Co-reporter:Shawn C. Eady, Molly M. MacInnes, and Nicolai Lehnert
Inorganic Chemistry October 2, 2017 Volume 56(Issue 19) pp:11654-11654
Publication Date(Web):September 21, 2017
DOI:10.1021/acs.inorgchem.7b01589
A series of cobalt bis(benzenedithiolate) complexes with varying benzenedithiolate (general abbreviation: bdt2–) ring substitutions (S2C6X42–) were prepared and adsorbed on inexpensive electrodes composed of (a) reduced graphene oxide (RGO) electrodeposited on fluorine-doped tin oxide (FTO) and (b) highly ordered pyrolytic graphite (HOPG). The catalyst-adsorbed electrodes are characterized by X-ray photoelectron spectroscopy. Catalyst loading across the ligand series improved notably with increasing halide substitution [from 2.7 × 10–11 mol cm–2 for TBA[Co(S2C6H4)2] (1) to 6.22 × 10–10 mol cm–2 for TBA[Co(S2C6Cl4)2] (3)] and increasing ring size of the benzenedithiolate ligand [up to 3.10 × 10–9 mol cm–2 for TBA[Co(S2C10H6)2] (6)]. Electrocatalytic analysis of the complexes immobilized on HOPG elicits a reductive current response indicative of dihydrogen generation in the presence of mildly acidic aqueous solutions (pH 2–4) of trifluoroacetic acid, with overpotentials of around 0.5 V versus SHE (measured vs platinum). Rate constant (kobs) estimates resulting from cyclic voltammetry analysis range from 24 to 230 s–1 with the maximum kobs for TBA[Co(S2C6H2Cl2)2] (2) at an overpotential of 0.59 V versus platinum. Controlled-potential electrolysis studies performed in 0.5 M H2SO4 at −0.5 V versus SHE show impressive initial rate constants of over 500 s–1 under bulk electrolysis conditions; however, steady catalyst deactivation over an 8 h period is observed, with turnover numbers reaching 9.1 × 106. Electrolysis studies reveal that halide substitution is a central factor in improving the turnover stability, whereas the ring size is less of a factor in optimizing the long-term stability of the heterogeneous catalyst manifolds. Catalyst deactivation is likely caused by catalyst desorption from the electrode surfaces.
Co-reporter:Manish Jana, Nabhendu Pal, Corey J. White, Claudia Kupper, Franc Meyer, Nicolai Lehnert, and Amit Majumdar
Journal of the American Chemical Society October 18, 2017 Volume 139(Issue 41) pp:14380-14380
Publication Date(Web):September 27, 2017
DOI:10.1021/jacs.7b08855
Reaction of [Fe2(N-Et-HPTB)(CH3COS)](BF4)2 (1) with (NO)(BF4) produces a nonheme mononitrosyl diiron(II) complex, [Fe2(N-Et-HPTB)(NO)(DMF)3](BF4)3 (2). Complex 2 is the first example of a [FeII{Fe(NO)}7] species and is also the first example of a mononitrosyl diiron(II) complex that mediates the reduction of NO to N2O. This work describes the selective synthesis, detailed characterization and NO reduction activity of 2 and thus provides new insights regarding the mechanism of flavodiiron nitric oxide reductases.
Co-reporter:Kiyoshi Fujisawa;Shoko Soma;Haruka Kurihara;Hai T. Dong;Max Bilodeau
Dalton Transactions 2017 vol. 46(Issue 39) pp:13273-13289
Publication Date(Web):2017/10/10
DOI:10.1039/C7DT01565H
The cobalt–nitrosyl complex [Co(NO)(L3)] is supported by a highly hindered tridentate nitrogen ligand, hydrotris(3-tertiary butyl-5-isopropyl-1-pyrazolyl)borate (denoted as L3−), and shows a linear Co–N–O unit. This complex was prepared by the reaction of the potassium salt of L3− with the cobalt–nitrosyl precursor [Co(NO)2(tmeda)](BPh4) (tmeda = N,N,N,′N′-tetramethylethylenediamine). The obtained cobalt–nitrosyl complex as well as the corresponding products from the reaction with dioxygen, [Co(η2-O2N)(L3)] and [Co(η2-O2NO)(L3)], were characterised by X-ray crystallography and a number of spectroscopic methods including IR/far-IR, UV-Vis, and NMR spectroscopy. We also performed MCD measurements and DFT calculations to further elucidate the electronic structure of [Co(NO)(L3)] and the optical properties of the complex. The MCD spectra reveal two NO-to-Co charge-transfer transitions with strong excited state displacements that give rise to vibrational progressions in the MCD spectra, indicative of a very covalent Co–NO bond. These results provide new insight into the properties of the Co–NO bond and the electron distribution in the complex, which is best described as [CoII(NO−)(L3)].
Co-reporter:Shawn C. Eady, Molly M. MacInnes, and Nicolai Lehnert
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 36) pp:23624
Publication Date(Web):August 18, 2016
DOI:10.1021/acsami.6b05159
Heterogeneous dihydrogen production manifolds comprised of bulk graphite, pencil graphite, graphite powder in Nafion films, graphene, and glassy carbon electrodes with adsorbed proton reduction catalyst TBA[Co(S2C6Cl2H2)2] have been prepared and tested for their efficiency to generate dihydrogen in acidic aqueous media. The catalyst adsorbed on these inexpensive graphitic surfaces consistently displays similar electrocatalytic profiles compared to the same catalyst on highly ordered pyrolytic graphite (HOPG) supports, including high activity in moderately acidic aqueous solutions (pH < 4), moderate overpotentials (0.42 V vs platinum), and some of the highest reported initial turnover frequencies under electrolysis conditions (96 s–1). The exceptions are glassy carbon and single-layer graphene surfaces, which only weakly adsorb the catalyst, with no sustained catalytic current upon acid addition. In particular, the improved stability and good activity observed for the catalyst adsorbed on graphite powder embedded in a Nafion film shows that this is a promising H2 production system that can be assembled at minimal cost and effort.Keywords: dithiolate complexes; graphitic materials; heterogeneous catalysis; hydrogen production; sustainable catalysis
Co-reporter:Amy L. Speelman, Bo Zhang, Alexey Silakov, Kelsey M. Skodje, E. Ercan Alp, Jiyong Zhao, Michael Y. Hu, Eunsuk Kim, Carsten Krebs, and Nicolai Lehnert
Inorganic Chemistry 2016 Volume 55(Issue 11) pp:5485-5501
Publication Date(Web):May 20, 2016
DOI:10.1021/acs.inorgchem.6b00510
Dinitrosyl iron complexes (DNICs) are among the most abundant NO-derived cellular species. Monomeric DNICs can exist in the {Fe(NO)2}9 or {Fe(NO)2}10 oxidation state (in the Enemark–Feltham notation). However, experimental studies of analogous DNICs in both oxidation states are rare, which prevents a thorough understanding of the differences in the electronic structures of these species. Here, the {Fe(NO)2}9 DNIC [Fe(dmp)(NO)2](OTf) (1; dmp = 2,9-dimethyl-1,10-phenanthroline) is synthesized from a ferrous precursor via an unusual pathway, involving disproportionation of an {FeNO}7 complex to yield the {Fe(NO)2}9 DNIC and a ferric species, which is subsequently reduced by NO gas to generate a ferrous complex that re-enters the reaction cycle. In contrast to most {Fe(NO)2}9 DNICs with neutral N-donor ligands, 1 exhibits high solution stability and can be characterized structurally and spectroscopically. Reduction of 1 yields the corresponding {Fe(NO)2}10 DNIC [Fe(dmp)(NO)2] (2). The Mössbauer isomer shift of 2 is 0.08 mm/s smaller than that of 1, which indicates that the iron center is slightly more oxidized in the reduced complex. The nuclear resonance vibrational spectra (NRVS) of 1 and 2 are distinct and provide direct experimental insight into differences in bonding in these complexes. In particular, the symmetric out-of-plane Fe–N–O bending mode is shifted to higher energy by 188 cm–1 in 2 in comparison to 1. Using quantum chemistry centered normal coordinate analysis (QCC-NCA), this is shown to arise from an increase in Fe–NO bond order and a stiffening of the Fe(NO)2 unit upon reduction of 1 to 2. DFT calculations demonstrate that the changes in bonding arise from an iron-centered reduction which leads to a distinct increase in Fe–NO π-back-bonding in {Fe(NO)2}10 DNICs in comparison to the corresponding {Fe(NO)2}9 complexes, in agreement with all experimental findings. Finally, the implications of the electronic structure of DNICs for their reactivity are discussed, especially with respect to N–N bond formation in NO reductases.
Co-reporter:Corey J. White
PNAS 2016 113 (51 ) pp:14474-14476
Publication Date(Web):2016-12-20
DOI:10.1073/pnas.1617953114
Co-reporter:Amy L. Speelman;Dr. Bo Zhang; Carsten Krebs; Nicolai Lehnert
Angewandte Chemie 2016 Volume 128( Issue 23) pp:6797-6800
Publication Date(Web):
DOI:10.1002/ange.201601742
Abstract
Although the interaction of low-spin ferric complexes with nitric oxide has been well studied, examples of stable high-spin ferric nitrosyls (such as those that could be expected to form at typical non-heme iron sites in biology) are extremely rare. Using the TMG3tren co-ligand, we have prepared a high-spin ferric NO adduct ({FeNO}6 complex) via electrochemical or chemical oxidation of the corresponding high-spin ferrous NO {FeNO}7 complex. The {FeNO}6 compound is characterized by UV/Visible and IR spectroelectrochemistry, Mössbauer and NMR spectroscopy, X-ray crystallography, and DFT calculations. The data show that its electronic structure is best described as a high-spin iron(IV) center bound to a triplet NO− ligand with a very covalent iron−NO bond. This finding demonstrates that this high-spin iron nitrosyl compound undergoes iron-centered redox chemistry, leading to fundamentally different properties than corresponding low-spin compounds, which undergo NO-centered redox transformations.
Co-reporter:Ashley B. McQuarters;Subhra Samanta;Pradip Kumar Das;Abhishek Dey
PNAS 2016 Volume 113 (Issue 24 ) pp:6611-6616
Publication Date(Web):2016-06-14
DOI:10.1073/pnas.1600525113
CytP450s have a cysteine-bound heme cofactor that, in its as-isolated resting (oxidized) form, can be conclusively described
as a ferric thiolate species. Unlike the native enzyme, most synthetic thiolate-bound ferric porphyrins are unstable in air
unless the axial thiolate ligand is sterically protected. Spectroscopic investigations on a series of synthetic mimics of
cytP450 indicate that a thiolate-bound ferric porphyrin coexists in organic solutions at room temperature (RT) with a thiyl-radical
bound ferrous porphyrin, i.e., its valence tautomer. The ferric thiolate state is favored by greater enthalpy and is air stable.
The ferrous thiyl state is favored by entropy, populates at RT, and degrades in air. These ground states can be reversibly
interchanged at RT by the addition or removal of water to the apolar medium. It is concluded that hydrogen bonding and local
electrostatics protect the resting oxidized cytP450 active site from degradation in air by stabilizing the ferric thiolate
ground state in contrast to its synthetic analogs.
Co-reporter:Ashley B. McQuarters;Amy L. Speelman
JBIC Journal of Biological Inorganic Chemistry 2016 Volume 21( Issue 8) pp:997-1008
Publication Date(Web):2016 December
DOI:10.1007/s00775-016-1396-1
Second coordination sphere (SCS) effects in proteins are modulated by active site residues and include hydrogen bonding, electrostatic/dipole interactions, steric interactions, and π-stacking of aromatic residues. In Cyt P450s, extended H-bonding networks are located around the proximal cysteinate ligand of the heme, referred to as the ‘Cys pocket’. These hydrogen bonding networks are generally believed to regulate the Fe–S interaction. Previous work identified the S(Cys) → Fe σ CT transition in the high-spin (hs) ferric form of Cyt P450cam and corresponding Cys pocket mutants by low-temperature (LT) MCD spectroscopy [Biochemistry 50:1053, 2011]. In this work, we have investigated the effect of the hydrogen bond from W409 to the axial Cys ligand of the heme in the hs ferric state (with H4B and l-Arg bound) of rat neuronal nitric oxide synthase oxygenase construct (nNOSoxy) using MCD spectroscopy. For this purpose, wt enzyme and W409 mutants were investigated where the H-bonding network with the axial Cys ligand is perturbed. Overall, the results are similar to Cyt P450cam and show the intense S(Cys) → Fe σ CT band in the LT MCD spectrum at about 27,800 cm−1, indicating that this feature is a hallmark of {heme-thiolate} active sites. The discovery of this MCD feature could constitute a new approach to classify {heme-thiolate} sites in hs ferric proteins. Finally, the W409 mutants show that the hydrogen bond from this group only has a small effect on the Fe–S(Cys) bond strength, at least in the hs ferric form of the protein studied here.
Co-reporter:Amy L. Speelman;Dr. Bo Zhang; Carsten Krebs; Nicolai Lehnert
Angewandte Chemie International Edition 2016 Volume 55( Issue 23) pp:6685-6688
Publication Date(Web):
DOI:10.1002/anie.201601742
Abstract
Although the interaction of low-spin ferric complexes with nitric oxide has been well studied, examples of stable high-spin ferric nitrosyls (such as those that could be expected to form at typical non-heme iron sites in biology) are extremely rare. Using the TMG3tren co-ligand, we have prepared a high-spin ferric NO adduct ({FeNO}6 complex) via electrochemical or chemical oxidation of the corresponding high-spin ferrous NO {FeNO}7 complex. The {FeNO}6 compound is characterized by UV/Visible and IR spectroelectrochemistry, Mössbauer and NMR spectroscopy, X-ray crystallography, and DFT calculations. The data show that its electronic structure is best described as a high-spin iron(IV) center bound to a triplet NO− ligand with a very covalent iron−NO bond. This finding demonstrates that this high-spin iron nitrosyl compound undergoes iron-centered redox chemistry, leading to fundamentally different properties than corresponding low-spin compounds, which undergo NO-centered redox transformations.
Co-reporter:Andrew P. Hunt and Nicolai Lehnert
Accounts of Chemical Research 2015 Volume 48(Issue 7) pp:2117
Publication Date(Web):June 26, 2015
DOI:10.1021/acs.accounts.5b00167
