Co-reporter:Miao Zhan, Yufang Deng, Lifeng Zhao, Guoyi Yan, Fangying Wang, Ye Tian, Lanxi Zhang, Hongxia Jiang, and Yuanwei Chen
Journal of Medicinal Chemistry May 11, 2017 Volume 60(Issue 9) pp:4023-4023
Publication Date(Web):April 14, 2017
DOI:10.1021/acs.jmedchem.7b00357
Dysfunctional signaling of the PI3K/AKT/mTOR pathway in cancer and its crucial role in cell growth and survival have made it a much desired target for cancer therapeutics. A series of dimorpholine substituted thienopyrimidine derivatives had been prepared and evaluated in vitro and in vivo. Among them, compound 14o was identified as a dual Class I PI3K and mTOR kinase inhibitor, which had an approximately 8-fold improvement in mTOR inhibition relative to the class I PI3K inhibitor 1 (pictilisib, GDC-0941). Western blot analysis confirmed the 14o mechanistic modulation of the cellular PI3K/AKT/mTOR pathway through inhibiting phosphorylation of both AKT and S6 in human cancer cell lines. In addition, 14o demonstrated significant efficacy in SKOV-3 and U87MG tumor xenograft models without causing significant weight loss and toxicity.
Co-reporter:Xintuo Yang, Xuehai Pang, Lei Fan, Xinghai Li, Yuanwei Chen
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 9(Issue 9) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.bmcl.2017.03.041
•Sulfonamide showed the hURAT1 inhibition activity.•Structure-activity relationship of sulfonamide as inhibitors of hURAT1.•Compounds 9b and 19b demonstrated moderate PK profile in rats.This letter presents synthesis and structure-activity relationship study of sulfonamide derivatives as inhibitors of Human Uric Acid Transporter 1 (hURAT1). Among all tested sulfonamide derivatives, compounds 9b, 16i and 19b exhibited excellent inhibition activity with IC50 value of 10, 2, and 83 nM, respectively. In addition, compounds 9b and 19b demonstrated moderate PK profile in rats.Download high-res image (52KB)Download full-size image
Co-reporter:Xuehai Pang, Yingwei Wang, Yuanwei Chen
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 12(Issue 12) pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.bmcl.2017.04.071
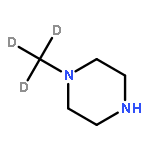
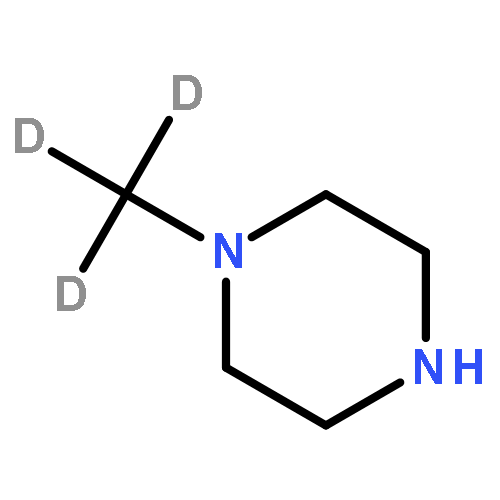
A series of deuterated apalutamide were designed and prepared. Compared to its prototype compound 18, deuterated analogues 19 and 21 showed obviously higher plasma concentrations and better PK parameters after oral administration in mice. In rats, N-trideuteromethyl compound 19 displayed 1.8-fold peak concentration (Cmax), and nearly doubled its drug exposure in plasma (AUC0–∞) compared to compound 18. Unsurprisingly, compounds 18 and 19 had similar affinity for AR in vitro. In summary, the deuteration strategy could obviously improve PK parameters of apalutamide.Download high-res image (74KB)Download full-size image
Co-reporter:Yingwei Wang, Yuanwei Chen
Tetrahedron Letters 2017 Volume 58, Issue 16(Issue 16) pp:
Publication Date(Web):19 April 2017
DOI:10.1016/j.tetlet.2017.02.065
•A three-component reaction of α,γ-dialkylallenoate esters, isatin derivatives with amino acids.•High yields (up to 92%) and diastereoselectivities (up to >20:1).•Two contiguous quaternary stereogenic centers are formed in target products.•A stereochemical model was proposed to rationalize the observed stereoselectivity.Novel polycyclic spiropyrrolidine oxindoles that bear two vicinal quaternary centers and an exocyclic CC double bond have been synthesized by a three-component reaction involving α,γ-dialkylallenoate esters, isatin derivatives and amino acids, by way of an endo-selective 1,3-dipolar cycloaddition. These reactions proceed in high yields and diastereoselectivities. The structures and relative stereochemistry of products were confirmed by NMR, HRMS and X-ray crystallography.Download high-res image (101KB)Download full-size image
Co-reporter:Yingwei Wang;Yufang Deng;Xuehai Pang;Jiang Yu;Lei Fan;Yuanwei Chen;Lifeng Zhao
RSC Advances (2011-Present) 2017 vol. 7(Issue 51) pp:31866-31874
Publication Date(Web):2017/06/21
DOI:10.1039/C7RA02142A
Enzalutamide (ENT) is an approved drug for the treatment of castration resistant prostate cancer (CRPC). Despite its success, the duration of response in patients is still limited with drug resistance. More robust CRPC drugs with novel structural motifs are urgently needed. Here, we designed and synthesized a series of 1-hydroxyl or 1-amino-2,2,2-trifluoro-1-ethyl compounds as isosteres to replace the amide group of ENT. Among the compounds prepared and tested, compund 13b is 2-fold more potent than ENT against LNCaP-AR cells. Western blot analysis showed that 13b dose-dependently inhibits the expression of the prostate-specific antigen (PSA). Further in vivo efficacy studies established that 13b has anti-tumor activity with oral administration at 15 mg kg−1 once daily.
