Co-reporter:Iuliia A. Gracheva, Iuliia V. Voitovich, Vladimir I. Faerman, Nikolay S. Sitnikov, Ekaterina V. Myrsikova, Hans-Gunther Schmalz, Elena V. Svirshevskaya, Alexey Yu Fedorov
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.11.020
•A series of furan-based allocolchicinoids was prepared from commercial colchicine.•Compounds exhibited excellent cytotoxicity towards epithelial cell lines.•14a and 7a induced cell accumulation in the G2/M phase in 2D and 3D cultures.•14a and 7a induce a panel of interphase effects.A series of furan-based allocolchicinoids was prepared from commercially available colchicine via a nine-step reaction sequence. Cytostatic activity, cell cycle arrest, apoptosis, tubulin and F-actin expression were studied in vitro in 2D and 3D cultures of normal and tumor epithelial keratinocytes, endothelial and mesenchymal cells. Among the prepared furanoallocolchicine analogues, 14a and 7a displayed the most pronounced anti-cancer activity. These compounds induced two types of effects: (a) cell cycle arrest in the G2/M phase as a direct consequence of effective tubulin binding (metaphase effect), and (b) pronounced cell stress (as evidenced by the overexpression of tubulin and F-actin), which was caused by the hyperpolarization of mitochondrial and lysosomal membranes (interphase effect).Download high-res image (130KB)Download full-size image
Co-reporter:Persefoni Thomopoulou, Julia Sachs, Nicole Teusch, Aruljothi Mariappan, Jay Gopalakrishnan, and Hans-Günther Schmalz
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 2) pp:188
Publication Date(Web):December 29, 2015
DOI:10.1021/acsmedchemlett.5b00418
A series of new colchicinoids with a variable triazole unit at C-7 was synthesized through Cu(I)-catalyzed 1,3-dipolar cycloaddition (click-chemistry) of a colchicine-derived azide with various alkynes and the cytotoxicity against THP-1 and Jurkat cancer cell lines was used for structural optimization. Three particularly active compounds (IC50 ≤ 5 nM) were additionally investigated with respect to their efficacy against relevant solid tumor cell lines (HeLa, A549, and SK MES 1). Besides distorting the microtubule morphology by tubulin depolymerization, one compound also exhibited a pronounced centrosome declustering effect in triple negative breast cancer cells (MDA-MB-231) and nonsmall cell lung cancer cells (H1975).Keywords: antitumoral compounds; click chemistry; Colchicine; resistance; tubulin
Co-reporter:Thomas Kerl;Florian Berger ;Dr. Hans-Günther Schmalz
Chemistry - A European Journal 2016 Volume 22( Issue 9) pp:2935-2938
Publication Date(Web):
DOI:10.1002/chem.201505118
Abstract
The first total synthesis of houttuynoid B, a powerful antiviral flavonoid glycoside from the Chinese plant Houttuynia cordata, is described. In a key step, a Baker–Venkataraman rearrangement employing an already glycosylated substrate was used to efficiently set up the fully functionalized carbon skeleton. The required benzofuran building block was prepared through a domino Sonogashira coupling/5-endo-dig cyclization and converted into a stable 1-hydroxybenzotriazole-derived active ester prior to linking with a galactosylated hydroxyacetophenone unit. The elaborated synthesis requires only nine steps (11 % overall yield) along the longest linear sequence and paves the way for the preparation of structurally related compounds for further biological evaluation.
Co-reporter:Dr. Sohajl Movahhed;Julia Westphal;Dr. Mehmet Dindaro&x11f;lu;Dr. Anna Falk ;Dr. Hans-Günther Schmalz
Chemistry - A European Journal 2016 Volume 22( Issue 22) pp:7381-7384
Publication Date(Web):
DOI:10.1002/chem.201601283
Abstract
An efficient and practical protocol for the enantioselective cobalt-catalyzed hydrovinylation of vinylarenes with ethylene at low (1.2 bar) pressure has been developed. As precatalysts, stable [L2CoCl2] complexes are employed that are activated in situ with Et2AlCl. A modular chiral TADDOL-derived phosphine–phosphite ligand was identified that allows the conversion of a broad spectrum of substrates, including heterocyclic vinylarenes and vinylferrocene, to smoothly afford the branched products with up to 99 % ee and virtually complete regioselectivity. Even polar functional groups, such as OH, NH2, CN, and CO2R, are tolerated.
Co-reporter:Robert Opitz;Matthias Müller;Cédric Reuter;Matthias Barone;Arne Soicke;Yvette Roske;Kirill Piotukh;Peter Huy;Michael Beyermann;Burkhard Wiesner;Monika Beerbaum;Christian Freund;Peter Schmieder;Rudolf Volkmer;Hartmut Oschkinat;Hans-Günther Schmalz;Ronald Kühne
PNAS 2015 Volume 112 (Issue 16 ) pp:5011-5016
Publication Date(Web):2015-04-21
DOI:10.1073/pnas.1422054112
Small-molecule competitors of protein–protein interactions are urgently needed for functional analysis of large-scale genomics
and proteomics data. Particularly abundant, yet so far undruggable, targets include domains specialized in recognizing proline-rich
segments, including Src-homology 3 (SH3), WW, GYF, and Drosophila enabled (Ena)/vasodilator-stimulated phosphoprotein (VASP) homology 1 (EVH1) domains. Here, we present a modular strategy
to obtain an extendable toolkit of chemical fragments (ProMs) designed to replace pairs of conserved prolines in recognition
motifs. As proof-of-principle, we developed a small, selective, peptidomimetic inhibitor of Ena/VASP EVH1 domain interactions.
Highly invasive MDA MB 231 breast-cancer cells treated with this ligand showed displacement of VASP from focal adhesions,
as well as from the front of lamellipodia, and strongly reduced cell invasion. General applicability of our strategy is illustrated
by the design of an ErbB4-derived ligand containing two ProM-1 fragments, targeting the yes-associated protein 1 (YAP1)-WW
domain with a fivefold higher affinity.
Co-reporter:Dr. Nikolay S. Sitnikov;Yingchun Li;Danfeng Zhang;Dr. Benito Yard;Dr. Hans-Günther Schmalz
Angewandte Chemie 2015 Volume 127( Issue 42) pp:12489-12493
Publication Date(Web):
DOI:10.1002/ange.201502445
Abstract
Wir berichten über die Entwicklung Protease-aktivierter Kohlenmonoxid-freisetzender Moleküle (CORMs). Die Tragfähigkeit des gewählten Konzeptes wurde durch die Synthese von Verbindungen belegt, bei denen eine η4-Oxydien-Fe(CO)3-Substruktur mit einer durch Penicillin-G-Amidase (PGA) spaltbaren Einheit über einen “selbstlösenden” Linker verknüpft ist. Die Geschwindigkeit der PGA-induzierten Hydrolyse wurde per HPLC und die nachfolgende CO-Freisetzung durch Headspace-GC bestimmt. Bei gemeinsamer Gabe eines unserer CORMs und PGA konnten die Inhibition der Entzündungsantwort sowie die Induktion der Expression von Hämoxygenase-1 in humanen Endothelzellen als typische biologische Effekte von CO in vitro nachgewiesen werden. Auf diese Weise wurde eine vielversprechende Basis für die zukünftige Entwicklung Protease-spezifischer CORMs für potenzielle klinische Anwendungen geschaffen.
Co-reporter:Dr. Nikolay S. Sitnikov;Yingchun Li;Danfeng Zhang;Dr. Benito Yard;Dr. Hans-Günther Schmalz
Angewandte Chemie International Edition 2015 Volume 54( Issue 42) pp:12314-12318
Publication Date(Web):
DOI:10.1002/anie.201502445
Abstract
Protease-triggered CO-releasing molecules (CORMs) were developed. The viability of the approach was demonstrated through the synthesis of compounds consisting of an η4-oxydiene–Fe(CO)3 moiety connected to a penicillin G amidase (PGA)-cleavable unit through a self-immolative linker. The rate of PGA-induced hydrolysis was investigated by HPLC analysis and the subsequent CO release was quantitatively assessed through headspace gas chromatography. In an in vitro assay with human endothelial cells, typical biological effects of CO, that is, inhibition of the inflammatory response and the induction of heme oxygenase-1 expression, were observed only upon co-administration of the CORM and PGA. This work forms a promising basis for the future development of protease-specific CORMs for potential medicinal applications.