The question of why mammalian systems use nitric oxide (NO), a potentially hazardous and toxic diatomic, as a signaling molecule to mediate important functions such as vasodilation (blood pressure control) and nerve signal transduction initially perplexed researchers when this discovery was made in the 1980s. Through extensive research over the past two decades, it is now well rationalized why NO is used in vivo for these signaling functions, and that heme proteins play a dominant role in NO signaling in mammals. Key insight into the properties of heme-nitrosyl complexes that make heme proteins so well poised to take full advantage of the unique properties of NO has come from in-depth structural, spectroscopic, and theoretical studies on ferrous and ferric heme-nitrosyls. This Account highlights recent findings that have led to greater understanding of the electronic structures of heme-nitrosyls, and the contributions that model complex studies have made to elucidate Fe–NO bonding are highlighted. These results are then discussed in the context of the biological functions of heme-nitrosyls, in particular in soluble guanylate cyclase (sGC; NO signaling), nitrophorins (NO transport), and NO-producing enzymes.Central to this Account is the thermodynamic σ-trans effect of NO, and how this relates to the activation of the universal mammalian NO sensor sGC, which uses a ferrous heme as the high affinity “NO detection unit”. It is shown via detailed spectroscopic and computational studies that the strong and very covalent Fe(II)–NO σ-bond is at the heart of the strong thermodynamic σ-trans effect of NO, which greatly weakens the proximal Fe–NHis (or Fe–SCys) bond in six-coordinate ferrous heme-nitrosyls. In sGC, this causes the dissociation of the proximally bound histidine ligand upon NO binding to the ferrous heme, inducing a significant conformational change that activates the sGC catalytic domain for the production of cGMP. This, in turn, leads to vasodilation and nerve signal transduction. Studies on ferrous heme-nitrosyl model complexes have allowed for a quantification of this thermodynamic σ-trans effect of NO, through the use of high-resolution crystal structures, binding constant studies, single-crystal vibrational spectroscopy and density functional theory (DFT) calculations. These studies have further identified the singly occupied molecular orbital (SOMO) of the NO complexes as the key MO that mediates the thermodynamic σ-trans effect of NO.In comparison to ferrous heme-nitrosyls, ferric heme-nitrosyls display thermodynamically much weaker Fe–NO bonds (from NO binding constants), but at the same time much stronger Fe–NO bonds in their ground states (from vibrational spectroscopy). Using spectroscopic investigations coupled to DFT calculations, this apparent contradiction has been rationalized with the involvement of at least three different electronic states in the binding/dissociation of NO to/from ferric hemes. This is of key significance for the release of NO from NO-producing enzymes like NOS, and further forms the basis for ferric hemes to serve as NO transporters in biological systems.
Co-reporter:Nicolai Lehnert;Jonas C. Peters
Inorganic Chemistry 2015 Volume 54(Issue 19) pp:9229-9233
Publication Date(Web):October 5, 2015
DOI:10.1021/acs.inorgchem.5b02124
Co-reporter:Mary Grace I. Galinato, Sarah E. J. Bowman, Jesse G. Kleingardner, Sherri Martin, Jiyong Zhao, Wolfgang Sturhahn, E. Ercan Alp, Kara L. Bren, and Nicolai Lehnert
Biochemistry 2015 Volume 54(Issue 4) pp:1064-1076
Publication Date(Web):December 22, 2014
DOI:10.1021/bi501430z
Cytochrome c (Cyt c) has a heme covalently bound to the polypeptide via a Cys-X-X-Cys-His (CXXCH) linker that is located in the interface region for protein–protein interactions. To determine whether the polypeptide matrix influences iron vibrational dynamics, nuclear resonance vibrational spectroscopy (NRVS) measurements were performed on 57Fe-labeled ferric Hydrogenobacter thermophilus cytochrome c-552, and variants M13V, M13V/K22M, and A7F, which have structural modifications that alter the composition or environment of the CXXCH pentapeptide loop. Simulations of the NRVS data indicate that the 150–325 cm–1 region is dominated by NHis–Fe–SMet axial ligand and polypeptide motions, while the 325–400 cm–1 region shows dominant contributions from ν(Fe–NPyr) (Pyr = pyrrole) and other heme-based modes. Diagnostic spectral signatures that directly relate to structural features of the heme active site are identified using a quantum chemistry-centered normal coordinate analysis (QCC-NCA). In particular, spectral features that directly correlate with CXXCH loop stiffness, the strength of the Fe–His interaction, and the degree of heme distortion are identified. Cumulative results from our investigation suggest that compared to the wild type (wt), variants M13V and M13V/K22M have a more rigid CXXCH pentapeptide segment, a stronger Fe–NHis interaction, and a more ruffled heme. Conversely, the A7F variant has a more planar heme and a weaker Fe–NHis bond. These results are correlated to the observed changes in reduction potential between wt protein and the variants studied here. Implications of these results for Cyt c biogenesis and electron transfer are also discussed.
Co-reporter:Shawn C. Eady, Sabrina L. Peczonczyk, Stephen Maldonado and Nicolai Lehnert
Chemical Communications 2014 vol. 50(Issue 59) pp:8065-8068
Publication Date(Web):04 Jun 2014
DOI:10.1039/C4CC02920H
A heterogeneous dihydrogen (H2) production system has been attained by simply soaking electrodes made from electro-deposited graphene on FTO plated glass in solutions of a cobalt bis(dithiolate) compound. The resulting electrodes are active in weakly acidic aqueous solutions (pH > 3), have relatively low overpotentials (0.37 V versus platinum), show high catalytic rates (TOF > 1000 s−1), and are resistant to degradation by dioxygen.
Co-reporter:Timothy C. Berto, Nan Xu, Se Ryeon Lee, Anne J. McNeil, E. Ercan Alp, Jiyong Zhao, George B. Richter-Addo, and Nicolai Lehnert
Inorganic Chemistry 2014 Volume 53(Issue 13) pp:6398-6414
Publication Date(Web):June 27, 2014
DOI:10.1021/ic5002573
The detoxification of nitric oxide (NO) by bacterial NO reductase (NorBC) represents a paradigm of how NO can be detoxified anaerobically in cells. In order to elucidate the mechanism of this enzyme, model complexes provide a convenient means to assess potential reaction intermediates. In particular, there have been many proposed mechanisms that invoke the formation of a hyponitrite bridge between the heme b3 and nonheme iron (FeB) centers within the NorBC active site. However, the reactivity of bridged iron hyponitrite complexes has not been investigated much in the literature. The model complex {[Fe(OEP)]2(μ-N2O2)} offers a unique opportunity to study the electronic structure and reactivity of such a hyponitrite-bridged complex. Here we report the detailed characterization of {[Fe(OEP)]2(μ-N2O2)} using a combination of IR, nuclear resonance vibrational spectroscopy, electron paramagnetic resonance, and magnetic circular dichroism spectroscopy along with SQUID magnetometry. These results show that the ground-state electronic structure of this complex is best described as having two intermediate-spin (S = 3/2) iron centers that are weakly antiferromagnetically coupled across the N2O22– bridge. The analogous complex {[Fe(PPDME)]2(μ-N2O2)} shows overall similar properties. Finally, we report the unexpected reaction of {[Fe(OEP)]2(μ-N2O2)} in the presence and absence of 1-methylimidizole to yield [Fe(OEP)(NO)]. Density functional theory calculations are used to rationalize why {[Fe(OEP)]2(μ-N2O2)} cannot be formed directly by dimerization of [Fe(OEP)(NO)] and why only the reverse reaction is observed experimentally. These results thus provide insight into the general reactivity of hyponitrite-bridged iron complexes with general relevance for the N–N bond-forming step in NorBC.
Co-reporter:Ashley B. McQuarters and Nicolai Lehnert
Dalton Transactions 2014 vol. 43(Issue 37) pp:13835-13838
Publication Date(Web):30 Jul 2014
DOI:10.1039/C4DT01388C
Recently, a new {RuNO}6 complex, [Ru(L)(PPh3)(NO)(Cl)]2+ (where L = 1-phenyl-1-(pyridin-2-yl)-2-(pyridin-2-ylmethylene)hydrazine), was reported which exhibits a one-electron quasireversible oxidation. The oxidized product, [Ru(L)(PPh3)(NO)(Cl)]3+, was isolated and proposed to be a highly unusual {RuNO}5 complex. In this paper, we investigate the electronic structure of both of these ruthenium complexes by DFT calculations and find that the oxidized species is best described as a {RuNO}6 complex with a co-ligand radical. [Ru(L)(PPh3)(NO)(Cl)]2+ is therefore oxidized to [Ru(L+˙)(PPh3)(NO)(Cl)]3+, i.e. this is an interesting example of a complex with two non-innocent ligands simultaneously bound to a ruthenium center.
Co-reporter:Ashley B. McQuarters;Matthew W. Wolf;Andrew P. Hunt ;Dr. Nicolai Lehnert
Angewandte Chemie International Edition 2014 Volume 53( Issue 19) pp:4750-4752
Publication Date(Web):
DOI:10.1002/anie.201402404
Co-reporter:Ashley B. McQuarters;Matthew W. Wolf;Andrew P. Hunt ;Dr. Nicolai Lehnert
Angewandte Chemie 2014 Volume 126( Issue 19) pp:4846-4848
Publication Date(Web):
DOI:10.1002/ange.201402404
Co-reporter:Lina K. Blusch ; Kathryn E. Craigo ; Vlad Martin-Diaconescu ; Ashley B. McQuarters ; Eckhard Bill ; Sebastian Dechert ; Serena DeBeer ; Nicolai Lehnert ;Franc Meyer
Journal of the American Chemical Society 2013 Volume 135(Issue 37) pp:13892-13899
Publication Date(Web):August 15, 2013
DOI:10.1021/ja406176e
The Siamese-twin porphyrin (2H4) is a unique pyrazole-expanded porphyrin providing two adjacent cavities each offering an {N4} binding motif. It was previously found to form stable dicopper(II) or dinickel(II) complexes where both metal ions are nested in a porphyrin-like environment. This work addresses the rich redox chemistry of the dicopper complex 2Cu2 that originates from the redox synergy of two proximate metal ions in combination with the potentially non-innocent expanded porphyrin ligand. Complementing previous X-ray crystallographic and SQUID data for solid material, the electronic structure of parent 2Cu2 in solution was now investigated by MCD and EPR spectroscopy. This allowed the assignment of UV–vis absorptions and confirmed the drastic twist of the molecule with ferromagnetically coupled copper(II) ions. 2Cu2 was found to exhibit multiple redox events in the potential range from −2.4 to +1.7 V versus Fc/Fc+, and singly oxidized [2Cu2]+ as well as doubly oxidized [2Cu2]2+ were characterized in detail by various analytical and spectroscopic methods. [2Cu2]+ was found by EPR spectroscopy and DFT calculations to have an S = 1/2 ground state, while [2Cu2]2+ is diamagnetic. Single crystal X-ray crystallography of [2Cu2(acetone)2](BF4)2 revealed that the 2Cu2 core is structurally invariant upon two-fold oxidation, while XAS measurements at the Cu K-edge for 2Cu2 and [2Cu2(acetone)2](BF4)2 showed that the copper ions remain in the +2 oxidation state throughout. The combined experimental and computational evidence identified the Siamese-twin porphyrin as a multi-electron redox-active ligand with hidden non-innocence. Each ligand subunit upon oxidation forms a ligand-centered radical, though the spin vanishes because of covalency and strong antiferromagnetic coupling between the ligand radical and the proximate metal ion. Complexes of the Siamese-twin porphyrin may thus serve as a valuable bioinspired platform that combines both metal–ligand and two-metal-ion cooperativities for use in multi-electron processes.
Co-reporter:Timothy C. Berto, Amy L. Speelman, Sheng Zheng, Nicolai Lehnert
Coordination Chemistry Reviews 2013 Volume 257(Issue 1) pp:244-259
Publication Date(Web):1 January 2013
DOI:10.1016/j.ccr.2012.05.007
High-spin non-heme iron–nitrosyls are of direct interest to both the chemical and biological communities as these species exhibit interesting chemical properties and act as direct models for enzymatic intermediates. The electronic ground state of the ferrous NO complexes, {Fe–NO}7, is best described as high-spin FeIII antiferromagnetically coupled to NO−, generating the spectroscopically observed S = 3/2 ground state. These species have been identified as catalytically relevant to a variety of NO-reducing enzymes such as bacterial nitric oxide reductase (NorBC) and flavo(rubredoxin) nitric oxide reductase (FNOR). Recently, the corresponding one-electron reduced {Fe–NO}8 (nitroxyl) complexes have also been implicated as biologically significant species. In this review the available spectroscopic data for {Fe–NO}7 and {Fe–NO}8 mono- and dinuclear non-heme iron–nitrosyls are summarized, and the implications of these results with respect to the electronic structures and reactivities of these species, in particular towards NO reduction, are discussed.Graphical abstractHighlights► Non-heme high-spin {Fe–NO}7 complexes show S = 3/2 ground states where a high-spin Fe(III) is antiferromagnetically coupled to the bound NO− ligand. ► In these complexes, NO− acts mostly as a π donor ligand. The strength of this π bond is modulated by the effective nuclear charge of iron, as evident from a direct correlation of the Fe–NO and N–O stretching frequencies in a series of these compounds. ► The FeNO bond is very covalent in these complexes, such that the bound NO− ligand is not basic, and does not show radical-type reactivity. ► One-electron reduced non-heme high-spin {Fe–N(H)O}8 complexes are postulated as intermediates in bacterial respiratory NO reductases, but the properties of these complexes are not well defined. ► Dinuclear non-heme high-spin ({Fe–NO}7)2 complexes are key intermediates in bacterial scavenging NO reductases.
Co-reporter:Sheng Zheng ; Timothy C. Berto ; Eric W. Dahl ; Melissa B. Hoffman ; Amy L. Speelman
Journal of the American Chemical Society 2013 Volume 135(Issue 13) pp:4902-4905
Publication Date(Web):March 10, 2013
DOI:10.1021/ja309782m
Flavodiiron nitric oxide reductases (FNORs), found in many pathogenic bacteria, are able to detoxify NO by reducing it to N2O. In this way, FNORs equip these pathogens with immunity to NO, which is a central immune defense agent in humans. Hence, FNORs are thought to promote infection of the human body, leading to chronic diseases. Despite this importance of FNORs for bacterial pathogenesis, the mechanism of NO reduction by these enzymes is not well understood. Here we present the synthesis and spectroscopic characterization of the diiron dinitrosyl model complex [Fe2(BPMP)(OPr)(NO)2](BPh4)2. The crystal structure of this complex shows two end-on-coordinated {FeNO}7 units that, based on spectroscopic and electrochemical results, are only weakly electronically coupled. Importantly, reduction of this complex by two electrons leads to the clean formation of N2O in quantitative yield. This complex therefore represents the first example of a functional model system for FNORs. The results provide key mechanistic insight into the mechanism of FNORs and, in particular, represent strong support for the proposed “super-reduced” mechanism for these enzymes.