Co-reporter:Miao Zhan, Ren-Zhe Li, Ze-Dong Mou, Chao-Guo Cao, Jie Liu, Yuan-Wei Chen, and Dawen Niu
ACS Catalysis 2016 Volume 6(Issue 5) pp:3381
Publication Date(Web):April 22, 2016
DOI:10.1021/acscatal.6b00719
Described here is an enantioselective approach of making chiral, β-substituted homoallylic organoboronic esters. In the presence of LiOtBu and a catalytic amount of silver salt, commercial bis[(pinacolato)boryl]methane participated in the iridium-catalyzed asymmetric allylation reactions, delivered a “CH2B(pin)” group, and yielded the title compounds from allylic carbonates. The synthetic utility of the prepared chiral organoboronates was demonstrated by their conversion to other important classes of compounds.Keywords: bis[(pinacolato)boryl]methane; boronic esters; catalysis; enantioselective allylation; iridium; silver
Co-reporter:Shiwei Guo, Xuehai Pang, Lingling Peng, Miao Zhan, Lei Fan, Yu Gong, Fanyuan Kang, Yuanwei Chen
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 11) pp:2425-2428
Publication Date(Web):1 June 2015
DOI:10.1016/j.bmcl.2015.03.088
Tivozanib is a potent and selective tyrosine kinase inhibitor of vascular endothelial growth factor receptor-1(VEGFR1), -2(VEGFR2), and -3(VEGFR3). Analog of Tivozanib with deuterium-for-hydrogen replacement in metabolically active site was prepared and evaluated in vitro. Compared to its prototype, deuterated Tivozanib compound HC-1144 retained in vitro activity against VEGFR tyrosine kinases. In vivo pharmacokinetic studies indicated HC-1144 clearly altered the blood circulation behavior, which was proved by significantly prolonged blood circulation half life time (t1/2) and increased AUC0–∞. Therefore, HC-1144 has the potential to be a novel inhibitor against VEGFR tyrosine kinases with long-acting plasma exposure.
Co-reporter:Ruixue Xu;Miao Zhan;Lingling Peng;Xuehai Pang;Jun Yang;Tao Zhang;Hongxia Jiang;Lifeng Zhao;Yuanwei Chen
Journal of Labelled Compounds and Radiopharmaceuticals 2015 Volume 58( Issue 7) pp:308-312
Publication Date(Web):
DOI:10.1002/jlcr.3299
Nintedanib is a novel triple angiokinase inhibitor that inhibits three growth factors simultaneously. Deuterated derivatives of nintedanib at certain metabolically active sites were prepared and evaluated in vitro and in vivo. In particular, deuterated compound SKLB-C2202 had significantly improved pharmacokinetic properties compared with nintedanib. These efforts lay the foundation for further investigating the druggability of SKLB-C2202.
Co-reporter:Miao Zhan, Hongxia Jiang, Xuehai Pang, Tao Zhang, Ruixue Xu, Lifeng Zhao, Yu Liu, Yu Gong, Yuanwei Chen
Tetrahedron Letters 2014 Volume 55(Issue 36) pp:5070-5073
Publication Date(Web):3 September 2014
DOI:10.1016/j.tetlet.2014.07.071
A highly effective and operationally practical method for the regioselective deuteration of N-alkyl-substituted anilines employing Ru3(CO)12 (⩽1 mol %) as catalyst and D2O as deuterium source was described. A variety of N-alkyl-substituted anilines were efficiently deuterated (up to 98%) at the ortho and/or para position with respect to the nitrogen at neutral conditions. Under the present conditions, deuterated anilines can be easily obtained with simple extraction and evaporation. Substituents with aromatic methoxy groups would not influence the selectivity compared to previous method.