Co-reporter:Andreas Ole Termath;Janna Velder;René T. Stemmler;Thomas Netscher;Werner Bonrath;Hans-Günther Schmalz
European Journal of Organic Chemistry 2014 Volume 2014( Issue 16) pp:3337-3340
Publication Date(Web):
DOI:10.1002/ejoc.201402240
Abstract
A novel strategy for the total synthesis of α-tocopherol (“vitamin E”) was elaborated on the basis of the conjugate addition of AlMe3 (as a methyl anion equivalent) to a 2-substituted chromenone. Starting from trimethylhydroquinone and (R,R)-hexahydrofarnesol, the required chromenone substrate was efficiently prepared in a short sequence exploiting a TiCl4-mediated Fries rearrangement and a KOtBu-induced Baker–Venkatamaran rearrangement. The envisioned key step, which sets up the quaternary center at C2, was performed in virtually quantitative yield through Ni-catalyzed conjugate addition of AlMe3. However, this transformation, which likely proceeds through a radical mechanism, could not be rendered stereoselective by means of chiral ligands. Nevertheless, the elaborated synthesis of (2RS,4′R,8′R)-α-tocopherol (2-ambo-α-tocopherol) is efficient and challenges the future development of suitable protocols for the asymmetric 1,4-addition.
Co-reporter:Mehmet Dindaro&x11f;lu;Sema Akyol Dinçer ;Hans-Günther Schmalz
European Journal of Organic Chemistry 2014 Volume 2014( Issue 20) pp:4315-4326
Publication Date(Web):
DOI:10.1002/ejoc.201402326
Abstract
Starting from tartaric acid derived chiral diols or dicarboxylic acid dichlorides with either a 2,2-dimethyl-1,3-dioxolane (Taddol) or a 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane (Tatrol) core structure, and BH3-protected ortho-phosphanyl phenols, a set of fourteen new C2-symmetric diphosphine ligands was synthesized. In addition, three related ligands were obtained from ortho-diphenylphosphino-anilines. The fully characterized ligands were then tested in the Pd-catalyzed enantioselective O-allylation of 4-methoxyphenol using crotyl methyl carbonate as a reagent. In addition, a pseudo-intramolecular variant of the reaction, using crotyl 4-methoxyphenyl carbonate as a substrate, was studied. The so-called Trost ligand was used as a reference. Although the Trost ligand (3 mol-%) gave up to 84 % ee, one of the new ligands showed higher activity (50 % ee with 0.075 mol-%).
Co-reporter:Arne Soicke;Cédric Reuter;Matthias Winter;Jörg-Martin Neudörfl;Nils Schlörer;Ronald Kühne;Hans-Günther Schmalz
European Journal of Organic Chemistry 2014 Volume 2014( Issue 29) pp:6467-6480
Publication Date(Web):
DOI:10.1002/ejoc.201402737
Abstract
Polycyclic proline-derived scaffolds (ProMs) have recently demonstrated their value as conformationally defined dipeptide analogs for the modular construction of secondary structure mimetics, specifically interfering with PPII helix-mediated protein–protein interactions. We disclose the stereoselective synthesis of two new tricyclic amino acid scaffolds (ProM-4 and ProM-8) that differ from the first generation scaffold ProM-1 by the size of ring A. Conformational preferences and subtle structural differences of the three homologous scaffolds were analyzed by X-ray crystallography, computational calculations, and NMR spectroscopy. N-tert-butoxycarbonyl(Boc)-3-(1-propenyl)azetidine-2-carboxylic acid was prepared from L-aspartic acid through β-lactam intermediates. The corresponding piperidine-based building block rac-N-Boc-3-vinylpipecolic acid was synthesized by Cu-catalyzed 1,4-addition of vinyl-MgBr to methyl N-Boc-2,3-dehydropipecolate. Target molecules were prepared through peptide coupling of the respective ring A building blocks with cis-5-vinylproline tert-butyl ester and subsequent ring-closing metathesis. Selective deprotection of a tert-butyl carbamate (N-Boc protecting group) in the presence of a tert-butyl ester was achieved with trifluoroacetic acid at 0 °C.
Co-reporter:Cédric Reuter;Margarethe Kleczka;Sarah de Mazancourt;Jörg-Martin Neudörfl;Ronald Kühne;Hans-Günther Schmalz
European Journal of Organic Chemistry 2014 Volume 2014( Issue 13) pp:2664-2667
Publication Date(Web):
DOI:10.1002/ejoc.201301875
Abstract
Following a peptide coupling/metathesis-based strategy, the two diastereomeric scaffolds ProM-3 and ProM-7 were stereoselectively synthesized (as 9-fluorenylmethoxycarbonyl derivatives), and their configuration was unambiguously proven by means of X-ray crystallography. The required dehydroisoleucine building blocks were prepared by applying the enantioselective Kazmaier–Claisen rearrangement. The target compounds represent dipeptide analogs rigidified in a PPII helix conformation, which are of interest for the development of new proteomimetics that selectively bind to protein domains specialized in the recognition of ligands adopting a PPII helix secondary structure.
Co-reporter:Dr. Andreas Ole Termath;B.Sc. Hanna Sebode;M.Sc. Waldemar Schlundt;Dr. René T. Stemmler;Dr. Thomas Netscher;Priv.-Doz.Dr. Werner Bonrath;Dr. Hans-Günther Schmalz
Chemistry - A European Journal 2014 Volume 20( Issue 38) pp:12051-12055
Publication Date(Web):
DOI:10.1002/chem.201404379
Abstract
By introducing a disposable activating substituent at C-3, the asymmetric 1,4-addition to a notoriously unreactive 2-substituted chromenone was achieved with high levels of (2R)-stereoselectivity in the presence of a chiral CuI-phosphoramidite complex as a catalyst. This paved the way for an efficient and conceptually novel synthesis of (R,R,R)-α-tocopherol from readily available starting materials.
Co-reporter:Dr. Christoph Hirschhäuser;Dr. Juraj Velcicky;Dr. Daniel Schlawe;Dr. Erik Hessler;Dr. André Majdalani;Dr. Jörg-Martin Neudörfl;Dr.Dr. Aram Prokop;Dr. Thomas Wieder;Dr. Hans-Günther Schmalz
Chemistry - A European Journal 2013 Volume 19( Issue 39) pp:13017-13029
Publication Date(Web):
DOI:10.1002/chem.201301672
Abstract
The synthesis and stereochemical assignment of two classes of iron-containing nucleoside analogues, both of which contain a butadieneFe(CO)3 substructure, is described. The first type of compounds are Fe(CO)3-complexed 3′-alkenyl-2′,3′-dideoxy-2′,3′-dehydro nucleosides (2,5-dihydrofuran derivatives), from which the second class of compounds is derived by formal replacement of the ring oxygen atom by a CH2 group (carbocyclic nucleoside analogues). These compounds were prepared in a stereoselective manner through the metal-assisted introduction of the nucleobase. Whilst the furanoid intermediates were prepared from carbohydrates (such as methyl-glucopyranoside), the carbocyclic compounds were obtained by using an intramolecular Pauson–Khand reaction. Stereochemical assignments based on NMR and CD spectroscopy were confirmed by X-ray structural analysis. Biological investigations revealed that several of the complexes exhibited pronounced apoptosis-inducing properties (through an unusual caspase 3-independent but ROS-dependent pathway). Furthermore, some structure–activity relationships were identified, also as a precondition for the design and synthesis of fluorescent and biotin-labeled conjugates.