Co-reporter:Natalie A. Dixon, Ashley B. McQuarters, Jodi S. Kraus, Jonathan B. Soffer, Nicolai Lehnert, Reinhard Schweitzer-Stenner and Elizabeth T. Papish
Chemical Communications 2013 vol. 49(Issue 49) pp:5571-5573
Publication Date(Web):07 May 2013
DOI:10.1039/C3CC00036B
Complexes with bulky hydrotris(triazolyl)borate (Ttz) ligands, TtzCuCO, were used to probe how acids change the donor properties of Ttz ligands. (TtztBu,Me)CuCO shows four distinct protonation states and a gradual increase in the CO stretch. The increased electrophilic nature of the Cu center upon protonation leads to enhanced C–H activation catalysis.
Co-reporter:Lauren E. Goodrich, Saikat Roy, E. Ercan Alp, Jiyong Zhao, Michael Y. Hu, and Nicolai Lehnert
Inorganic Chemistry 2013 Volume 52(Issue 13) pp:7766-7780
Publication Date(Web):June 7, 2013
DOI:10.1021/ic400977h
Because of HNO’s emerging role as an important effector molecule in biology, there is great current interest in the coordination chemistry of HNO and its deprotonated form, the nitroxyl anion (NO–), with hemes. Here we report the preparation of four new ferrous heme-nitroxyl model complexes, {FeNO}8 in the Enemark–Feltham notation, using three electron-poor porphyrin ligands and the bis-picket fence porphyrin H2[3,5-Me-BAFP] (3,5-Me-BAFP2– = 3,5-methyl-bis(aryloxy)-fence porphyrin dianion). Electrochemical reduction of [Fe(3,5-Me-BAFP)(NO)] (1-NO) induces a shift of ν(N–O) from 1684 to 1466 cm–1, indicative of formation of [Fe(3,5-Me-BAFP)(NO)]− (1-NO–), and similar results are obtained with the electron-poor hemes. These results provide the basis to analyze general trends in the properties of ferrous heme-nitroxyl complexes for the first time. In particular, we found a strong correlation between the electronic structures of analogous {FeNO}7 and {FeNO}8 complexes, which we analyzed using density functional theory (DFT) calculations. To further study their reactivity, we have developed a new method for the preparation of bulk material of pure heme {FeNO}8 complexes via corresponding [Fe(porphyrin)]− species. Reaction of [Fe(To-F2PP)(NO)]− (To-F2PP2– = tetra(ortho-difluorophenyl)porphyrin dianion) prepared this way with acetic acid generates the corresponding {FeNO}7 complex along with the release of H2. Importantly, this disproportionation can be suppressed when the bis-picket fence porphyrin complex [Fe(3,5-Me-BAFP)(NO)]− is used, and excitingly, with this system we were able to generate the first ferrous heme-NHO model complex reported to date. The picket fence of the porphyrin renders this HNO complex very stable, with a half-life of ∼5 h at room temperature in solution. Finally, with analogous {FeNO}8 and {FeNHO}8 complexes in hand, their biologically relevant reactivity toward NO was then explored.
Co-reporter:Lauren E. Goodrich, Nicolai Lehnert
Journal of Inorganic Biochemistry 2013 Volume 118() pp:179-186
Publication Date(Web):January 2013
DOI:10.1016/j.jinorgbio.2012.07.024
Soluble guanylate cyclase (sGC) is the primary mammalian nitric oxide (NO) sensor. Through the strong thermodynamic σ-trans effect of NO, binding of NO at the distal side of the ferrous heme induces cleavage of the proximal FeNHis bond, activating the catalytic domain of the enzyme. It has been proposed that nitroxyl (HNO) is also capable of activating sGC, but the key question remains as to whether HNO can induce cleavage of the FeNHis bond. Here we report calculated binding constants for 1-methylimidazole (MI) to [Fe(P)(X)] (P = porphine2 −) where X = NO, HNO, CO, and MI to evaluate the trans interaction of these groups, X, with the proximal imidazole (histidine) in sGC. Systematic assessment of DFT methods suggests that the prediction of accurate MI binding constants is critically dependent on the inclusion of van der Waals interactions (− D functionals). Calculated (B3LYP-D/TZVP) MI binding constants for X = NO and MI are 110 and 5.6 × 105 M− 1, respectively, predicted only one order of magnitude higher than the corresponding experimentally determined values. MI binding constants where X = HNO and CO are consistently predicted to be essentially equal and ~ six orders of magnitude larger than those of NO, indicating that CO and HNO mediate a weak thermodynamic trans effect in this system. Orbital analysis of the key σ-bonding orbital, π*h_dz2, and comparison of FeNMI bond lengths support this prediction. This suggests that HNO does not induce a σ-trans effect strong enough to promote cleavage of the FeNHis bond—a key step in the activation of sGC.Comparison of the thermodynamic σ-trans effect of NO and HNO in ferrous porphyrin 1-methylimidazole complexes as models for the sensing heme in sGC is presented. Decrease in dz2 character in the key π*h_dz2 orbital of the HNO relative to the NO complex indicates a significantly weakened trans effect of HNO relative to NO.Highlights► Soluble guanylate cyclase (sGC) is the primary sensor for NO in mammalian systems. ► Activation of sGC is mediated by the strong thermodynamic σ trans effect of NO. ► DFT is used to compare the thermodynamic trans effect for NO, HNO, CO and imidazole. ► DFT methods are calibrated for the calculation of weak binding constants Keq. ► Inclusion of van der Waals interactions is critical to determine accurate ΔG values.
Co-reporter:Deidra L. Gerlach;Dimitri Coucouvanis
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 22-23) pp:3883-3890
Publication Date(Web):
DOI:10.1002/ejic.201300308
Abstract
In this paper, we present a new design for biologically inspired models for the active site of assimilatory sulfite and nitrite reductases (aSIR and aNIR), which consists of a siroheme that is directly linked to a [4Fe–4S] cubane cluster. The individual components used here to construct this model are a site-differentiated [4Fe–4S] cluster, a bifunctional bridging ligand, and a metalloporphyrin. We have prepared two new site-differentiated clusters, [Fe4S4(TriS)(SPy)] and [Fe4S4(TriS)(SEtIm)], which contain pyridine and imidazole linkers for the binding to a metalloporphyrin, and characterized these compounds, using UV/Vis, IR, and 1H-NMR spectroscopy, cyclic voltammetry (CV), and mass spectrometry. Titration experiments where then performed by using [Zn(TPP)] (TPP2– = meso-tetraphenylporphyrin dianion) and corresponding fluorinated derivatives to find the best [4Fe–4S]–heme combination for an optimal binding of the two components in solution. Excitingly, our results demonstrate the formation of the desired [4Fe–4S]–heme catalytic arrays in solution with high specificity. The best combination of cubane cluster and metalloporphyrin for future catalyst development corresponds to the complex (Bu4N)2[M(To-F2PP)–{Fe4S4(TriS)(SEtIm)}] {To-F2PP2– = meso-tetra(ortho-difluorophenyl)porphyrin dianion}. The binding between these components with M = Zn2+ was further confirmed by CV. Thus, we have created a new type of biologically inspired model system for the aSIR and aNIR active site that leads to a robust attachment of the individual components in solution.
Co-reporter:Deidra L. Gerlach;Dimitri Coucouvanis;Jeff Kampf
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 30) pp:5253-5264
Publication Date(Web):
DOI:10.1002/ejic.201300802
Abstract
Cuboidal iron–sulfur clusters, [4Fe–4S], are important electron-transfer (ET) sites in biology. In addition, more complex structures, usually consisting of modified or fused cubane clusters, are used as active sites in many important enzymes. For example, the Fe–Mo cofactor (FeMoco) of nitrogenase contains two fused cubanes. Here, we report the synthesis of three new para-pyridylthiolate ligated iron–sulfur cubane clusters, two single clusters (Bu4N)2[Fe4S4(SMePy)4] and (Bu4N)2[Fe4S4(SPy)4], and the sulfide-bridged double cubane (Bu4N)4[{Fe4S4(SPy)3}2S] with 4-pyridinethiolato exogenous ligands. The properties of these clusters were then explored by 1H NMR, IR, and UV/Vis spectroscopy, cyclic voltammetry (CV), and X-ray crystallography. Importantly, (Bu4N)4[{Fe4S4(SPy)3}2S] is the first example of a crystallographically characterized sulfide-bridged double cubane with all-thiolato exogenous ligands that is not supported by a large encapsulating ligand. This cluster shows a bridging Fe–S–Fe angle of 104°, its other structural parameters are in close agreement with those of the single-cluster analog (Bu4N)2[Fe4S4(SPy)4]. Finally, the one-electron-reduced forms of (Bu4N)2[Fe4S4(SPy)4] and (Bu4N)4[{Fe4S4(SPy)3}2S] were studied by low-temperature electron paramagnetic resonance (EPR) spectroscopy. Both clusters exhibit reversible one-electron reductions at –401 and –528 mV [vs. the normal hydrogen electrode (NHE)], respectively. The one-electron-reduced forms of both clusters show S = 1/2 ground states as evident from EPR spectroscopy at liquid-helium temperature. The temperature-dependent data for the double cubane further indicate that the extra electron is trapped in one of the clusters of the dimer and that a low-lying excited state is likely present in this complex, close in energy to the ground state.
Co-reporter:Amy L. Speelman ;Dr. Nicolai Lehnert
Angewandte Chemie International Edition 2013 Volume 52( Issue 47) pp:12283-12287
Publication Date(Web):
DOI:10.1002/anie.201305291
Co-reporter:Amy L. Speelman ;Dr. Nicolai Lehnert
Angewandte Chemie 2013 Volume 125( Issue 47) pp:12509-12513
Publication Date(Web):
DOI:10.1002/ange.201305291
Co-reporter:Dr. Nan Xu;Lauren E. Goodrich;Dr. Nicolai Lehnert;Dr. Douglas R. Powell;Dr. George B. Richter-Addo
Angewandte Chemie International Edition 2013 Volume 52( Issue 14) pp:3896-3900
Publication Date(Web):
DOI:10.1002/anie.201208063
Co-reporter:Mukesh Kumar, Natalie A. Dixon, Anna C. Merkle, Matthias Zeller, Nicolai Lehnert, and Elizabeth T. Papish
Inorganic Chemistry 2012 Volume 51(Issue 13) pp:7004-7006
Publication Date(Web):June 6, 2012
DOI:10.1021/ic300160c
Hydrotris(triazolyl)borate (Ttz) ligands form CuNOx (x = 2, 3) complexes for structural and functional models of copper nitrite reductase. These complexes have distinct properties relative to complexes of hydrotris(pyrazolyl)borate (Tp) and neutral tridentate N-donor ligands. The electron paramagnetic resonance spectra of five-coordinate copper complexes show rare nitrogen superhyperfine couplings with the Ttz ligand, indicating strong σ donation. The copper(I) nitrite complex [PPN]+[(TtztBu,Me)CuINO2]− has been synthesized and characterized and allows for the stoichiometric reduction of NO2– to NO with H+ addition. Anionic Cu(I) nitrite complexes are unusual and are stabilized here for the first time because Ttz is a good π acceptor.
Co-reporter:Anna C. Merkle and Nicolai Lehnert
Dalton Transactions 2012 vol. 41(Issue 12) pp:3355-3368
Publication Date(Web):15 Sep 2011
DOI:10.1039/C1DT11049G
The reduction of nitrite to nitric oxide in dissimilatory denitrification is carried out by copper nitrite reductases (CuNIRs) via a type 2 copper site. Extended studies on CuNIRs in combination with model complexes have allowed for the establishment of two potential mechanisms for this transformation. Recent experimental and computational results have revealed further details of this process. In addition, the interaction of NO with copper sites has recently gained much attention. This review discusses recent results in the context of the known coordination chemistry of CuNIRs.
Co-reporter:Anna C. Merkle, Ashley B. McQuarters and Nicolai Lehnert
Dalton Transactions 2012 vol. 41(Issue 26) pp:8047-8059
Publication Date(Web):01 May 2012
DOI:10.1039/C2DT30464C
In this paper, the synthesis, structural and spectroscopic characterization of a series of new Ru(III)–nitrosyls of {RuNO}6 type with the coligand TPA (tris(2-pyridylmethyl)amine) are presented. The complex [Ru(TPA)Cl2(NO)]ClO4 (2) was prepared from the Ru(III) precursor [Ru(TPA)Cl2]ClO4 (1) by simple reaction with NO gas. This led to the surprising displacement of one of the pyridine (py) arms of TPA by NO (instead of the substitution of a chloride anion by NO), as confirmed by X-ray crystallography. NO complexes where TPA serves as a tetradentate ligand were obtained by reacting the new Ru(II) precursor [Ru(TPA)(NO2)2] (3) with a strong acid. This leads to the dehydration of nitrite to NO+, and the formation of the {RuNO}6 complex [Ru(TPA)(ONO)(NO)](PF6)2 (4), which was also structurally characterized. Derivatives of 4 where nitrite is replaced by urea (5) or water (6) were also obtained. The nitrosyl complexes obtained this way were then further investigated using IR and FT-Raman spectroscopy. Complex 2 with the two anionic chloride coligands shows the lowest N–O and highest Ru–NO stretching frequencies of 1903 and 619 cm−1 of all the complexes investigated here. Complexes 5 and 6 where TPA serves as a tetradentate ligand show ν(N–O) at higher energy, 1930 and 1917 cm−1, respectively, and ν(Ru–NO) at lower energy, 577 and 579 cm−1, respectively, compared to 2. These vibrational energies, as well as the inverse correlation of ν(N–O) and ν(Ru–NO) observed along this series of complexes, again support the Ru(II)–NO+ type electronic structure previously proposed for {RuNO}6 complexes. Finally, we investigated the photolability of the Ru–NO bond upon irradiation with UV light to determine the quantum yields (ϕ) for NO photorelease in complexes 2, 4, 5, and additional water-soluble complexes [Ru(H2edta)(Cl)(NO)] (7) and [Ru(Hedta)(NO)] (8). Although {RuNO}6 complexes are frequently proposed as NO delivery agents in vivo, studies that investigate how ϕ is affected by the solvent water are lacking. Our results indicate that neutral water is not a solvent that promotes the photodissociation of NO, which would present a major obstacle to the goal of designing {RuNO}6 complexes as photolabile NO delivery agents in vivo.
Co-reporter:Mary Grace I. Galinato;Jesse G. Kleingardner;E. Ercan Alp;Sarah E. J. Bowman;Jiyong Zhao;Kara L. Bren
PNAS 2012 Volume 109 (Issue 23 ) pp:8896-8900
