.com/ccimg/74200/74191-85-8_b.png)







![5-[2-(3-hydroxy-5-methoxyphenyl)ethyl]-2-methoxyphenol](http://img.cochemist.com/ccimg/67900/67884-30-4.png)
![5-[2-(3-hydroxy-5-methoxyphenyl)ethyl]-2-methoxyphenol](http://img.cochemist.com/ccimg/67900/67884-30-4_b.png)




























![(1S)-1-(3-FURYL)-2-[(1S,4AS,8AS)-5,5,8A-TRIMETHYL-2-METHYLENEDECA<WBR />HYDRO-1-NAPHTHALENYL]ETHANOL](http://img.cochemist.com/ccimg/34200/34169-70-5.png)
![(1S)-1-(3-FURYL)-2-[(1S,4AS,8AS)-5,5,8A-TRIMETHYL-2-METHYLENEDECA<WBR />HYDRO-1-NAPHTHALENYL]ETHANOL](http://img.cochemist.com/ccimg/34200/34169-70-5_b.png)
![2-Propenoic acid,2-methyl-,(3aR,4S,9R,11E,12aR)-2,3,3a,4,5,9,10,12a-octahydro-11-methyl-3-methylene-2,7-dioxo-7H-9,6-methenofuro[2,3-f]oxacycloundecin-4-ylester](http://img.cochemist.com/ccimg/29400/29307-03-7.png)
![2-Propenoic acid,2-methyl-,(3aR,4S,9R,11E,12aR)-2,3,3a,4,5,9,10,12a-octahydro-11-methyl-3-methylene-2,7-dioxo-7H-9,6-methenofuro[2,3-f]oxacycloundecin-4-ylester](http://img.cochemist.com/ccimg/29400/29307-03-7_b.png)




















![(1R,3aR,5aR,5bR,7aR,11aR,11bR,13aR,13bR)-3a,5a,5b,8,8,11a-Hexamethyl-1-(prop-1-en-2-yl)icosahydro-1H-cyclopenta[a]chrysen-9-yl acetate](http://img.cochemist.com/ccimg/1700/1617-68-1.png)
![(1R,3aR,5aR,5bR,7aR,11aR,11bR,13aR,13bR)-3a,5a,5b,8,8,11a-Hexamethyl-1-(prop-1-en-2-yl)icosahydro-1H-cyclopenta[a]chrysen-9-yl acetate](http://img.cochemist.com/ccimg/1700/1617-68-1_b.png)