Co-reporter:Dipl.-Chem. Anna Falk;B.Sc. Anna-Lena Göderz ;Dr. Hans-Günther Schmalz
Angewandte Chemie International Edition 2013 Volume 52( Issue 5) pp:1576-1580
Publication Date(Web):
DOI:10.1002/anie.201208082
Co-reporter:Mehmet Dindaroğlu, Sema Akyol, Hamza Şimşir, Jörg-Martin Neudörfl, Anthony Burke, Hans-Günther Schmalz
Tetrahedron: Asymmetry 2013 Volume 24(Issue 11) pp:657-662
Publication Date(Web):15 June 2013
DOI:10.1016/j.tetasy.2013.04.008
By using (R,R,R,R)-2,3-dimethoxy-2,3-dimethyl-1,4-dioxane-5,6-bis-diphenylmethanol (TARTROL) as a chiral building block, a set of six modular phosphine–phosphite ligands (with a 1,2-phenylene backbone) were synthesized and evaluated in the Cu-catalyzed asymmetric 1,4-addition of Grignard reagents to cyclohexenone. Ligands with bulky substituents at the ortho- and para-positions to the chiral phosphite moiety were found to be the most selective affording the 1,4-addition products with enantioselectivities of up to 84% ee.(2R,3R,5R,6R)-5,6-Dimethoxy-5,6-dimethyl-1,4-dioxane-2,3-dicarboxylic acid dimethyl esterC12H20O8[α]58920=-140.6 (c 1.0, CHCl3), [α]54620=-166.0 (c 1.0, CHCl3), [α]40520=-324.7 (c 1.0, CHCl3), [α]36520=-413.8 (c 1.0, CHCl3).Absolute configuration: (2R,3R,5R,6R)(2R,3R,5R,6R)-2,3-Bis(hydroxydiphenylmethyl)-5,6-dimethoxy-5,6-dimethyl-1,4-dioxaneC34H36O6[α]58920=+50.6 (c 1.0, CHCl3), [α]54620=+62.0 (c 1.0, CHCl3), [α]40520=+154.1 (c 1.0, CHCl3), [α]36520=+226.0 (c 1.0, CHCl3)Absolute configuration: (2R,3R,5R,6R)(2R,3R,4aR,9aR)-7-(2,4-Di-tert-butyl-6-diphenyl-phosphino-phenoxy)-2,3-dimethoxy-2,3-dimethyl-5,5,9,9-tetraphenyl-hexahydro-[1,4]dioxino[2,3-e][1,3,2]-dioxaphosphepineC60H64O7P2[α]58920=+125.7 (c 0.5, CHCl3), [α]54620=+153.5 (c 0.5, CHCl3), [α]40520=+450.2 (c 0.5, CHCl3), [α]36520=+764.0 (c 0.5, CHCl3)Absolute configuration: (2R,3R,4aR,9aR)(2R,3R,4aR,9aR)-7-(2-Diphenylphosphino-4,6-di-tert-pentyl-phenoxy)-2,3-dimethoxy-2,3-dimethyl-5,5,9,9-tetraphenyl-hexahydro-[1,4]dioxino[2,3-e][1,3,2]-dioxaphosphepineC62H68O7P2[α]58920=+79.5 (c 0.5, CHCl3), [α]54620=+99.8 (c 0.5, CHCl3), [α]40520=+296.5 (c 0.5, CHCl3)Absolute configuration: (2R,3R,4aR,9aR)(2R,3R,4aR,9aR)-7-(2-Diphenylphosphino-6-tert-pentyl-phenoxy)-2,3-dimethoxy-2,3-dimethyl-5,5,9,9-tetraphenyl-hexahydro-[1,4]dioxino[2,3-e][1,3,2]-dioxaphosphepineC57H58O7P2[α]58920=+137.1 (c 0.5, CHCl3), [α]54620=+169.0 (c 0.5, CHCl3), [α]40520=+470.0 (c 0.5, CHCl3), [α]36520=+777.0 (c 0.5, CHCl3)Absolute configuration: (2R,3R,4aR,9aR)(2R,3R,4aR,9aR)-7-(2-tert-Butyl-6-diphenylphosphino-phenoxy)-2,3-dimethoxy-2,3-dimethyl-5,5,9,9-tetraphenyl-hexahydro-[1,4]dioxino[2,3-e][1,3,2]-dioxaphosphepineC56H56O7P2[α]58920=+189.3 (c 0.5, CHCl3), [α]54620=+230.7 (c 0.5, CHCl3), [α]40520=+650.9 (c 0.5, CHCl3)Absolute configuration: (2R,3R,4aR,9aR)(2R,3R,4aR,9aR)-7-(3-Diphenylphosphino-biphenyl-2-yloxy)-2,3-dimethoxy-2,3-dimethyl-5,5,9,9-tetraphenyl-hexahydro-[1,4]dioxino[2,3-e][1,3,2]-dioxaphosphepineC58H52O7P2[α]58920=+219.1 (c 0.5, CHCl3), [α]54620=+269.4 (c 0.5, CHCl3)Absolute configuration: (2R,3R,4aR,9aR)(2R,3R,4aR,9aR)-7-(2-Diphenylphosphino-6-methyl-phenoxy)-2,3-dimethoxy-2,3-dimethyl-5,5,9,9-tetraphenyl-hexahydro-[1,4]dioxino[2,3-e][1,3,2]-dioxaphosphepineC53H50O7P2[α]58920=+113.3 (c 0.5, CHCl3), [α]54620+138.5 (c 0.5, CHCl3), [α]40520= +375.1 (c 0.5, CHCl3), [α]36520= +598.2 (c 0.5, CHCl3)Absolute configuration: (2R,3R,4aR,9aR)
Co-reporter:Svetlana Botov, Eleni Stamellou, Steffen Romanski, Miguel Guttentag, Roger Alberto, Jörg-Martin Neudörfl, Benito Yard, and Hans-Günther Schmalz
Organometallics 2013 Volume 32(Issue 13) pp:3587-3594
Publication Date(Web):June 17, 2013
DOI:10.1021/om301233h
Novel η4-acyloxy-cyclohexadiene-Fe(CO)3 complexes (with variable length of the acyloxy chain) were synthesized as potential enzyme-triggered carbon monoxide (CO)-releasing molecules (ET-CORMs). The molecular structure of two complexes was additionally confirmed by X-ray crystallography. The enzyme-triggered CO-releasing activity of the compounds was assessed under physiological conditions (37 °C, 0.1 M phosphate buffer, pH = 7.4) by headspace gas chromatography (GC) and additionally by means of a myoglobin assay (UV). The relative rate of CO release and the amount of liberated CO were found to depend on the length of the acyloxy chain and its position at the diene unit (outer or inner position). Some of the new ET-CORMs exhibited very good biological activity as assessed in different cellular assays (cytotoxicity, protective effect against hypothermia-associated cell damage, and inhibition of TNF-α-mediated VCAM-1 expression).
Co-reporter:Dipl.-Chem. Anna Falk;B.Sc. Anna-Lena Göderz ;Dr. Hans-Günther Schmalz
Angewandte Chemie 2013 Volume 125( Issue 5) pp:1617-1621
Publication Date(Web):
DOI:10.1002/ange.201208082
Co-reporter:Dario C. Gerbino, Daniel Augner, Nikolay Slavov, and Hans-Günther Schmalz
Organic Letters 2012 Volume 14(Issue 9) pp:2338-2341
Publication Date(Web):April 22, 2012
DOI:10.1021/ol300757m
The surprisingly facile conversion (isomerization) of 2-formyl-arylketones into 3-substituted phthalides, as observed for the marine natural product pestalone and its per-O-methylated derivative, was investigated using a series of simple 2-acylbenzaldehydes as substrates. The transformation generally proceeds smoothly in DMSO, either in a Cannizarro–Tishchenko-type reaction under nucleophile catalysis (NaCN) or under photochemical conditions (DMSO, 350 nm).
Co-reporter:Darius P. Kranz, Slim Chiha, Aike Meier zu Greffen, Jörg-Martin Neudörfl, and Hans-Günther Schmalz
Organic Letters 2012 Volume 14(Issue 14) pp:3692-3695
Publication Date(Web):July 5, 2012
DOI:10.1021/ol301532w
The BF3·Et2O-promoted reaction of 3β-acetoxy-5,19-cyclo-pregnan-6β-ol-20-one with different nucleophiles was investigated. B-homo steroids (3β-acetoxy-B-homo-6a-β-alkoxy-pregna-5(10)-en-20-ones) were obtained with primary and secondary alcohols, while the reaction with common carboxylic acids selectively afforded the corresponding 3β-acetoxy-6β-(acyloxymethyl)-pregna-5(10)-en-20-ones. The transformations are supposed to proceed via the rearrangement of a cyclopropyl-methyl cation (bicyclobutonium) intermediate, which is regioselectively opened in dependence on the nucleophile employed. The method represents an efficient, diversity-oriented entry to new B-ring-modified steroids, which are of potential pharmaceutical relevance.
Co-reporter:Wibke Lölsberg, Susen Werle, Jörg-Martin Neudörfl, and Hans-Günther Schmalz
Organic Letters 2012 Volume 14(Issue 23) pp:5996-5999
Publication Date(Web):November 13, 2012
DOI:10.1021/ol302898h
A short and enantioselective total synthesis of helioporins C and E, which are bioactive marine diterpenes containing a serrulatane or amphilectane skeleton, was elaborated. The chirogenic step, i.e. a Cu(I)-catalyzed allylic alkylation of a cinnamyl chloride with methylmagnesium bromide, proceeded with virtually complete enantioselectivity (99% ee) in the presence of a chiral phosphine-phosphite ligand. The other stereocenters were diastereoselectively established through Me2AlCl-mediated cationic cyclization and Ir-catalyzed hydrogenation.