Publication Date(Web):2012-06-05
DOI:10.1073/pnas.1200345109
The active site of cytochrome c (Cyt c) consists of a heme covalently linked to a pentapeptide segment (Cys-X-X-Cys-His), which provides a link between the heme
and the protein surface, where the redox partners of Cyt c bind. To elucidate the vibrational properties of heme c, nuclear resonance vibrational spectroscopy (NRVS) measurements were performed on 57Fe-labeled ferric Hydrogenobacter thermophilus cytochrome c552, including 13C8-heme–, 13C515N-Met–, and 13C15N-polypeptide (pp)–labeled samples, revealing heme-based vibrational modes in the 200- to 450-cm−1 spectral region. Simulations of the NRVS spectra of H. thermophilus cytochrome c552 allowed for a complete assignment of the Fe vibrational spectrum of the protein-bound heme, as well as the quantitative determination
of the amount of mixing between local heme vibrations and pp modes from the Cys-X-X-Cys-His motif. These results provide the
basis to propose that heme-pp vibrational dynamic couplings play a role in electron transfer (ET) by coupling vibrations of
the heme directly to vibrations of the pp at the protein–protein interface. This could allow for the direct transduction of
the thermal (vibrational) energy from the protein surface to the heme that is released on protein/protein complex formation,
or it could modulate the heme vibrations in the protein/protein complex to minimize reorganization energy. Both mechanisms
lower energy barriers for ET. Notably, the conformation of the distal Met side chain is fine-tuned in the protein to localize
heme-pp mixed vibrations within the 250- to 400-cm−1 spectral region. These findings point to a particular orientation of the distal Met that maximizes ET.
Co-reporter:Corinne D. Sulok, Jonathan L. Bauer, Amy L. Speelman, Birgit Weber, Nicolai Lehnert
Inorganica Chimica Acta 2012 380() pp: 148-160
Publication Date(Web):
DOI:10.1016/j.ica.2011.09.039
Co-reporter:Aaron S. Rury, Lauren E. Goodrich, Mary Grace I. Galinato, Nicolai Lehnert, and Roseanne J. Sension
The Journal of Physical Chemistry A 2012 Volume 116(Issue 32) pp:8321-8333
Publication Date(Web):August 8, 2012
DOI:10.1021/jp304667t
We report evidence for the formation of long-lived photoproducts following excitation of iron(III) tetraphenylporphyrin chloride (Fe(III)TPPCl) in a 1:1 glass of toluene and CH2Cl2 at 77 K. The formation of these photoproducts is dependent on solvent environment and temperature, appearing only in the presence of toluene. No long-lived product is observed in neat CH2Cl2 solvent. A 2-photon absorption model is proposed to account for the power-dependent photoproduct populations. The products are formed in a mixture of spin states of the central iron(III) metal atom. Metastable six-coordinate high-spin and low-spin complexes and a five-coordinate high-spin complex of iron(III) tetraphenylporphyrin are assigned using structure-sensitive vibrations in the resonance Raman spectrum. These species appear in conjunction with resonantly enhanced toluene solvent vibrations, indicating that the Fe(III) compound formed following photoexcitation recruits a toluene ligand from the surrounding environment. Low-temperature transient absorption (TA) measurements are used to explain the dependence of product formation on excitation frequency in this photochemical model. The six-coordinate photoproduct is initially formed in the high-spin Fe(III) state, but population relaxes into both high-spin and low-spin state at 77 K. This is the first demonstration of coupling between the optical and magnetic properties of an iron-centered porphyrin molecule.
Co-reporter:Timothy C. Berto ; Melissa B. Hoffman ; Yuki Murata ; Kira B. Landenberger ; E. Ercan Alp ; Jiyong Zhao
Journal of the American Chemical Society 2011 Volume 133(Issue 42) pp:16714-16717
Publication Date(Web):June 1, 2011
DOI:10.1021/ja111693f
The detoxification of nitric oxide (NO) by bacterial NO reductase (NorBC) has gained much attention as this reaction provides a paradigm as to how NO can be detoxified anaerobically in cells. However, a clear mechanistic picture of how the heme/non-heme active site of NorBC activates NO is lacking, mostly as a result of insufficient knowledge about the properties of the non-heme iron(II)–NO adduct. Here we report the first biomimetic model complexes for this species that closely resemble the coordination environment found in the protein, using the ligands BMPA-Pr and TPA. The systematic investigation of these compounds allowed us to gain key insight into the electronic structure and geometric properties of high-spin non-heme iron(II)–NO adducts. In particular, we show how small changes in the ligand environment of iron could be used by NorBC to greatly modulate the properties, and hence, the reactivity of this species.
Co-reporter:Timothy C. Berto and Nicolai Lehnert
Inorganic Chemistry 2011 Volume 50(Issue 16) pp:7361-7363
Publication Date(Web):July 11, 2011
DOI:10.1021/ic2003854
The role of NO and nitrite-bound methemoglobin (HbIIINO2–) in hypoxic signaling is highly controversial. One provoking possibility is that hemoglobin (Hb) functions as a nitrite anhydrase, producing N2O3 (from nitrite) as an NO carrier. The ability of Hb to generate N2O3 would provide an intriguing means of NO release from red blood cells. We have investigated this proposed new reactivity of Hb using density functional theory (DFT) calculations. For this purpose, models of the Hb/myoglobin (Mb) active site have been constructed. Our results show that the O-bound (nitrito) form of Hb/MbIIINO2– is essential for the formation of N2O3. The formation and release of N2O3 is shown to be energetically favorable by 1–3 kcal/mol, indicating that the anhydrase function of Hb/Mb is biologically feasible.
Co-reporter:Anna C. Merkle ; Nicole L. Fry ; Pradip K. Mascharak
Inorganic Chemistry 2011 Volume 50(Issue 23) pp:12192-12203
Publication Date(Web):October 31, 2011
DOI:10.1021/ic201967f
The Mn-nitrosyl complexes [Mn(PaPy3)(NO)](ClO4) (1; PaPy3– = N,N-bis(2-pyridylmethyl)amine-N-ethyl-2-pyridine-2-carboxamide) and [Mn(PaPy2Q)(NO)](ClO4) (2, PaPy2Q– = N,N-bis(2-pyridylmethyl)amine-N-ethyl-2-quinoline-2-carboxamide) show a remarkable photolability of the NO ligand upon irradiation of the complexes with UV–vis–NIR light [Eroy-Reveles, A. A.; Leung, Y.; Beavers, C. M.; Olmstead, M. M.; Mascharak, P. K. J. Am. Chem. Soc.2008, 130, 4447]. Here we report detailed spectroscopic and theoretical studies on complexes 1 and 2 that provide key insight into the mechanism of NO photolabilization in these compounds. IR- and FT-Raman spectroscopy show N–O and Mn–NO stretching frequencies in the 1720–1750 and 630–650 cm–1 range, respectively, for these Mn-nitrosyls. The latter value for ν(Mn–NO) is one of the highest transition-metal–NO stretching frequencies reported to this date, indicating that the Mn–NO bond is very strong in these complexes. The electronic structure of 1 and 2 is best described as Mn(I)–NO+, where the Mn(I) center is in the diamagnetic low-spin state and the NO+ ligand forms two very strong π backbonds with the dxz and dyz orbitals of the metal. This explains the very strong Mn–NO bonds observed in these complexes, which even supersede the strengths of the Fe– and Ru–NO bonds in analogous (isoelectronic) Fe/Ru(II)–NO+ complexes. Using time-dependent density functional theory (TD-DFT) calculations, we were able to assign the electronic spectra of 1 and 2, and to gain key insight into the mechanism of NO photorelease in these complexes. Upon irradiation in the UV region, NO is released because of the direct excitation of dπ_π* → π*_dπ charge transfer (CT) states (direct mechanism), which is similar to analogous NO adducts of Ru(III) and Fe(III) complexes. These are transitions from the Mn–NO bonding (dπ_π*) into the Mn–NO antibonding (π*_dπ) orbitals within the Mn–NO π backbond. Since these transitions lead to the population of Mn–NO antibonding orbitals, they promote the photorelease of NO. In the case of 1 and 2, further transitions with distinct dπ_π* → π*_dπ CT character are observed in the 450–500 nm spectral range, again promoting photorelease of NO. This is confirmed by resonance Raman spectroscopy, showing strong resonance enhancement of the Mn–NO stretch at 450–500 nm excitation. The extraordinary photolability of the Mn-nitrosyls upon irradiation in the vis–NIR region is due to the presence of low-lying dxy → π*_dπ singlet and triplet excited states. These have zero oscillator strengths, but can be populated by initial excitation into dxy → L(Py/Q_π*) CT transitions between Mn and the coligand, followed by interconversion into the dxy → π*_dπ singlet excited states. These show strong spin–orbit coupling with the analogous dxy → π*_dπ triplet excited states, which promotes intersystem crossing. TD-DFT shows that the dxy → π*_dπ triplet excited states are indeed found at very low energy. These states are strongly Mn–NO antibonding in nature, and hence, promote dissociation of the NO ligand (indirect mechanism). The Mn-nitrosyls therefore show the long sought-after potential for easy tunability of the NO photorelease properties by simple changes in the coligand.
Co-reporter:
Biochemistry 2011 Volume 50(Issue 6) pp:1053-1069
Publication Date(Web):December 15, 2010
DOI:10.1021/bi101911y
Although extensive research has been performed on various cytochrome P450s, especially Cyt P450cam, there is much to be learned about the mechanism of how its functional unit, a heme b ligated by an axial cysteine, is finely tuned for catalysis by its second coordination sphere. Here we study how the hydrogen-bonding network affects the proximal cysteine and the Fe−S(Cys) bond in ferric Cyt P450cam. This is accomplished using low-temperature magnetic circular dichroism (MCD) spectroscopy on wild-type (wt) Cyt P450cam and on the mutants Q360P (pure ferric high-spin at low temperature) and L358P where the “Cys pocket” has been altered (by removing amino acids involved in the hydrogen-bonding network), and Y96W (pure ferric low-spin). The MCD spectrum of Q360P reveals fourteen electronic transitions between 15200 and 31050 cm−1. Variable-temperature variable-field (VTVH) saturation curves were used to determine the polarizations of these electronic transitions with respect to in-plane (xy) and out-of-plane (z) polarization relative to the heme. The polarizations, oscillator strengths, and TD-DFT calculations were then used to assign the observed electronic transitions. In the lower energy region, prominent bands at 15909 and 16919 cm−1 correspond to porphyrin (P) → Fe charge transfer (CT) transitions. The band at 17881 cm−1 has distinct sulfur S(π) → Fe CT contributions. The Q band is observed as a pseudo A-term (derivative shape) at 18604 and 19539 cm−1. In the case of the Soret band, the negative component of the expected pseudo A-term is split into two features due to mixing with another π → π* and potentially a P → Fe CT excited state. The resulting three features are observed at 23731, 24859, and 25618 cm−1. Most importantly, the broad, prominent band at 28570 cm−1 is assigned to the S(σ) → Fe CT transition, whose intensity is generated through a multitude of CT transitions with strong iron character. For wt, Q360P, and L358P, this band occurs at 28724, 28570, and 28620 cm−1, respectively. The small shift of this feature upon altering the hydrogen bonds to the proximal cysteine indicates that the role of the Cys pocket is not primarily for electronic fine-tuning of the sulfur donor strength but is more for stabilizing the proximal thiolate against external reactants (NO, O2, H3O+), and for properly positioning cysteine to coordinate to the iron center. This aspect is discussed in detail.
Co-reporter:Mary Grace I. Galinato;C. Matthew Whaley;Dean Roberts;Peng Wang
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 7) pp:1147-1154
Publication Date(Web):
DOI:10.1002/ejic.201001037
Abstract
The mechanism of hydrogen production in [FeFe] hydrogenase remains elusive. However, a species featuring a terminal hydride bound to the distal Fe is thought to be the key intermediate leading to hydrogen production. In this study, density functional theory (DFT) calculations on the terminal (H-term) and bridging (μ-H) hydride isomers of (μ-edt)[Fe2(PMe3)4(CO)2H]+ are presented in order to understand the factors affecting their propensity for protonation. Relative to H-term, μ-H is 12.7 kcal/mol more stable, which contributes to its decreased reactivity towards an acid. Potential energy surface (PES) calculations for the reaction of the H-term isomer with 4-nitropyridinium, a proton source, further reveal a lower activation energy barrier (14.5 kcal/mol) for H-term than for μ-H (29 kcal/mol). Besides these energetic considerations, the H-term isomer displays a key molecular orbital (MO <139>) that has a relatively strong hydride (1s) contribution (23 %), which is not present in the μ-H isomer. This indicates a potential orbital control of the reaction of the hydride complexes with acid. The lower activation energy barrier and this key MO together control the overall catalytic activity of (μ-edt)[Fe2(PMe3)4(CO)2(H-term)]+. Lastly, Raman and IR spectroscopy were performed in order to probe the ν(Fe-H) stretching mode of the two isomers and their deuterated counterparts. A ν(Fe-H) stretching mode was observed for the μ-H complex at 1220 cm–1. However, the corresponding mode is not observed for the less stable H-term isomer.
Co-reporter:Deidra L. Gerlach ;Dr. Nicolai Lehnert
Angewandte Chemie 2011 Volume 123( Issue 35) pp:8133-8135
Publication Date(Web):
DOI:10.1002/ange.201102979
Co-reporter:Deidra L. Gerlach ;Dr. Nicolai Lehnert
Angewandte Chemie International Edition 2011 Volume 50( Issue 35) pp:7984-7986
Publication Date(Web):
DOI:10.1002/anie.201102979
Co-reporter:Chuanjiang Hu ; Corinne D. Sulok ; Florian Paulat ; Nicolai Lehnert ; Anna I. Twigg ; Michael P. Hendrich ; Charles E. Schulz ;W. Robert Scheidt
Journal of the American Chemical Society 2010 Volume 132(Issue 11) pp:3737-3750
Publication Date(Web):March 1, 2010
DOI:10.1021/ja907584x
