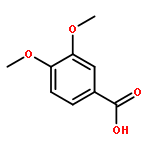
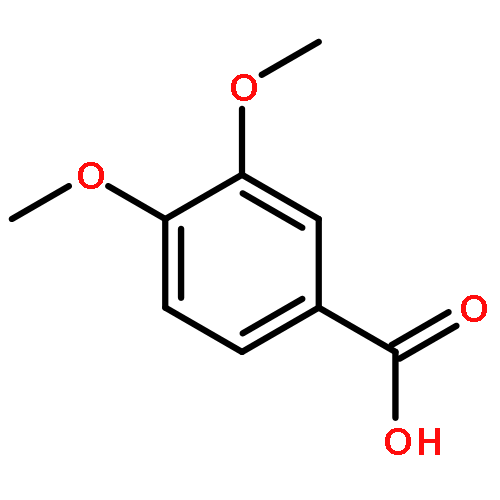
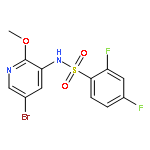
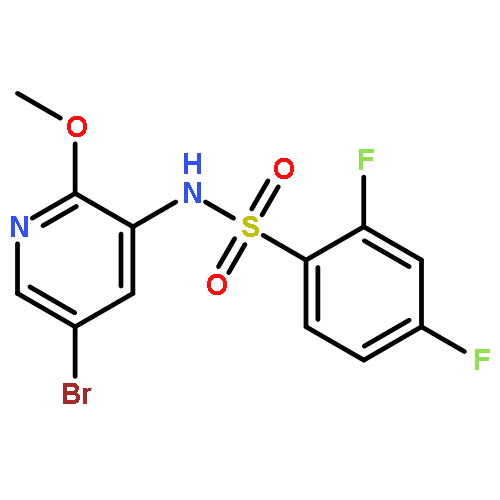
Co-reporter:Jinsong Han, Ying Chen, Chao Yang, Ting Liu, Mingping Wang, Haojie Xu, Ling Zhang, Canhui Zheng, Yunlong Song, Ju Zhu
European Journal of Medicinal Chemistry 2016 Volume 122() pp:684-701
Publication Date(Web):21 October 2016
DOI:10.1016/j.ejmech.2016.06.030
•A series of arylsulfonamides were synthesized as PI3K/mTOR inhibitors.•A two-step structure-based optimization led to the discovery of 13b.•The 13b showed potent broad-spectrum cytotoxic activities and superior PK profiles.•The 13b showed higher potency and less toxicity in vivo than GSK2126458.The phosphoinositide 3-kinase (PI3K) family is one of the most frequently activated enzymes in a wide range of human cancers; thus, inhibition of PI3K represents a promising strategy for cancer therapy. Herein, a series of benzylamine substituted arylsulfonamides were designed and synthesized as dual PI3K/mTOR inhibitors using a strategy integrating focused library design and virtual screening, resulting in the discovery of 13b (NSC765844). The compound 13b exhibits highly potent enzyme inhibition with IC50s of 1.3, 1.8, 1.5, 3.8 and 3.8 nM for PI3Kα, β, γ, δ, and mTOR, respectively. 13b was further evaluated in NCI by an in vitro cytotoxic screening program. Broad-spectrum antitumor activities with mean GI50 value of 18.6 nM against approximately 60 human tumor cell lines were found. 13b displayed favorable physicochemical properties and superior pharmacokinetic profiles for animal studies. It significantly inhibited tumor growth when administered orally in an A549 non-small-cell lung carcinoma xenograft and BEL7404 human hepatocellular carcinoma xenograft models. On the basis of its excellent in vivo efficacy and superior pharmacokinetic profiles, 13b has been selected for further preclinical investigation as a promising anticancer drug candidate.
Co-reporter:Ying Chen, Ling Zhang, Chao Yang, Jinsong Han, Chongqing Wang, Canhui Zheng, Youjun Zhou, Jiaguo Lv, Yunlong Song, Ju Zhu
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 5) pp:957-966
Publication Date(Web):1 March 2016
DOI:10.1016/j.bmc.2016.01.008
The phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway is related to cellular activities. Abnormalities of this signaling pathway were discovered in various cancers, including hepatocellular carcinoma (HCC). The PI3K/mTOR dual inhibitors were proposed to have enhanced antitumor efficacies by targeting multiple points of the signaling pathway. We synthesized a series of propynyl-substituted benzenesulfonamide derivatives as PI3K/mTOR dual inhibitors. Compound 7k (NSC781406) was identified as a highly potent dual inhibitor, which exhibited potent tumor growth inhibition in the hepatocellular carcinoma BEL-7404 xenograft model. Compound 7k may be a potential therapeutic drug candidate for HCC.