Co-reporter:Steffen Romanski, Birgit Kraus, Miguel Guttentag, Waldemar Schlundt, Hannelore Rücker, Andreas Adler, Jörg-Martin Neudörfl, Roger Alberto, Sabine Amslinger and Hans-Günther Schmalz
Dalton Transactions 2012 vol. 41(Issue 45) pp:13862-13875
Publication Date(Web):16 May 2012
DOI:10.1039/C2DT30662J
A series of η4-acyloxycyclohexadiene–Fe(CO)3 complexes was prepared and fully characterized by spectroscopic methods including single crystal X-ray diffraction. For this purpose a new synthetic access to differently acylated 1,3- and 1,5-dienol–Fe(CO)3 complexes was developed. The enzymatically triggered CO release from these compounds was monitored (detection of CO through GC and/or by means of a myoglobin assay) and the anti-inflammatory effect of the compounds was assessed by a cellular assay based on the inhibition of NO-production by inducible NO synthase (iNOS). It was demonstrated that the properties (rate of esterase-triggered CO release, iNOS inhibition, cytotoxicity) of the complexes strongly depend on the substitution pattern of the π-ligand and the nature of the acyloxy substituent.
Co-reporter:Qaseem Naeemi;Mehmet Dindaro&x11f;lu;Darius P. Kranz;Janna Velder ;Hans-Günther Schmalz
European Journal of Organic Chemistry 2012 Volume 2012( Issue 6) pp:1179-1185
Publication Date(Web):
DOI:10.1002/ejoc.201101258
Abstract
Asymmetric conjugate additions (1,4-additions) of aryl-Grignard reagents to cyclohex-2-enone, currently a more or less unsolved challenge, were investigated. For this purpose, a small library of phenol-derived chiral phosphane-phosphite ligands containing TADDOL- or BINOL-based phosphite moieties was evaluated. These ligands are easily prepared by a short modular scheme previously developed in this laboratory. Two particularly powerful ligands (4a and 4b, both TADDOL-derived and each possessing a bulky tert-butyl substituent ortho to the phosphite group) were identified. Conditions were optimized with use of the addition of (4-methoxyphenyl)magnesium bromide to cyclohexenone as a standard reaction system. Under optimized conditions [CuBr·SMe2 (4 mol-%), ligand 4a (6 mol-%), 2-methyl-THF, –78 °C, slow addition of Grignard reagent] the 1,4-product was obtained with high enantioselectivity (up to 95 % ee) and good regioselectivity (r.r. = 90:10). The scope of the method was probed with different aryl-Grignard reagents. It was found that reagents with electron-donating substituents in meta- or para-positions performed particularly well, whereas the presence of F or CF3 substituents led to decreased ee values. Only ortho-substituted aryl-Grignard reagents did not give rise to useful results. A series of phosphane-phosphite ligands were also tested in the Rh-catalyzed 1,4-addition of phenylboronic acid to cyclohexenone, but enantioselectivities did not exceed 70 % ee in this case.
Co-reporter:Andreas Ole Termath;Stefanie Ritter;Marcel König;Darius Paul Kranz;Jörg-Martin Neudörfl;Aram Prokop;Hans-Günther Schmalz
European Journal of Organic Chemistry 2012 Volume 2012( Issue 24) pp:4501-4507
Publication Date(Web):
DOI:10.1002/ejoc.201200677
Abstract
A synthetic approach towards (5R)-5-methyl-6-oxa-desacetamido colchicine as a conformationally defined non-natural colchicine analog with a modified B-ring was undertaken. The synthetic strategy was based on a Rh-catalyzed cascade reaction involving a [5+2] cycloaddition of a carbonyl ylide intermediate as a key step, in which both seven-membered rings of the polycyclic framework are formed in a single operation. Starting from 2-iodo-3,4,5-trimethoxy-acetophenone, an upper side-chain was constructed through enantioselective CBS reduction (up to 75 % ee) and propargylation, while a lower succinoyl side-chain was attached either throughiodine–magnesium–copper exchange and subsequent reaction with methyl 4-chloro-4-oxobutanoate, or by Pd-catalyzed Stille cross-coupling with 2-tributylstannyl-5-methoxyfuran followed by hydrolytic furan-opening. Treatment of an α-diazoketone intermediate with Rh2(OAc)4 (3 mol-%)initiated the diastereoselective key cyclization cascade (≥97:3 dr). Treatment of the cycloadduct 3 with Et2AlCl afforded an interesting 11,12-dihydrocolchicine analog 24, which, however, could not be oxidized to the corresponding tropolone. Structural assignments were confirmed by X-ray crystallography. While compounds 3 and 24 did not exhibit noteworthy cytotoxic activity by themselves, they were found to strongly enhance the cytostatic (apoptosis-inducing) activity of doxorubicin against resistant Nalm-6 cells (i.e., in a synergy effect).
Co-reporter:Michal Drusan;Wibke Lölsberg;Andrea &x160;kvorcová;Hans-Günther Schmalz;Radovan &x160;ebesta
European Journal of Organic Chemistry 2012 Volume 2012( Issue 31) pp:6285-6290
Publication Date(Web):
DOI:10.1002/ejoc.201200729
Abstract
Copper(I) complexes of TADDOL-based phosphane–phosphite ligands catalyze enantioselective conjugate additions of various Grignard reagents to cyclic enones. Through trapping of the resulting chiral magnesium enolates with N-benzylidene-4-methylbenzenesulfonamide (as an imine) in one-pot procedures, the corresponding Mannich products (i.e., β-aminocarbonyl compounds) were obtained in good yields and high enantiomeric purities, although with only low diastereoselectivities.
Co-reporter:Aroonchai Saiai, Harald Bielig, Janna Velder, Jörg-Martin Neudörfl, Maureen Menning, Thomas A. Kufer and Hans-Günther Schmalz
MedChemComm 2012 vol. 3(Issue 11) pp:1377-1385
Publication Date(Web):24 Sep 2012
DOI:10.1039/C2MD20221B
Small molecules, which specifically inhibit NOD2 signalling, are of high interest in the search for new anti-inflammatory drugs. In this context, the previously discovered hydrophenalene–Cr(CO)3 complex 1 was taken as a lead and new structural analogues were prepared by means of chemical synthesis. The NOD2-inhibiting activity of the compounds was determined with cell-based reporter assays employing human embryonic kidney cells. As a result of the study, some clear structure activity relationships (SARs) could be identified. The best compounds were active at low μM concentration.
Co-reporter:Dipl.-Chem. Darius Paul Kranz;Dr. Axel Georg Griesbeck;Dr. Ronald Alle;Dr. Raul Perez-Ruiz;Dr. Jörg Martin Neudörfl;Dr. Klaus Meerholz;Dr. Hans-Günther Schmalz
Angewandte Chemie International Edition 2012 Volume 51( Issue 24) pp:6000-6004
Publication Date(Web):
DOI:10.1002/anie.201201222
Co-reporter:Steffen Romanski, Hannelore Rücker, Eleni Stamellou, Miguel Guttentag, Jörg-Martin Neudörfl, Roger Alberto, Sabine Amslinger, Benito Yard, and Hans-Günther Schmalz
Organometallics 2012 Volume 31(Issue 16) pp:5800-5809
Publication Date(Web):June 5, 2012
DOI:10.1021/om300359a
A series of racemic phosphoryloxy-substituted (η4-cyclohexadiene)Fe(CO)3 complexes was synthesized by exploiting the O-phosphorylation of (dienol)Fe(CO)3 intermediates generated in situ from the corresponding triisopropylsiloxy-protected complexes. The phosphorylated products were fully characterized by spectroscopic methods, including single-crystal X-ray diffraction in four cases. Monodeprotection of two dimethyl phosphate derivatives with trimethylamine led to the tetramethylammonium salts of the (cyclohexadienyl methyl phosphate)Fe(CO)3 complexes. These compounds are the first water-soluble enzyme-trigged CO-releasing molecules (ET-CORMs). The phosphatase-induced CO release was monitored by means of GC. The biological activity was assessed in different cellular assays. The compounds were shown to be only slightly toxic, and a moderate anti-inflammatory potential was determined in an assay based on the inhibition of inducible NO synthase (iNOS)-induced NO production.
Co-reporter:Nikolay Sitnikov;Dr. Janna Velder;Liliane Abodo;Nicole Cuvelier;Dr. Jörg Neudörfl;Dr. Aram Prokop;Dr. Günter Krause;Dr. Aleksey Y. Fedorov;Dr. Hans-Günther Schmalz
Chemistry - A European Journal 2012 Volume 18( Issue 38) pp:12096-12102
Publication Date(Web):
DOI:10.1002/chem.201200083
Abstract
A series of novel pyrrolo-allocolchicine derivatives (containing a 1-methyl-1H-indol-5-yl moiety replacing ring C) was synthesized. The tetracyclic ring system was constructed by Suzuki–Miyaura cross-coupling of a 1-methylindole-5-boronate with an ortho-iodo-dihydrocinnamic acid derivative and subsequent intramolecular Friedel–Crafts acylation. After reduction of the resulting ketone, the nitrogen functionality was introduced in a Mitsunobu-type reaction by using zinc azide followed by LiAlH4 reduction. Structural assignments were supported by X-ray crystallography. The compounds synthesized were then tested against BJAB tumor cells and found to exhibit pronounced cytotoxic activity (proliferation inhibition and apoptosis induction). The ketone 24 b was even active at sub-nanomolar concentration. In addition, the antitumor potential of the compounds was confirmed by using B lymphoid cell lines.