We report detailed studies on two S = 2 electronic states of high-spin iron(II) porphyrinates. These two states are exemplified by the five-coordinate derivatives with either neutral imidazole or anionic imidazolate as the axial ligand. The application of several physical methods all demonstrate distinctive differences between the two states. These include characteristic molecular structure differences, Mössbauer spectra, magnetic circular dichroism spectroscopy, and integer-spin EPR spectral distinctions. These distinctions are supported by DFT calculations. The two states are characterized by very different spatial properties of the doubly occupied orbital of the high-spin that are consonant with the physical properties.
Co-reporter:Nicolai Lehnert ; J. Timothy Sage ; Nathan Silvernail ; W. Robert Scheidt ; E. Ercan Alp ; Wolfgang Sturhahn ;Jiyong Zhao
Inorganic Chemistry 2010 Volume 49(Issue 15) pp:7197-7215
Publication Date(Web):June 29, 2010
DOI:10.1021/ic1010677
This paper presents oriented single-crystal Nuclear Resonance Vibrational Spectroscopy (NRVS) data for the six-coordinate (6C) ferrous heme-nitrosyl model complex [57Fe(TPP)(MI)(NO)] (1; TPP2− = tetraphenylporphyrin dianion; MI = 1-methylimidazole). The availability of these data enables for the first time the detailed simulation of the complete NRVS data, including the porphyrin-based vibrations, of a 6C ferrous heme-nitrosyl, using our quantum chemistry centered normal coordinate analysis (QCC-NCA). Importantly, the Fe−NO stretch is split by interaction with a porphyrin-based vibration into two features, observed at 437 and 472 cm−1. The 437 cm−1 feature is strongly out-of-plane (oop) polarized and shows a 15N18O isotope shift of 8 cm−1 and is therefore assigned to ν(Fe−NO). The admixture of Fe−N−O bending character is small. Main contributions to the Fe−N−O bend are observed in the 520−580 cm−1 region, distributed over a number of in-plane (ip) polarized porphyrin-based vibrations. The main component, assigned to δip(Fe−N−O), is identified with the feature at 563 cm−1. The Fe−N−O bend also shows strong mixing with the Fe−NO stretching internal coordinate, as evidenced by the oop NRVS intensity in the 520−580 cm−1 region. Very accurate normal mode descriptions of ν(Fe−NO) and δip(Fe−N−O) have been obtained in this study. These results contradict previous interpretations of the vibrational spectra of 6C ferrous heme-nitrosyls where the higher energy feature at ∼550 cm−1 had usually been associated with ν(Fe−NO). Furthermore, these results provide key insight into NO binding to ferrous heme active sites in globins and other heme proteins, in particular with respect to (a) the effect of hydrogen bonding to the coordinated NO and (b) changes in heme dynamics upon NO coordination. [Fe(TPP)(MI)(NO)] constitutes an excellent model system for ferrous NO adducts of myoglobin (Mb) mutants where the distal histidine (His64) has been removed. Comparison to the reported vibrational data for wild-type (wt) Mb−NO then shows that the effect of H bonding to the coordinated NO is weak and mostly leads to a polarization of the π/π* orbitals of bound NO. In addition, the observation that δip(Fe−N−O) does not correlate well with ν(N−O) can be traced back to the very mixed nature of this mode. The Fe−N(imidazole) stretching frequency is observed at 149 cm−1 in [Fe(TPP)(MI)(NO)], and spectral changes upon NO binding to five-coordinate ferrous heme active sites are discussed. The obtained high-quality force constants for the Fe−NO and N−O bonds of 2.57 and 11.55 mdyn/Å can further be compared to those of corresponding 5C species, which allows for a quantitative analysis of the σ trans interaction between the proximal imidazole (His) ligand and NO. This is key for the activation of the NO sensor soluble guanylate cyclase. Finally, DFT methods are calibrated against the experimentally determined vibrational properties of the Fe−N−O subunit in 1. DFT is in fact incapable of reproducing the vibrational energies and normal mode descriptions of the Fe−N−O unit well, and thus, DFT-based predictions of changes in vibrational properties upon heme modification or other perturbations of these 6C complexes have to be treated with caution.
Co-reporter:Mary Grace I. Galinato, C. Matthew Whaley and Nicolai Lehnert
Inorganic Chemistry 2010 Volume 49(Issue 7) pp:3201-3215
Publication Date(Web):March 12, 2010
DOI:10.1021/ic9022135
Research on simple [FeFe] hydrogenase model systems of type (μ-S2R)[Fe(CO)3]2 (R = C2H4 (edt), C3H6 (pdt)) which have been shown to function as robust electrocatalysts for proton reduction, provides a reference to understand the electronic and vibrational properties of the active site of [FeFe] hydrogenases and of more sophisticated model systems. In this study, the solution and solid state Raman spectra of (μ-edt)[Fe(CO)3]2 and of the corresponding 13CO-labeled complex are presented and analyzed in detail, with focus on the ν(C═O) and ν(Fe−CO)/δ(Fe−C═O) vibrational regions. These regions are specifically important as vibrations involving CO ligands serve as probes for the “electron richness” of low-valent transition metal centers and the geometric structures of the complexes. The obtained vibrational spectra have been completely assigned in terms of the ν(C═O), ν(Fe−CO), and δ(Fe−C═O) modes, and the force constants of the important C═O and Fe−CO bonds have been determined using our Quantum Chemistry Centered Normal Coordinate Analysis (QCC-NCA). In the 400−650 cm−1 region, fifteen mixed ν(Fe−CO)/δ(Fe−C═O) modes have been identified. The most prominent Raman peaks at 454, 456, and 483 cm−1 correspond to a combination of ν(Fe−CO) stretching and δ(Fe−C═O) linear bending modes. The less intense peaks at 416 cm−1 and 419 cm−1 correspond to pure δ(Fe−C═O) linear bends. In the ν(C═O) region, the ν(C═O) normal modes at lower energy (1968 and 1964 cm−1) are almost pure equatorial (eq) ν(C═O)eq stretching vibrations, whereas the remaining four ν(C═O) normal modes show dominant (C═O)eq (2070 and 1961 cm−1) and (C═O)ax (2005 and 1979 cm−1; ax = axial) contributions. Importantly, an inverse correlation between the f(C═O)ax/eq and f(Fe−CO)ax/eq force constants is obtained, in agreement with the idea that the Fe(I)−CO bond in these types of complexes is dominated by π-backdonation. Compared to the reduced form of [FeFe] hydrogenase (Hred), the ν(C═O) vibrational frequencies of (μ-edt)[Fe(CO)3]2 are higher in energy, indicating that the dinuclear iron core in (μ-edt)[Fe(CO)3]2 is less electron rich compared to Hred in the actual enzyme. Finally, quantum yields for the photodecomposition of (μ-edt)[Fe(CO)3]2 have been determined.
Co-reporter:Nicolai Lehnert ; Mary Grace I. Galinato ; Florian Paulat ; George B. Richter-Addo ; Wolfgang Sturhahn ; Nan Xu ;Jiyong Zhao
Inorganic Chemistry 2010 Volume 49(Issue 9) pp:4133-4148
Publication Date(Web):March 26, 2010
DOI:10.1021/ic902181e
This study presents Nuclear Resonance Vibrational Spectroscopy (NRVS) data on the five-coordinate (5C) ferrous heme−nitrosyl complex [Fe(OEP)(NO)] (1, OEP2− = octaethylporphyrinato dianion) and the corresponding 15N18O labeled complex. The obtained spectra identify two isotope sensitive features at 522 and 388 cm−1, which shift to 508 and 381 cm−1, respectively, upon isotope labeling. These features are assigned to the Fe−NO stretch ν(Fe−NO) and the in-plane Fe−N−O bending mode δip(Fe−N−O), the latter has been unambiguously assigned for the first time for 1. The obtained NRVS data were simulated using our quantum chemistry centered normal coordinate analysis (QCC-NCA). Since complex 1 can potentially exist in 12 different conformations involving the FeNO and peripheral ethyl orientations, extended density functional theory (DFT) calculations and QCC-NCA simulations were performed to determine how these conformations affect the NRVS properties of [Fe(OEP)NO]. These results show that the properties and force constants of the FeNO unit are hardly affected by the conformational changes involving the ethyl substituents. On the other hand, the NRVS-active porphyrin-based vibrations around 340−360, 300−320, and 250−270 cm−1 are sensitive to the conformational changes. The spectroscopic changes observed in these regions are due to selective mechanical couplings of one component of Eu-type (in ideal D4h symmetry) porphyrin-based vibrations with the in-plane Fe−N−O bending mode. This leads to the observed variations in Fe(OEP) core mode energies and NRVS intensities without affecting the properties of the FeNO unit. The QCC-NCA simulated NRVS spectra of 1 show excellent agreement with experiment, and indicate that conformer F is likely present in the samples of this complex investigated here. The observed porphyrin-based vibrations in the NRVS spectra of 1 are also assigned based on the QCC-NCA results. The obtained force constants of the Fe−NO and N−O bonds are 2.83−2.94 (based on the DFT functional applied) and about 12.15 mdyn/Å, respectively. The electronic structures of 5C ferrous heme−nitrosyls in different model complexes are then analyzed, and variations in their properties based on different porphyrin substituents are explained. Finally, the shortcomings of different DFT functionals in describing the axial FeNO subunit in heme−nitrosyls are elucidated.
Co-reporter:Nan Xu ; Lauren E. Goodrich ; Nicolai Lehnert ; Douglas R. Powell ;George B. Richter-Addo
Inorganic Chemistry 2010 Volume 49(Issue 10) pp:4405-4419
Publication Date(Web):April 14, 2010
DOI:10.1021/ic901751z
Nitrosamines are well-known for their toxic and carcinogenic properties. The metabolic activation of nitrosamines occurs via interaction with the heme-containing cytochrome P450 enzymes. We report the preparation and structural characterization of a number of nitrosamine adducts of synthetic iron porphyrins. The reactions of the cations [(por)Fe(THF)2]ClO4 (por = TPP, TTP, OEP) with dialkylnitrosamines (R2NNO; R2 = Me2, Et2, (cyclo-CH2)4, (cyclo-CH2)5, (PhCH2)2) in toluene generate the six-coordinate high-spin (S = 5/2) [(por)Fe(ONNR2)2]ClO4 compounds and a five-coordinate intermediate-spin (S = 3/2) [(OEP)Fe(ONNMe2)]ClO4 derivative in 57−72% yields (TPP = 5,10,15,20-tetraphenylporphyrinato dianion, TTP = 5,10,15,20-tetra-p-tolylporphyrinato dianion, OEP = 2,3,7,8,12,13,17,18-octaethylporphyrinato dianion). The N−O and N−N vibrations of the coordinated nitrosamine groups in [(por)Fe(ONNR2)2]ClO4 occur in the 1239−1271 cm−1 range. Three of the six-coordinate [(por)Fe(ONNR2)2]ClO4 compounds and one five-coordinate [(OEP)Fe(ONNMe2)]ClO4 compound have been characterized by single crystal X-ray crystallography. All the nitrosamine ligands in these complexes bind to the ferric centers via a sole η1-O binding mode. No arylnitrosamine adducts were obtained from the reactions of the precursor compounds [(por)Fe(THF)2]ClO4 with three arylnitrosamines (Ph2NNO, Ph(Me)NNO, Ph(Et)NNO). However, prolonged exposure of [(por)Fe(THF)2]ClO4 to these arylnitrosamines resulted in the formation of the known five-coordinate (por)Fe(NO) derivatives. The latter (por)Fe(NO) compounds were obtained more readily by the reactions of the three arylnitrosamines with the four-coordinate (por)FeII precursors.
Co-reporter:Lauren E. Goodrich ; Florian Paulat ; V. K. K. Praneeth
Inorganic Chemistry 2010 Volume 49(Issue 14) pp:6293-6316
Publication Date(Web):July 12, 2010
DOI:10.1021/ic902304a
This review summarizes recent developments in the investigation of the electronic structures, spectroscopic properties, and reactivities of ferrous and ferric heme-nitrosyls and how this relates to important biological processes. Ferrous heme-nitrosyls show interesting variations in electronic structure as a function of the different types of proximal ligands, as is evident from electron paramagnetic resonance, magnetic circular dichroism, and vibrational spectroscopy. In particular, coordination of imidazoles like histidine (His) increases the radical character on NO and, in this way, could help activate the bound NO for catalysis. Vice versa, the bound NO ligand imposes a strong σ trans effect on the proximal His, which, in the case of soluble guanylate cyclase (sGC), the biological NO sensor protein, induces breaking of the FeII−His bond and activates the protein. The possibility of sGC activation by HNO is also discussed. Finally, the properties of ferrous heme-nitrosyls with proximal cysteinate (Cys) coordination are evaluated. It has been known for some time that ferric heme-nitrosyls are intrinsically more labile than their ferrous counterparts, but the underlying reasons for this observation have not been clarified. New results show that this property relates to the presence of a low-lying excited state that is dissociative with respect to the FeIII−NO bond. On the other hand, the ground state of these complexes is best described as FeII−NO+, which shows a very strong Fe−NO bond, as is evident from vibrational spectroscopy. NO, therefore, is a weak ligand to ferric heme, which, at the same time, forms a strong Fe−NO bond. This is possible because the thermodynamic weakness and spectroscopic strength of the Fe−NO bond relate to the properties of different electronic states. Thiolate coordination to ferric hemes leads to a weakening of both the Fe−NO and N−O bonds as a function of the thiolate donor strength. This observation can be explained by a σ backbond into the σ* orbital of the Fe−N−O unit that is mediated by the thiolate σ-donor orbital via orbital mixing. This is a new interaction in heme-nitrosyl that has not been observed before. This also induces a bending of the Fe−N−O subunit in these cases. New spectroscopic data on a corresponding model complex are included in this paper. Finally, the mechanism of NO reduction by cytochrome P450nor is elucidated based on recent density functional theory results.
Co-reporter:Nicolai Lehnert ;W. Robert Scheidt
Inorganic Chemistry 2010 Volume 49(Issue 14) pp:6223-6225
Publication Date(Web):July 12, 2010
DOI:10.1021/ic101080g
Co-reporter:Timothy C. Berto ; V. K. K. Praneeth ; Lauren E. Goodrich
Journal of the American Chemical Society 2009 Volume 131(Issue 47) pp:17116-17126
Publication Date(Web):November 6, 2009
DOI:10.1021/ja904368n