Co-reporter:Miao Zhan;Tao Zhang;Haoxi Huang;Yongmei Xie;Yuanwei Chen
Journal of Labelled Compounds and Radiopharmaceuticals 2014 Volume 57( Issue 8) pp:533-539
Publication Date(Web):
DOI:10.1002/jlcr.3210
A simple, cost-effective method for deuteration of carbonyl compounds employing pyrrolidine as catalyst and D2O as deuterium source was described. High degree of deuterium incorporation (up to 99%) and extensive functional group tolerance were achieved. It is the first time that secondary amines are used as catalysts for H/D exchange of carbonyl compounds, which also allow the deuteration of complex pharmaceutically interesting substrates. A possible catalytic mechanism, based on the hydrolysis of 1-pyrrolidino-1-cyclohexene, for this pyrrolidine-catalyzed H/D exchange reaction has been proposed.
.jpg)
![methyl-1 -(chloroacetyl)-3- [methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate](http://img.cochemist.com/ccimg/1174400/1174335-83-1.png)
![methyl-1 -(chloroacetyl)-3- [methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate](http://img.cochemist.com/ccimg/1174400/1174335-83-1_b.png)


![N-[2-Chloro-4-[(6,7-dimethoxy-4-quinolyl)oxy]phenyl]-N'-(5-methyl-3-isoxazolyl)urea](http://img.cochemist.com/ccimg/475200/475108-18-0.png)
![N-[2-Chloro-4-[(6,7-dimethoxy-4-quinolyl)oxy]phenyl]-N'-(5-methyl-3-isoxazolyl)urea](http://img.cochemist.com/ccimg/475200/475108-18-0_b.png)








![Benzenamine, 4-[(4-methoxyphenyl)methyl]-N,N-dimethyl-](http://img.cochemist.com/ccimg/53100/53039-60-4.png)
![Benzenamine, 4-[(4-methoxyphenyl)methyl]-N,N-dimethyl-](http://img.cochemist.com/ccimg/53100/53039-60-4_b.png)
![1-Propanaminium, N,N-dimethyl-N-[2-[(2-methyl-1-oxo-2-propen-1-yl)oxy]ethyl]-3-sulfo-, inner salt, homopolymer](/data/chemimg/3779600/41488-70-4.png)
![1-Propanaminium, N,N-dimethyl-N-[2-[(2-methyl-1-oxo-2-propen-1-yl)oxy]ethyl]-3-sulfo-, inner salt, homopolymer](/data/chemimg/3779600/41488-70-4_b.png)
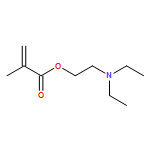
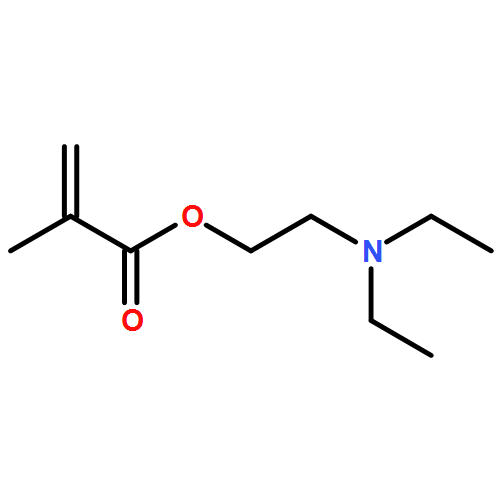
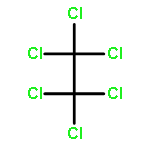
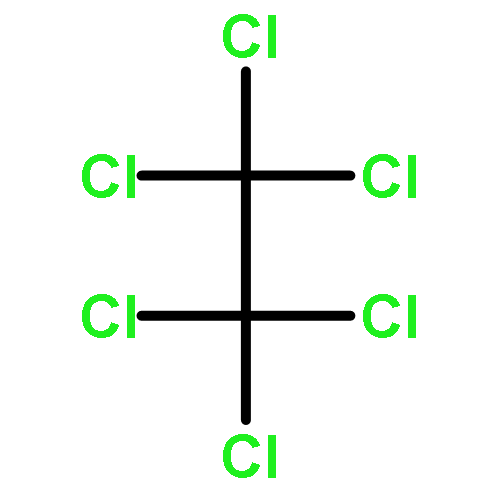




![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)