Co-reporter:Juraj Velcicky ; Arne Soicke ; Roland Steiner ;Hans-Günther Schmalz
Journal of the American Chemical Society 2011 Volume 133(Issue 18) pp:6948-6951
Publication Date(Web):April 19, 2011
DOI:10.1021/ja201743j
A one-pot protocol for the cyanomethylation of aryl halides through a palladium-catalyzed reaction with isoxazole-4-boronic acid pinacol ester was developed. Mechanistically, the reaction proceeds through (1) Suzuki coupling, (2) base-induced fragmentation, and (3) deformylation as shown by characterization of all postulated intermediates. Under optimized conditions (PdCl2dppf, KF, DMSO/H2O, 130 °C) a broad spectrum of aryl bromides could be converted into arylacetonitriles with up to 88% yield.
Co-reporter:Peter Huy, Jörg-Martin Neudörfl, and Hans-Günther Schmalz
Organic Letters 2011 Volume 13(Issue 2) pp:216-219
Publication Date(Web):December 15, 2010
DOI:10.1021/ol102613z
A practical four-step synthesis of 3-alkyl-, vinyl-, and aryl-substituted proline derivatives, which are important building blocks for conformationally restrained peptide analogs, was developed. The method relies on a Cu-catalyzed 1,4-addition of Grignard reagents to N-protected 2,3-dehydroproline esters, efficiently prepared in a new one-pot protocol. The 1,4-addition products are obtained with good trans-selectivity (dr 5:1 to 25:1). A nonracemic sample of N-Cbz-3-vinylproline (74% ee) was obtained using Evans oxazolidinone as a chiral auxiliary.
Co-reporter:Daniel Augner, Dario C. Gerbino, Nikolay Slavov, Jörg-Martin Neudörfl, and Hans-Günther Schmalz
Organic Letters 2011 Volume 13(Issue 19) pp:5374-5377
Publication Date(Web):September 8, 2011
DOI:10.1021/ol202271k
A unique reactivity pattern, first observed in the conversion of the marine natural product pestalone into pestalachloride A, was investigated. It was shown that 2-formyl-arylketones smoothly react with ammonia and primary amines, respectively, under mild conditions to afford 3-substituted isoindolinones in high yield. The reaction represents a new option for the derivatization (N-capping) of primary amines. As the substrates are readily accessible the methodology opens a short and modular access to pharmaceutically relevant substituted isoindolinones.
Co-reporter:Anna Falk;Lukas Fiebig;Jörg-Martin Neudörfl;Andreas Adler ;Hans-Günther Schmalz
Advanced Synthesis & Catalysis 2011 Volume 353( Issue 18) pp:3357-3362
Publication Date(Web):
DOI:10.1002/adsc.201100658
Abstract
The use of modular α,α,α′,α′-tetraaryl-1,3-dioxolane-4,5-dimethanol (TADDOL)- and 1,1′-bi-2-naphthol (BINOL)-derived phosphine-phosphite ligands (L2*) in the asymmetric rhodium-catalyzed intramolecular [4+2] cycloaddition (“neutral” Diels–Alder reaction) of (E,E)-1,6,8-decatriene derivatives (including a 4-oxa and a 4-aza analogue) was investigated. Initial screening of a small ligand library led to the identification of a most promising, TADDOL-derived ligand bearing a phenyl group adjacent to the phosphite moiety at the arene backbone. In the course of further optimization studies, the formation of a new, more selective catalyst species during the reaction time was observed. By irradiating the pre-catalyst with microwaves prior to substrate addition high enantioselectivities (up to 93% ee) were achieved. The new cyclization protocol was successfully applied to all three substrates investigated to give the bicyclic products in good yield and selectivity. 31P NMR and ESI-MS measurements indicated the formation of a [Rh(L2*)2]+ species as the more selective (pre-) catalyst.
Co-reporter:Nina Kausch-Busies;Benjamin Kater;Jörg M. Neudörfl;Aram Prokop;Hans-Günther Schmalz
European Journal of Organic Chemistry 2011 Volume 2011( Issue 6) pp:1133-1139
Publication Date(Web):
DOI:10.1002/ejoc.201001445
Abstract
As previous studies in the field of bioorganometallic chemistry have unveiled, metal-containing analogues of natural products frequently exhibit surprising biological activities. In this context, a synthetic approach to analogues of the eicosanoids 5-HETE and 8-HETE possessing a 1,3-butadiene–Fe(CO)3 substructure was elaborated. The chosen structures are characterized by a central (2E,4Z)-hexa-2,4-dien-1-ol–Fe(CO)3 moiety (as the potential “metal pharmacophore”) and carry a lipophilic ω-6-substituent as well as a hydrophilic α-side chain (carboxylic acid). Using the cationic complex tricarbonyl[η5-1-(methoxycarbonyl)pentadienyl]iron hexafluorophosphate (rac-4) as a starting material, the synthesis of a simplified Fe(CO)3-complexed HETE analogue (rac-3) was achieved by exploiting the addition of an alkynyl Cu species to rac-4. The carbon skeleton was completed through diastereo(Ψ-exo)selective addition of a titanium–zinc organyl [prepared from ethyl 4-iodobutyrate, activated Zn powder, and Ti(OiPr)3Cl] as a butyrate D4 synthon to an aldehyde function by using 2-Me-THF as the solvent of choice. The (sensitive) target compound rac-3 was shown to induce apoptosis at moderate concentrations in cancer cells (BJAB and Nalm-6).
Co-reporter:Darius Paul Kranz;Aike Meier zu Greffen;Sherif El Sheikh;Jörg Martin Neudörfl;Hans-Günther Schmalz
European Journal of Organic Chemistry 2011 Volume 2011( Issue 15) pp:2860-2866
Publication Date(Web):
DOI:10.1002/ejoc.201100020
Abstract
Attempts to convert 3β-acetoxy-5,19-cyclo-pregna-6,20-dione (8) into a cyclocitrinol-related steroid with a bicyclo[4.4.1]-undecane AB-ring substructure through a Lewis acid-assisted fragmentation failed. Instead, treating 8 with an excess of Ac2O and BF3·Et2O afforded B-homo steroid 9 in 90 % yield. This unexpected rearrangement is assumed to proceed via an intermediate bicyclobutonium cation. Remarkably, tricyclo[4.4.1.0]undecane-1-one (rac-13), prepared as a model substrate, did not react under the same conditions. However, by screening various Lewis acids, stannous triflate was identified as a particularly efficient reagent for this and related Lewis acid-assisted ring-opening reactions of cyclopropyl ketones in the presence of Ac2O, and even catalytic amounts of Sn(OTf)2 (5 mol-%) proved to be effective.
Co-reporter:Nina Kausch-Busies;Jörg Martin Neudörfl;Pascal Wefelmeier;Aram Prokop;Hartmut Kühn;Hans-Günther Schmalz
European Journal of Organic Chemistry 2011 Volume 2011( Issue 24) pp:4634-4644
Publication Date(Web):
DOI:10.1002/ejoc.201100003
Abstract
The stereoselective synthesis of a set of four stable 5-hydroxyeicosatetraenoic acid (HETE) analogues with a ferrocene backbone is described. The common substructure of all HETEs, the (2E,4Z)-1-hydroxyhexadiene moiety, was formally replaced by a ferrocenylmethanol fragment. Whereas the hydrophilic side chain remained the same as that of 5-HETE (butyrate), the lipophilic side chain was simplified by replacing the “natural” (Z,Z)-1,4-decadiene side chain with (Z)-1-heptene, 1-heptyne, 1-octyne, or phenylacetylene. As a key building block, Kagan's chiral acetal (derived from ferrocenecarbaldehyde) was used for the stereoselective generation of the planar-chiral substructure through diastereoselective ortho-lithiation and subsequent formylation, methoxycarbonylation or iodination, respectively. The lipophilic side chain was installed either by a “salt-free” Wittig reaction or (better) by Pd-catalyzed Sonogashira coupling. The (Z)-selective semi-reduction of a (hindered) triple bond was achieved with NaBH4, PdI2 and PEG200. The introduction of the hydrophilic side chain was found to proceed with complete diastereoselectivity through addition of a titanium–zinc organyl [prepared from ethyl 4-iodobutyrate, activated Zn powder and Ti(OiPr)3Cl] as a butyrate D4 synthon to an aldehyde function using 2-methyltetrahydrofuran (2-Me-THF) as a solvent. The expected (Rp,5S) configuration of the products was confirmed by X-ray crystallography of an intermediate and by chemical correlation and NMR spectroscopy of a Mosher-type derivative. Preliminary biological experiments revealed that compound 4 [with a (Z)-heptenyl side chain] displayed significant apoptosis induction, whereas analogues with an alkynyl side chain were inactive. None of the HETE analogues prepared showed significant inhibition of lipoxygenase (15-LOX) or cyclooxygenase (COX-2).