Co-reporter:Mingping Wang, Wei Tian, Chongqing Wang, Shihai Lu, Chao Yang, Juan Wang, Yunlong Song, Youjun Zhou, Ju Zhu, Zhiyu Li, Canhui Zheng
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 21) pp:5207-5211
Publication Date(Web):1 November 2016
DOI:10.1016/j.bmcl.2016.09.061
The anti-apoptotic Bcl-2 proteins are attractive targets for anti-cancer drug development, and the discovery of their selective inhibitors has become a research focus. In this Letter, obvious differences in the P1 pocket of the active site between Bcl-2, Bcl-xL, and Mcl-1 proteins were proposed by the structural comparison of these proteins. As a result, the groups in their inhibitors binding to the P1 pockets may have significant effect on the selectivity for these proteins. Based on this hypothesis, five types of derivatives of the lead compound B-1 were designed, and several highly selective inhibitors of Bcl-xL (E-1) or Mcl-1 proteins (G) were found. The selective inhibitors of Mcl-1 protein found in this Letter provide new structural types for the development of novel antitumor agents.
Co-reporter:Chao Yang, Hui Chen, Shihai Lu, Meng Zhang, Wei Tian, Mingping Wang, Ling Zhang, Yunlong Song, Aijun Shen, Youjun Zhou, Ju Zhu, Canhui Zheng
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 15) pp:3464-3467
Publication Date(Web):1 August 2016
DOI:10.1016/j.bmcl.2016.06.043
The luteolin from Flos Chrysanthemi was found to directly bind to the Bcl-2 protein and inhibit the tumor cell growth in our previous study. However, it has been shown to possess wide and week biological activities. In this study, a series of derivatives of luteolin were designed and synthesized, and their tumor cell growth inhibitory activities were evaluated against human leukemia cell line HL-60. The results showed that compounds 1B-2, 2A-3, and 2B-5, with hydrophobic substituted benzyl groups introduced to B ring and hydrogen or methyl introduced to 7-OH group of luteolin, exhibited the strongest inhibitory activity with the IC50 lower than 10 μM, which were significantly more potent than luteolin. The studies presented here offer a good example for modifications of flavones to improve their tumor cell growth inhibitory activities.
Co-reporter:Can-Hui Zheng, Meng Zhang, Hui Chen, Chong-Qing Wang, Min-Min Zhang, Jun-Hang Jiang, Wei Tian, Jia-Guo Lv, Tie-Jun Li, Ju Zhu, You-Jun Zhou
Bioorganic & Medicinal Chemistry Letters 2014 24(19) pp: 4672-4677
Publication Date(Web):
DOI:10.1016/j.bmcl.2014.08.034
Co-reporter:Can-Hui Zheng, Hui Yang, Meng Zhang, Shi-Hai Lu, Duo Shi, Juan Wang, Xiu-Hua Chen, Xiao-Hui Ren, Jia Liu, Jia-Guo Lv, Ju Zhu, You-Jun Zhou
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 1) pp:39-44
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmcl.2011.11.101
On the basis of the comparison of the structure of the Bim BH3: Bcl-xL complex and that of the ABT-737: Bcl-xL complex, a series of class A compounds were designed. These compounds had the basic skeleton of ABT-737 and the h2 residues of Bim BH3. These residues had shown themselves to be relevant to Bim BH3’s broad-spectrum binding properties in saturation mutagenesis assays. Unlike ABT-737, which is a selective inhibitor of anti-apoptotic members of the Bcl-2 protein family, the class A compounds showed broad-spectrum binding activity to target proteins similar to those of Bim BH3 peptide. Then class B compounds were synthesized by modifying the structure of the most effective class A compound, A-4. Most of these class B compounds showed better binding affinity to the target proteins than the class A compounds had. They also showed themselves more effective than ABT-737 at inhibiting growth in multiple tumor cell lines known to express Bcl-xL, Bcl-2, and Mcl-1 proteins at high levels. Compounds B-11 and B-12 had the strongest anti-tumor activity of any compounds we produced. This study suggests that it is feasible to design small-molecule inhibitors based on the structure of Bim BH3, which shows broad-spectrum binding to Bcl-xL, Bcl-2, and Mcl-1 proteins. Our results also suggest that the broad-spectrum properties of small-molecule inhibitors binding to target proteins play a critical role in inhibiting the growth of many tumor cells. Finally, our study provides a series of lead compounds that merit further research into anti-cancer therapeutics.A series of class A compounds which have the basic skeleton of ABT-737 and the h2 residues of Bim BH3 were designed, and showed broad-spectrum binding activity to target proteins similar to that of Bim BH3 peptide. Then class B compounds were synthesized by modifying the structure of the most effective class A compound, A-4, and showed better binding affinity to the target proteins.