A series of substituted tetraphenylporphyrin type macrocycles (TMP or To-F2PP) with covalently attached N-donor ligands (pyridine or imidazole linker) have been synthesized. Linkers with varying chain lengths and designs have been applied to systematically investigate the effect of chain length and rigidity on the binding affinity of the linker to the corresponding Fe(II)−NO heme complexes. The binding of the linker is monitored in solution using a variety of spectroscopic methods including UV−vis absorption, EPR, and IR spectroscopy. Both the N−O stretching frequency and the imidazole 14N hyperfine coupling constants show a good correlation with the Fe−(N-donor) bond strength in these systems. The complexes with covalently attached pyridyl and alkyl imidazole ligands only exhibit weak interactions of the linker with iron(II). However, the stable six-coordinate complex [Fe(To-F2PP-BzIM)(NO)] (4) is obtained when a rigid benzyl linker is applied. This complex exhibits typical properties of six-coordinate ferrous heme-nitrosyls in which an N-donor ligand is bound trans to NO, including the Soret band at 427 nm and the typical nine line 14N hyperfine splitting in the EPR spectrum. A crystal structure has been obtained for the corresponding zinc complex. Here, we report the first systematic study on the requirements for the formation of stable six-coordinate ferrous heme nitrosyl complexes in solution at room temperature in the absence of excess axial N-donor ligand.
Co-reporter:Anna C. Merkle and Nicolai Lehnert
Inorganic Chemistry 2009 Volume 48(Issue 24) pp:11504-11506
Publication Date(Web):November 25, 2009
DOI:10.1021/ic9018376
Density functional theory calculations were used to investigate the binding mode of copper(I) nitrosyl (Cu(I)−NO) in copper nitrite reductase (CuNIR). The end-on Cu(I)−NO geometry (2) was found to be the global energy minimum, while the side-on binding mode (1) corresponds to a local minimum. Isoleucine-257 severely interacts sterically with the Cu(I)−NO unit when bound end-on but not in the side-on case. In addition, the side-on geometry is also stabilized by a hydrogen bond between aspartic acid-98 and NO, estimated to be ∼3 kcal/mol. The steric constraint of the CuNIR active site is mainly responsible for the observed side-on coordination of NO in the CuNIR crystal structure. We speculate that a small conformational change of the active site that slightly changes the position of isoleucine-257 would allow NO to bind end-on. This explains the observed end-on binding of NO to copper(I) when CuNIR is in solution.
Co-reporter:Florian Paulat
Inorganic Chemistry 2008 Volume 47(Issue 11) pp:4963-4976
Publication Date(Web):April 25, 2008
DOI:10.1021/ic8002838
High-spin (hs) ferric heme centers occur in the catalytic or redox cycles of many metalloproteins and exhibit very complicated magnetic circular dichroism (MCD) and UV−vis absorption spectra. Therefore, detailed assignments of the MCD spectra of these species are missing. In this study, the electronic spectra (MCD and UV−vis) of the five-coordinate hs ferric model complex [Fe(TPP)(Cl)] are analyzed and assigned for the first time. A correlated fit of the absorption and low-temperature MCD spectra of [Fe(TPP)(Cl)] lead to the identification of at least 20 different electronic transitions. The assignments of these spectra are based on the following: (a) variable temperature and variable field saturation data, (b) time-dependent density functional theory calculations, (c) MCD pseudo A-terms, and (d) correlation to resonance Raman (rRaman) data to validate the assignments. From these results, a number of puzzling questions about the electronic spectra of [Fe(TPP)(Cl)] are answered. The Soret band in [Fe(TPP)(Cl)] is split into three components because one of its components is mixed with the porphyrin A2u⟨72⟩ → Eg⟨82/83⟩ (π → π*) transition. The broad, intense absorption feature at higher energy from the Soret band is due to one of the Soret components and a mixed σ and π chloro to iron CT transition. The high-temperature MCD data allow for the identification of the Qv band at 20 202 cm−1, which corresponds to the C-term feature at 20 150 cm−1. Q is not observed but can be localized by correlation to rRaman data published before. Finally, the low energy absorption band around 650 nm is assigned to two P → Fe charge transfer transitions, one being the long sought after A1u(HOMO) → dπ transition.
Co-reporter:Florian Paulat, Timothy C. Berto, Serena DeBeer George, Lauren Goodrich, V. K. K. Praneeth, Corinne D. Sulok and Nicolai Lehnert
Inorganic Chemistry 2008 Volume 47(Issue 24) pp:11449-11451
Publication Date(Web):November 8, 2008
DOI:10.1021/ic801626w
This Communication addresses a long-standing problem: the exact vibrational assignments of the low-energy modes of the Fe−N−O subunit in six-coordinate ferrous heme nitrosyl model complexes. This problem is addressed using nuclear resonance vibrational spectroscopy (NRVS) coupled to 15N18O isotope labeling and detailed simulations of the obtained data. Two isotope-sensitive features are identified at 437 and 563 cm−1. Normal coordinate analysis shows that the 437 cm−1 mode corresponds to the Fe−NO stretch, whereas the 563 cm−1 band is identified with the Fe−N−O bend. The relative NRVS intensities of these features determine the degree of vibrational mixing between the stretch and the bend. The implications of these results are discussed with respect to the trans effect of imidazole on the bound NO. In addition, a comparison to myoglobin-NO (Mb-NO) is made to determine the effect of the Mb active site pocket on the bound NO.
Co-reporter:Florian Paulat, Nicolai Lehnert, Yoko Ishikawa, Ken-Ichi Okamoto, Kiyoshi Fujisawa
Inorganica Chimica Acta 2008 Volume 361(Issue 4) pp:901-915
Publication Date(Web):3 March 2008
DOI:10.1016/j.ica.2007.05.027
Starting from copper(II) hydrotris(pyrazolyl)borate precursors and substituted hydrazines, we were able to synthesize mononuclear and binuclear copper(I)–diazene complexes for the first time. The mechanism of these reactions corresponds to a simple hydrazine oxidation by the copper(II) centers. Binuclear diazene complexes contain the central Cu(I)–NRNR–Cu(I) unit, which we have structurally and spectroscopically characterized. Interestingly, usage of a sterically demanding hydrotris(pyrazolyl)borate ligand that prevents the dimer formation, leads to decomposition of the diazene complex and the isolation of a copper(I)–hydrazine complex. The same is observed when a tris(pyrazolyl)methane ligand is applied. Finally, using 1,1-diphenylhydrazine or the corresponding monophenyl derivative, we were able to obtain mononuclear Cu(I)–diazene complexes. In this case, the phenyl substituent(s) prevent the dimerization, but also stabilize the diazene ligand. Mononuclear and binuclear Cu(I)–diazene complexes are characterized by their intense reddish to purple color, which is caused by an intense Cu-d to diazene-π∗ transition in the visible region. Resonance Raman spectra of the diazene complexes show the Cu–N and N–N stretching vibrations in the 500 cm−1 and 1350–1400 cm−1 energy regions, respectively, in agreement with the diazene description of the complexes. The copper(I)–diazene bond in these complexes is dominated by π backbonding.Mononuclear and binuclear diazene and hydrazine complexes of copper(I) have been prepared for the first time and structurally and spectroscopically characterized. Examples for mononuclear complexes are shown in the figure. For this purpose, hydrotris(pyrazolyl)borate coligands have been applied. The diazene complexes show purple to red colors, unusual for copper(I). These colors are caused by intense Cu(I)-d to diazene-πv∗ CT transitions.
Co-reporter:Shawn C. Eady, Tanya Breault, Levi Thompson and Nicolai Lehnert
Dalton Transactions 2016 - vol. 45(Issue 3) pp:NaN1151-1151
Publication Date(Web):2015/11/23
DOI:10.1039/C5DT03744A
Penta-coordinate iron carbonyl complexes that are built around the rigid Fe(PNP) motif (PNP = (C6H5)2PN(R)P(C6H5)2) are synthesized and structurally and spectroscopically characterized. These complexes allow for facile customization of the secondary ligand sphere with various types of linkers and functional groups. The new [Fe(S2C6H4)(PNP)(CO)] complexes show dihydrogen production electrocatalysis at overpotentials of 0.09–0.21 V vs. Pt under mildly acidic conditions (and activities from 0.28 to 3.51 s−1). The most active compound exhibits a turnover frequency (TOF) of 3.51 s−1 at an overpotential of only 0.15 V vs. Pt. Trends in activity are further analyzed. It is found that (a) a decrease in overpotential correlates strongly with the electron withdrawing strength of the PNP amine substituent, and (b) aliphatic substituents give comparatively higher TOFs than aromatic ones. An EC-type mechanism is shown to proceed by initial reduction of the FeII metal center and proposed formation of an FeIII hydride intermediate. Analogous cobalt compounds were synthesized and characterized, but were found to have low stability in acidic media under turnover conditions, and hence, are unsuitable as catalysts for proton reduction.
Co-reporter:Anna C. Merkle, Ashley B. McQuarters and Nicolai Lehnert
Dalton Transactions 2012 - vol. 41(Issue 26) pp:NaN8059-8059
Publication Date(Web):2012/05/01
DOI:10.1039/C2DT30464C
In this paper, the synthesis, structural and spectroscopic characterization of a series of new Ru(III)–nitrosyls of {RuNO}6 type with the coligand TPA (tris(2-pyridylmethyl)amine) are presented. The complex [Ru(TPA)Cl2(NO)]ClO4 (2) was prepared from the Ru(III) precursor [Ru(TPA)Cl2]ClO4 (1) by simple reaction with NO gas. This led to the surprising displacement of one of the pyridine (py) arms of TPA by NO (instead of the substitution of a chloride anion by NO), as confirmed by X-ray crystallography. NO complexes where TPA serves as a tetradentate ligand were obtained by reacting the new Ru(II) precursor [Ru(TPA)(NO2)2] (3) with a strong acid. This leads to the dehydration of nitrite to NO+, and the formation of the {RuNO}6 complex [Ru(TPA)(ONO)(NO)](PF6)2 (4), which was also structurally characterized. Derivatives of 4 where nitrite is replaced by urea (5) or water (6) were also obtained. The nitrosyl complexes obtained this way were then further investigated using IR and FT-Raman spectroscopy. Complex 2 with the two anionic chloride coligands shows the lowest N–O and highest Ru–NO stretching frequencies of 1903 and 619 cm−1 of all the complexes investigated here. Complexes 5 and 6 where TPA serves as a tetradentate ligand show ν(N–O) at higher energy, 1930 and 1917 cm−1, respectively, and ν(Ru–NO) at lower energy, 577 and 579 cm−1, respectively, compared to 2. These vibrational energies, as well as the inverse correlation of ν(N–O) and ν(Ru–NO) observed along this series of complexes, again support the Ru(II)–NO+ type electronic structure previously proposed for {RuNO}6 complexes. Finally, we investigated the photolability of the Ru–NO bond upon irradiation with UV light to determine the quantum yields (ϕ) for NO photorelease in complexes 2, 4, 5, and additional water-soluble complexes [Ru(H2edta)(Cl)(NO)] (7) and [Ru(Hedta)(NO)] (8). Although {RuNO}6 complexes are frequently proposed as NO delivery agents in vivo, studies that investigate how ϕ is affected by the solvent water are lacking. Our results indicate that neutral water is not a solvent that promotes the photodissociation of NO, which would present a major obstacle to the goal of designing {RuNO}6 complexes as photolabile NO delivery agents in vivo.
Co-reporter:Anna C. Merkle and Nicolai Lehnert
Dalton Transactions 2012 - vol. 41(Issue 12) pp:NaN3368-3368
Publication Date(Web):2011/09/15
DOI:10.1039/C1DT11049G
The reduction of nitrite to nitric oxide in dissimilatory denitrification is carried out by copper nitrite reductases (CuNIRs) via a type 2 copper site. Extended studies on CuNIRs in combination with model complexes have allowed for the establishment of two potential mechanisms for this transformation. Recent experimental and computational results have revealed further details of this process. In addition, the interaction of NO with copper sites has recently gained much attention. This review discusses recent results in the context of the known coordination chemistry of CuNIRs.
Co-reporter:Ashley B. McQuarters and Nicolai Lehnert
Dalton Transactions 2014 - vol. 43(Issue 37) pp:NaN13838-13838
Publication Date(Web):2014/07/30
DOI:10.1039/C4DT01388C
Recently, a new {RuNO}6 complex, [Ru(L)(PPh3)(NO)(Cl)]2+ (where L = 1-phenyl-1-(pyridin-2-yl)-2-(pyridin-2-ylmethylene)hydrazine), was reported which exhibits a one-electron quasireversible oxidation. The oxidized product, [Ru(L)(PPh3)(NO)(Cl)]3+, was isolated and proposed to be a highly unusual {RuNO}5 complex. In this paper, we investigate the electronic structure of both of these ruthenium complexes by DFT calculations and find that the oxidized species is best described as a {RuNO}6 complex with a co-ligand radical. [Ru(L)(PPh3)(NO)(Cl)]2+ is therefore oxidized to [Ru(L+˙)(PPh3)(NO)(Cl)]3+, i.e. this is an interesting example of a complex with two non-innocent ligands simultaneously bound to a ruthenium center.
Co-reporter:Natalie A. Dixon, Ashley B. McQuarters, Jodi S. Kraus, Jonathan B. Soffer, Nicolai Lehnert, Reinhard Schweitzer-Stenner and Elizabeth T. Papish
Chemical Communications 2013 - vol. 49(Issue 49) pp:NaN5573-5573
Publication Date(Web):2013/05/07
DOI:10.1039/C3CC00036B
Complexes with bulky hydrotris(triazolyl)borate (Ttz) ligands, TtzCuCO, were used to probe how acids change the donor properties of Ttz ligands. (TtztBu,Me)CuCO shows four distinct protonation states and a gradual increase in the CO stretch. The increased electrophilic nature of the Cu center upon protonation leads to enhanced C–H activation catalysis.
Co-reporter:Shawn C. Eady, Sabrina L. Peczonczyk, Stephen Maldonado and Nicolai Lehnert
Chemical Communications 2014 - vol. 50(Issue 59) pp:NaN8068-8068
Publication Date(Web):2014/06/04
DOI:10.1039/C4CC02920H
A heterogeneous dihydrogen (H2) production system has been attained by simply soaking electrodes made from electro-deposited graphene on FTO plated glass in solutions of a cobalt bis(dithiolate) compound. The resulting electrodes are active in weakly acidic aqueous solutions (pH > 3), have relatively low overpotentials (0.37 V versus platinum), show high catalytic rates (TOF > 1000 s−1), and are resistant to degradation by dioxygen.
.jpg)