Co-reporter:Dipl.-Chem. Steffen Romanski;Dr. Birgit Kraus;Dr. Ulrich Schatzschneider;Dr. Jörg-Martin Neudörfl;Dr. Sabine Amslinger;Dr. Hans-Günther Schmalz
Angewandte Chemie International Edition 2011 Volume 50( Issue 10) pp:2392-2396
Publication Date(Web):
DOI:10.1002/anie.201006598
Co-reporter:Qaseem Naeemi, Tobias Robert, Darius P. Kranz, Janna Velder, Hans-Günther Schmalz
Tetrahedron: Asymmetry 2011 Volume 22(Issue 8) pp:887-892
Publication Date(Web):30 April 2011
DOI:10.1016/j.tetasy.2011.04.018
A library of chiral phosphine–phosphite ligands was evaluated in the Cu-catalyzed asymmetric 1,4-addition of Grignard reagents to cyclopentenone, cycloheptenone and 5,6-dihydro-2H-pyran-2-one. TADDOL-based ligands 1a and 1b with a bulky substituent at the ortho-position to the chiral phosphite moiety gave rise to the 1,4-addition products with high enantioselectivities (up to 93% ee). In addition to ethyl-MgBr (as a standard alkyl nucleophile) phenyl- and isopropenyl-MgBr could also be employed. In the case of cyclopentenone, the use of chlorotrimethylsilane as an additive led to improved regio- and enantioselectivities.3-Ethyl-cyclopentanoneC7H12O93% ee[α]58925=+90 (c 1, CHCl3)Source of chirality: enantioselective catalysis: (R,R)-taddolAbsolute configuration: (R)(R)-3-(2-Propenyl)-cyclopentanoneC8H12O89% ee[α]58920=+87.8 (c 0.32, CHCl3)Source of chirality: enantioselective catalysis: (R,R)-taddolAbsolute configuration: (R)(R)-3-Phenyl-cyclopentanoneC12H12O57% ee[α]58920=+47.2 (c 1, CH2Cl2)Source of chirality: enantioselective catalysis: (R,R)-taddolAbsolute configuration: (R)(S)-3-Ethyl-cycloheptanoneC9H16O91% ee[α]58920=+62.1 (c 1.1, CH3CN)Source of chirality: enantioselective catalysis: (R,R)-taddolAbsolute configuration: (S)(R)-3-(2-Propenyl)-cycloheptanoneC10H16O86% ee[α]58920=+68.2 (c 1.1, CH2Cl2)Source of chirality: enantioselective catalysis: (R,R)-taddolAbsolute configuration: (R)(R)-3-Phenyl-cycloheptanoneC13H16O90% ee[α]58920=+54.2 (c 0.9, CH2Cl2)Source of chirality: enantioselective catalysis: (R,R)-taddolAbsolute configuration: (R)(S)-4-Ethyl-tetrahydro-pyrane-2-oneC7H12O2[α]58920=-16.5 (c 0.95, CHCl3)Source of chirality: enantioselective catalysis: (R,R)-taddolAbsolute configuration: (S)
Co-reporter:Dr. Stefan Neufeind;Nils Hülsken;Dr. Jörg-Martin Neudörfl;Dr. Nils Schlörer;Dr. Hans-Günther Schmalz
Chemistry - A European Journal 2011 Volume 17( Issue 9) pp:2633-2641
Publication Date(Web):
DOI:10.1002/chem.201003166
Abstract
The structurally unique polyketide mumbaistatin is the strongest naturally occurring inhibitor of glucose-6-phosphate translocase-1 (G6P-T1), which is a promising target for drugs against type-2 diabetes mellitus and angiogenic processes associated with brain tumor development. Despite its high relevance, mumbaistatin has so far withstood all attempts towards its total synthesis. In the present study an efficient total synthesis of a deoxy-mumbaistatin analogue containing the complete carbon skeleton and a spirolactone motif closely resembling the natural product in its cyclized form was elaborated. Key steps of the synthesis are a Diels–Alder cycloaddition for the construction of the fully functionalized anthraquinone moiety and an anionic homo-Fries rearrangement to build up the tetra-ortho-substituted benzophenone core motif, from which a spiroketal lactone forms in a spontaneous process. The elaborated strategy opens an entry to a variety of new analogs of mumbaistatin and cyclo-mumbaistatin and may be exploited for the total synthesis of the natural product itself in the future.
Co-reporter:Dipl.-Chem. Cédric Reuter;Dr. Peter Huy;Dr. Jörg-Martin Neudörfl;Dr. Ronald Kühne;Dr. Hans-Günther Schmalz
Chemistry - A European Journal 2011 Volume 17( Issue 43) pp:12037-12044
Publication Date(Web):
DOI:10.1002/chem.201101704
Abstract
A practical and scalable synthesis of a Fmoc-protected tricyclic dipeptide mimetic (6), that is, a 1,4-diaza-tricyclo-[8.3.03, 7]-tridec-8-ene derivative resembling a rigidified di-L-proline in a polyproline type II (PPII) helix conformation, was developed. The strategy is based on a Ru-catalyzed ring-closing metathesis of a dipeptide (4) prepared by PyBOP coupling of cis-5-vinylproline tert-butylester (2) and trans-N-Boc-3-vinylproline (rac-3) followed by chromatographic diastereomer separation. Building block 2 was prepared from L-proline in six steps via electrochemical C5-methoxylation, cyanation and conversion of the nitrile into a vinyl substituent. Building block rac-3 was prepared in five steps exploiting a Cu-catalyzed 1,4-addition of vinyl-MgBr to a 2,3-dehydroproline derivative in the key step. In the course of the investigation subtle dependencies of protecting groups on the reactivity of the 2,3- and 2,5-disubstituted pyrrolidine derivatives were observed. The configuration and conformational preference of several intermediates were determined by X-ray crystallography. The developed synthesis allows the preparation of substantial amounts of 6, which will be used in the search for new small molecules for the modulation of protein–protein interactions involving prolin-rich motifs (PRDs).
Co-reporter:Wibke Lölsberg;Shute Ye ;Hans-Günther Schmalz
Advanced Synthesis & Catalysis 2010 Volume 352( Issue 11-12) pp:2023-2031
Publication Date(Web):
DOI:10.1002/adsc.201000213
Abstract
The copper(I)-catalysed SN2′-type allylic substitution of E-3-aryl-allyl chlorides (cinnamyl chlorides) using Grignard reagents represents a powerful method for the synthesis of compounds carrying a benzylic stereocentre. By screening a small library of modular chiral phosphine-phosphite ligands a new copper(I)-based catalyst system was identified which allows the performance of such reactions with exceptional high degrees of regio- and enantioselectivity. Best results were obtained using TADDOL-derived ligands (3 mol%), copper(I) bromide⋅dimethyl sulfide (CuBr⋅SMe2) (2.5 mol%) and methyl tert-butyl ether (MTBE) as a solvent. Various (1-alkyl-allyl)benzene derivatives were prepared with up to 99% ee (GC) in isolated yields of up to 99%. In most cases the product contained less than 3% of the linear regioisomer (except for ortho-substituted substrates). Both electron-rich and electron-deficient cinnamyl chlorides were successfully employed. The absolute configuration of the products was assigned by comparison of experimental and calculated CD spectra. The substrates were prepared from the corresponding alcohols by reaction with thionyl chloride. Initially formed mixtures of regioisomeric allylic chlorides were homogenised by treatment with CuBr⋅SMe2 (2.5 mol%) in the presence of triphenyl phosphine (PPh3) (3 mol%) in MTBE at low temperature to give the pure linear isomers. In reactions with methylmagnesium bromide (MeMgBr) an ortho-diphenylphosphanyl-arylphosphite ligand with an additional phenyl substituent in ortho′-position at the aryl backbone proved to be superior. In contrast, best results were obtained in the case of higher alkyl Grignard reagents (such as ethyl-, n-butyl-, isopropyl-, and 3-butenylmagnesium bromides) with a related ligand carrying an isopropyl substituent in ortho′-position. The method was tested on a multi-mmol scale and is suited for application in natural product synthesis.