Co-reporter:Can-Hui Zheng;Jun Chen;Jia Liu;Xiao-Tian Zhou;Na Liu;Duo Shi;Jing-Jing Huang;Jia-Guo Lv;You-Jun Zhou
Archiv der Pharmazie 2012 Volume 345( Issue 6) pp:454-462
Publication Date(Web):
DOI:10.1002/ardp.201100169
Abstract
A series of 1-phenyl-3,4-dihydroisoquinoline derivatives and several 1-phenyl-1,2,3,4-tetrahydroisoquinoline, 1-phenyl-isoquinoline analogues were synthesized, and their cytotoxicity and tubulin polymerization inhibitory activity were evaluated. The 1-phenyl-3,4-dihydroisoquinoline compounds were found to be potential tubulin polymerization inhibitors. Compound 5n, bearing a 3′OH and 4′OCH3 substituted 1-phenyl B-ring, was shown to confer optimal bioactivity. The single-crystal structure of 5n was further determined by X-ray diffraction, and the binding mode of 5n to tubulin was obtained by molecular docking, which can explain the structure–activity relationships. The studies presented here provide a new structural type for the development of novel antitumor agents.
Co-reporter:Hui Tang, Canhui Zheng, Jiaguo Lv, Juan Wu, Yanan Li, Hui Yang, Bingyue Fu, Chuntong Li, Youjun Zhou, Ju Zhu
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 3) pp:979-982
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmcl.2009.12.050
A series of novel pyrazino[2,1-a]isoquinolin compounds were designed and synthesized, and their antifungal activities in vitro were evaluated. The results showed that all of the compounds exhibited antifungal activities. Some of them exhibited stronger antifungal activities than that of lead compounds and among them compound 11b was the most potent one, which showed more potent than that of the active control fluconazole to the four of the five tested fungi. The studies presented here provide a new structural type for the development of novel antifungal agents.The mode of action of compound 13a with the active site of CYP51 of Candida albicans is reported.


![1H-Imidazole, 1-[(4-bromophenyl)methyl]-2-methyl-](http://img.cochemist.com/ccimg/777900/777865-57-3.png)
![1H-Imidazole, 1-[(4-bromophenyl)methyl]-2-methyl-](http://img.cochemist.com/ccimg/777900/777865-57-3_b.png)









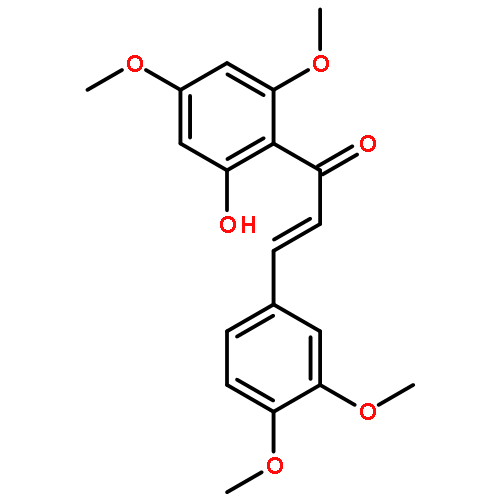
![2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenol](http://img.cochemist.com/ccimg/117100/117048-59-6.png)
![2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenol](http://img.cochemist.com/ccimg/117100/117048-59-6_b.png)
![(2,3-Dihydro-benzo[1,4]dioxin-6-yl)-methyl-amine](http://img.cochemist.com/ccimg/90700/90609-94-2.png)
![(2,3-Dihydro-benzo[1,4]dioxin-6-yl)-methyl-amine](http://img.cochemist.com/ccimg/90700/90609-94-2_b.png)








![ETHANOL, 2,2'-[[(4-BROMOPHENYL)METHYL]IMINO]BIS-](http://img.cochemist.com/ccimg/2800/2743-06-8.png)
![ETHANOL, 2,2'-[[(4-BROMOPHENYL)METHYL]IMINO]BIS-](http://img.cochemist.com/ccimg/2800/2743-06-8_b.png)


![2-[3,4-bis(acetyloxy)phenyl]-4-oxo-4H-chromene-5,7-diyl diacetate](http://img.cochemist.com/ccimg/1100/1061-93-4.png)
![2-[3,4-bis(acetyloxy)phenyl]-4-oxo-4H-chromene-5,7-diyl diacetate](http://img.cochemist.com/ccimg/1100/1061-93-4_b.png)