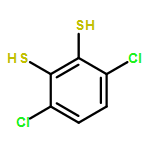
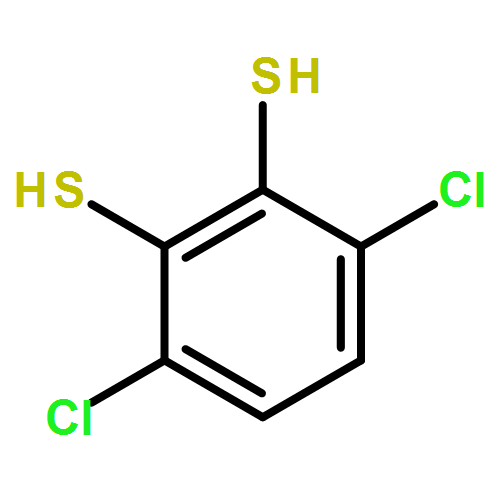




![2-{[(2E)-2-cyano-3-(4-methoxyphenyl)prop-2-enoyl]amino}benzamide](http://img.cochemist.com/ccimg/6200/6165-31-7.png)
![2-{[(2E)-2-cyano-3-(4-methoxyphenyl)prop-2-enoyl]amino}benzamide](http://img.cochemist.com/ccimg/6200/6165-31-7_b.png)

















