Co-reporter:Stephan Labsch, Shute Ye, Andreas Adler, Jörg-Martin Neudörfl, Hans-Günther Schmalz
Tetrahedron: Asymmetry 2010 Volume 21(13–14) pp:1745-1751
Publication Date(Web):14 July 2010
DOI:10.1016/j.tetasy.2010.05.019
The stereospecificity of the Au(I)-catalyzed reaction of 1-alkynyl-bicyclo[4.1.0]-heptan-2-ones with nucleophiles was investigated. The substrates were prepared in non-racemic form (up to 88% ee) through parallel kinetic resolution (CBS reduction) and reoxidation of the separated diastereomeric alcohols. The key Au-catalyzed reaction was then found to proceed without significant loss of absolute stereochemical information; this way, an achiral carbocationic intermediate could be excluded. Thus, a bicyclobutonium-type intermediate seems to be attacked in a SN2-type fashion by the nucleophile in accordance with computational predictions.(1R,2R,6S)-1-Phenylethynylbicyclo[4.1.0]heptan-2-olC15H16OEe 71%[α]λ (20 °C, CHCl3, c 1): [α]589 = 73.4, [α]546 = 88.4, [α]405 = 200.9, [α]365 = 280.4Source of chirality: parallel kinetic resolution of rac-1-(phenylethynyl)-bicyclo[4.1.0]heptan-2-one using (S)-CBS catalystAbsolute configuration: (1R,2R,6S)(1S,2R,6R)-1-Phenylethynylbicyclo[4.1.0]heptan-2-olC15H16OEe 88%[α]λ (20 °C, CHCl3, c 1): [α]589 = −90.5, [α]546 = −108.8, [α]405 = −242.9, [α]365 = −338.2, [α]334 = −470.1Source of chirality: parallel kinetic resolution of rac-1-(phenylethynyl)-bicyclo[4.1.0]heptan-2-one using (S)-CBS catalystAbsolute configuration: (1S,2R,6R)(1S,6R)-1-(Phenylethynyl)-bicyclo[4.1.0]heptan-2-oneC15H14OEe 86%[α]λ (20 °C, CHCl3, c 1): [α]589 = −23.2, [α]546 = −24.3, [α]405 = 16.6, [α]365 = 115.5, [α]334 = 419.3(1S,2R,6R)-1-Phenylethynylbicyclo[4.1.0]heptan-2-olAbsolute configuration: (1S,6R)(1R,2R,6S)-1-Hex-1-ynylbicyclo[4.1.0]heptan-2-olC13H20OEe could not be determined[α]λ (20 °C, CHCl3, c 1): [α]589 = 46.3, [α]546 = 83.6, [α]405 = 121.1, [α]365 = 163.0Source of chirality: parallel kinetic resolution of rac-1-hex-1-ynyl-bicyclo[4.1.0]heptan-2-one using (S)-CBS catalystAbsolute configuration: (1R,2R,6S)(1S,2R,6R)-1-Hex-1-ynylbicyclo[4.1.0]heptan-2-olC13H20OEe 81%[α]λ (20 °C, CHCl3, c 1): [α]589 = −38.1, [α]546 = −44.8, [α]405 = −88.3, [α]365 = −113.5Source of chirality: parallel kinetic resolution of rac-1-hex-1-ynyl-bicyclo[4.1.0]heptan-2-one using (S)-CBS catalystAbsolute configuration: (1S,2R,6R)(1S,6R)-1-Hex-1-ynyl-bicyclo[4.1.0]heptan-2-oneC13H18OEe 79%[α]λ (20 °C, CHCl3, c 1): [α]589 = −13.0, [α]546 = −13.8, [α]405 = −1.9, [α]365 = +29.6Source of chirality: (1S,2R,6R)-1-hex-1-ynylbicyclo[4.1.0]heptan-2-olAbsolute configuration: (1S,6R)(S)-2-Phenyl-5-methoxy-5,6,7,8-tetrahydro-4H-cyclohepta[b]furanC16H18O2Ee 83%[α]λ (20 °C, CHCl3, c 1): [α]589 = −3.2, [α]546 = −5.0, [α]405 = −27.2, [α]365 = −61.9Source of chirality: (1S,6R)-1-(phenylethynyl)-bicyclo[4.1.0]heptan-2-oneAbsolute configuration: (S)(S)-5-tert-Butoxy-5,6,7,8-tetrahydro-2-phenyl-4H-cyclohepta[b]furanC19H24O2Ee 83%[α]λ (20 °C, CHCl3, c 1): [α]589 = +7.1, [α]546 = +7.7, [α]405 = +4.6Source of chirality: (1S,6R)-1-(phenylethynyl)-bicyclo[4.1.0]heptan-2-oneAbsolute configuration: (S)(S)-5,6,7,8-Tetrahydro-2-phenyl-5-(prop-2-ynyloxy)-4H-cyclohepta[b]furanC18H18O2Ee 77%[α]λ (20 °C, CHCl3, c 1): [α]589 = −18.9, [α]546 = −23.5, [α]405 = −103.1Source of chirality: (1S,6R)-1-(phenylethynyl)-bicyclo[4.1.0]heptan-2-oneAbsolute configuration: (S)(S)-2-Butyl-5,6,7,8-tetrahydro-5-methoxy-4H-cyclohepta[b]furanC14H22O2Ee 79%[α]λ (20 °C, CHCl3, c 1): [α]589 = −2.9, [α]546 = −3.8, [α]405 = −13.8, [α]365 = −23.7Source of chirality: (1S,6R)-1-hex-1-ynyl-bicyclo[4.1.0]heptan-2-oneAbsolute configuration: (S)(S)-5-tert-Butoxy-2-butyl-5,6,7,8-tetrahydro-4H-cyclohepta[b]furanC17H28O2Ee could not be determined[α]λ (20 °C, CHCl3, c 1): [α]589 = +7.5; [α]546 = +9.2; [α]405 = +16.0Source of chirality: (1S,6R)-1-hex-1-ynyl-bicyclo[4.1.0]heptan-2-oneAbsolute configuration: (S)(S)-2-Butyl-5,6,7,8-tetrahydro-5-(prop-2-ynyloxy)-4H-cyclohepta[b]furanC16H22O2Ee 80%[α]λ (20 °C, CHCl3, c 1): [α]589 = −2.2, [α]546 = −2.7, [α]405 = −9.6, [α]365 = −17.2, [α]365 = −31.8Source of chirality: (1S,6R)-1-hex-1-ynyl-bicyclo[4.1.0]heptan-2-oneAbsolute configuration: (S)
Co-reporter:Dr. Jan Zaminer;Dr. Christoph Brockmann;Dipl.-Chem. Peter Huy;Dipl.-Biophys. Robert Opitz;Dipl.-Chem. Cédric Reuter;Dr. Michael Beyermann;Priv.-Doz.Dr. Christian Freund;Dipl.-Biochem. Matthias Müller;Dr. Hartmut Oschkinat;Dr. Ronald Kühne;Dr. Hans-Günther Schmalz
Angewandte Chemie International Edition 2010 Volume 49( Issue 39) pp:7111-7115
Publication Date(Web):
DOI:10.1002/anie.201001739
Co-reporter:Nikolay Slavov;Dr. Ján Cvengro&x161;;Dr. Jörg-Martin Neudörfl ;Dr. Hans-Günther Schmalz
Angewandte Chemie International Edition 2010 Volume 49( Issue 41) pp:7588-7591
Publication Date(Web):
DOI:10.1002/anie.201003755
Co-reporter:Nikolay Slavov;Dr. Ján Cvengro&x161;;Dr. Jörg-Martin Neudörfl ;Dr. Hans-Günther Schmalz
Angewandte Chemie 2010 Volume 122( Issue 41) pp:7751-7754
Publication Date(Web):
DOI:10.1002/ange.201003755
Co-reporter:Tobias Robert, Zohar Abiri, Jeroen Wassenaar, Albertus J. Sandee, Steffen Romanski, Jörg-Martin Neudörfl, Hans-Günther Schmalz and Joost N. H. Reek
Organometallics 2010 Volume 29(Issue 2) pp:478-483
Publication Date(Web):December 18, 2009
DOI:10.1021/om9009735
A small library of 17 modular and easily accessible phenol-derived chiral phosphine−phosphite ligands was evaluated in the asymmetric Rh-catalyzed hydroformylation of styrene. It was found that the stereochemical outcome of the reaction is highly dependent on the chiral phosphite moiety and the substituents on the phenolic backbone. Among the ligands studied, Taddol-based ligands of type 10 bearing bulky substituents in ortho-position to the phosphite performed best, with enantioselectivities of up to 85% ee and regioselectivities of ≥98:2. High-pressure NMR of the active catalyst [HRh(P−P)(CO)2] (P−P = 10h) revealed an equatorial-apical coordination of the ligand at rhodium. Temperature dependency of the coupling constants observed during the experiment indicates equilibrium between the two equatorial-apical isomers, with the isomer in which the phosphite occupies the equatorial position being the dominant species.