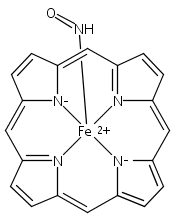

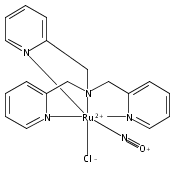
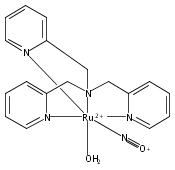
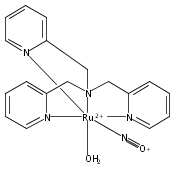
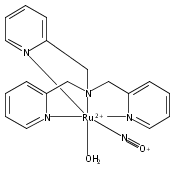
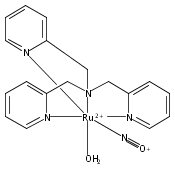





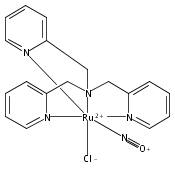
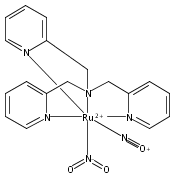






















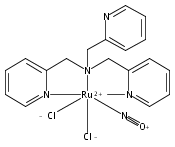




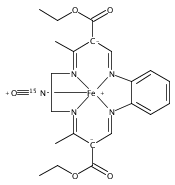











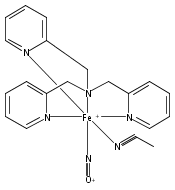
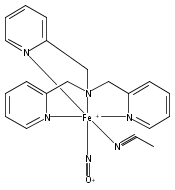








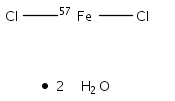
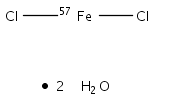
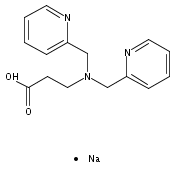





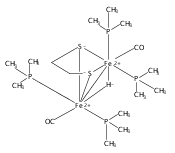
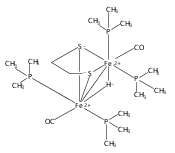






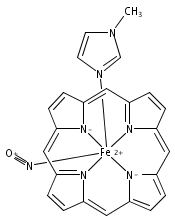




































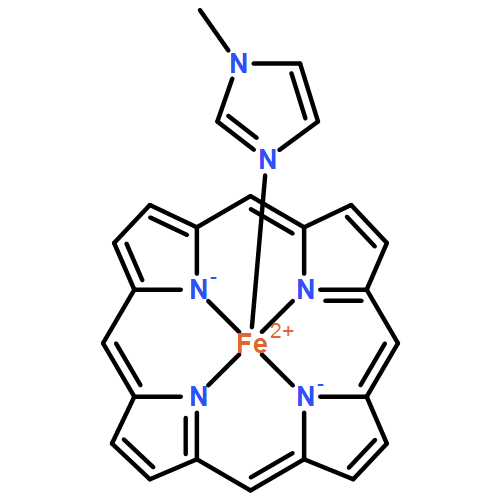


























![Phenol, 2,6-bis[[bis(2-pyridinylmethyl)amino]methyl]-4-methyl-](http://img.cochemist.com/ccimg/80600/80528-41-2.png)
![Phenol, 2,6-bis[[bis(2-pyridinylmethyl)amino]methyl]-4-methyl-](http://img.cochemist.com/ccimg/80600/80528-41-2_b.png)


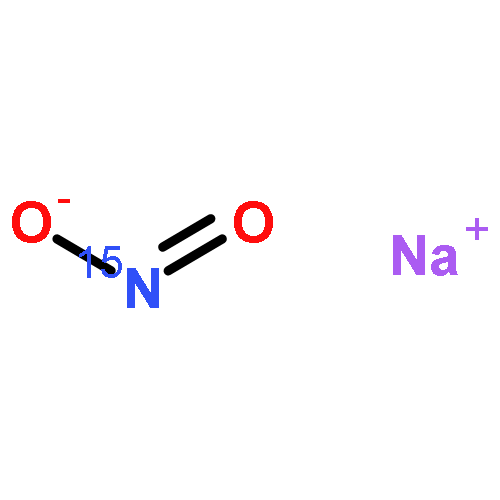


![Zinc, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/478800/14074-80-7.png)
![Zinc, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/478800/14074-80-7_b.png)









![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)