Co-reporter:Norman Nicolaus;Janet Zapke;Philipp Riesterer;Jörg-Martin Neudörfl Dr.;Aram Prokop Dr.Dr.;Hartmut Oschkinat Dr.;Hans-Günther Schmalz Dr.
ChemMedChem 2010 Volume 5( Issue 5) pp:661-665
Publication Date(Web):
DOI:10.1002/cmdc.201000063
Co-reporter:Harald Bielig;Dr. Janna Velder;Aroonchai Saiai;Maureen Menning;Dr. Sonja Meemboor;Dr. Wiltrud Kalka-Moll;Dr. Martin Krönke;Dr. Hans-Günther Schmalz;Dr. Thomas A. Kufer
ChemMedChem 2010 Volume 5( Issue 12) pp:2065-2071
Publication Date(Web):
DOI:10.1002/cmdc.201000320
Abstract
Inflammation is a hallmark of microbial infection in mammals and is the result of a pathogen-induced release of inflammatory effectors. In humans a variety of germ-line encoded receptors, so-called pattern-recognition receptors, respond to conserved signatures on invading pathogens, which results in the transcriptional activation of pro-inflammatory responses. Inflammation is often detrimental to the host and leads to tissue damage and/or systemic dysfunctions. Thus, specific inhibitors of these pathways are desirable for medical interventions. Herein we report on the synthesis and use of some chromium-containing compounds (areneCr(CO)3 complexes) with a core structure related to anti-inflammatory diterpenes produced by the sea whip Pseudopterogorgia elisabethae. By using cell-based reporter assays we identified complexes with a potent inhibitory activity on tumour necrosis factor (TNF), Toll-like receptor (TLR), and nucleotide binding domain, leucine-rich repeat-containing receptor (NLR) pathways. Moreover, we found one complex to be a specific inhibitor of inflammatory responses mediated by the NLR protein NOD2, a pivotal innate immune receptor involved in bacterial recognition. Synthesis and characterisation of a set of derivatives of this substance revealed structural requirements for NOD2 specificity. Taken together, our studies suggest this type of areneCr(CO)3 complex as a potential lead for the development of antiphlogistica and pharmacologically relevant NOD2 inhibitors.
Co-reporter:Andrea Hunold;Ines Neundorf;Philippe James;Jörg Neudörfl;Hans-Günther Schmalz
European Journal of Organic Chemistry 2009 Volume 2009( Issue 26) pp:4429-4440
Publication Date(Web):
DOI:10.1002/ejoc.200900552
Abstract
As a contribution to bioorganometallic chemistry, a dia- and enantioselective synthesis of novel carbocyclic amino acid analogues with a 1,2-ferroceno-fused cyclopentene backbone has been developed. Using two related planar-chiral intermediates, i.e. (1S,E)-ethyl-3-[2-(phenylsulfonylacetyl)ferrocen-1-yl]acrylate, (1S,E)-ethyl-3-[2-(methoxycarbonyl)ferrocen-1-yl]acrylate, stereoselective entries to different tBoc- or Fmoc-protected ferroceno-fused 1-amino-3-carboxyalkyl-2-cyclopentenes were elaborated. These compounds were then used for the preparation of metal-containing peptides by means of solid-phase peptide synthesis (SPPS). (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Ján Cvengro&x161; Dr.;Jutta Schütte Dipl.-Chem.;Nils Schlörer Dr.;Jörg Neudörfl Dr. ;Hans-Günther Schmalz Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 33) pp:6148-6151
Publication Date(Web):
DOI:10.1002/anie.200901837
Co-reporter:Ján Cvengro&x161; Dr.;Jutta Schütte Dipl.-Chem.;Nils Schlörer Dr.;Jörg Neudörfl Dr. ;Hans-Günther Schmalz Dr.
Angewandte Chemie 2009 Volume 121( Issue 33) pp:6264-6267
Publication Date(Web):
DOI:10.1002/ange.200901837
Co-reporter:Janna Velder;Tobias Robert;Ingo Weidner;Jörg-Martin Neudörfl;Johann Lex ;Hans-Günther Schmalz
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 9) pp:1309-1315
Publication Date(Web):
DOI:10.1002/adsc.200800146
Abstract
An efficient and modular approach to bidentate phosphine-phosphite ligands formally derived from a 6-alkyl-2-phosphanylphenol, a chiral diol and phosphorus trichloride has been developed. In a key step, a borane-protected phosphinite, prepared from an o-bromophenol by O-phosphanylation, is reacted with n-butyllithium to afford the corresponding ortho-phosphanylphenol (as the stable borane adduct) through bromine-lithium exchange and anionic migration rearrangement. Treatment with phosphorus trichloride in the presence of a base and subsequent reaction of the in situ formed dichlorophosphite with a chiral diol (such as TADDOL or BINOL) affords the target P,P ligands in good overall yield (up to 60% over 4 steps). In contrast to an earlier approach, the new methodology is very general and tolerates bulky ortho-substituents. The reliability of the operationally convenient protocol was demonstrated in the synthesis of a library of 16 new phosphine-phosphite ligands, starting from different ortho-alkylphenols. The modular concept opens a rapid access to a broad variety of ligands and might be useful in the search for and structural optimization of suitable ligands for specific chirogenic transition metal-catalyzed transformations.
Co-reporter:Tobias Robert Dipl.-Chem.;Janna Velder Dr. ;Hans-Günther Schmalz Dr.
Angewandte Chemie 2008 Volume 120( Issue 40) pp:7832-7835
Publication Date(Web):
DOI:10.1002/ange.200803247
Co-reporter:Tobias Robert Dipl.-Chem.;Janna Velder Dr. ;Hans-Günther Schmalz Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 40) pp:7718-7721
Publication Date(Web):
DOI:10.1002/anie.200803247
Co-reporter:Anja Pfletschinger;Johann Lex;Hans-Günther Schmalz;Ulrich Schneider
European Journal of Organic Chemistry 2007 Volume 2007(Issue 24) pp:3991-3998
Publication Date(Web):3 JUL 2007
DOI:10.1002/ejoc.200700342
The configurational stability of reactive intermediates (cation, radical and anion) derived from ethylcyclobutadiene–Fe(CO)3 by formal abstraction of a hydride, a hydrogen atom or a proton from the pseudobenzylic position was investigated. Density functional calculations (Becke3lyp) predicted that all these reactive intermediates can be regarded as planar chiral structures having a significant configurational stability. The racemization barriers were calculated to be 38.8 kcal mol–1 for the cationic, 16.9 kcal mol–1 for the radical and 44.8 kcal mol–1 for the anionic intermediate. In an experimental part of the study, derivatives of the 1-hydroxyethyl-substituted complex, which was enantioselectively prepared by CBS reduction of the acetyl complex, were subjected to SN1-type as well as single-electron-transfer-driven umpolung reactions. The observed stereospecificity of these transformations (retention of configuration) is in accordance with the predicted configurational stability of the involved reactive intermediates. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:E.E. Shults, J. Velder, H.-G. Schmalz, S.V. Chernov, T.V. Rubalova, Y.V. Gatilov, G. Henze, G.A. Tolstikov, A. Prokop
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 16) pp:4228-4232
Publication Date(Web):15 August 2006
DOI:10.1016/j.bmcl.2006.05.077
Pinusolide (1), a known platelet-activating factor (PAF) receptor binding antagonist, was synthesized from lambertianic acid (2), a labdane-type diterpene readily accessible in multigram quantities from the Siberian pine tree. It was shown that 1 not only decreases the proliferation activity of tumor cells at relatively low concentrations but specifically induces apoptosis at 100 μM via the mitochondrial pathway in the Burkitt lymphoma cell line BJAB. Also, using primary lymphoblasts and leukemic cells from children with acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML), a significant DNA fragmentation in pinusolide-treated cells could be detected in an ex vivo apoptosis assay.
Co-reporter:Junliang Zhang Dr.;Hans-Günther Schmalz Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 40) pp:
Publication Date(Web):20 SEP 2006
DOI:10.1002/anie.200601252
Ring round: 1-Acyl-1-alkynyl-cyclopropanes smoothly react with nucleophiles in the presence of a gold(I) catalyst to afford substituted furans in a fully atom-economical fashion (see scheme; Nu=nucleophile, Tf=trifluoromethanesulfonyl). The reaction tolerates a wide range of substrates.
Co-reporter:Daniel Schlawe Dr.;André Majdalani;Juraj Velcicky Dr.;Erik Heßler;Thomas Wieder Priv.-Doz. Dr.;Aram Prokop Dr. Dr.;Hans-Günther Schmalz Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 13) pp:
Publication Date(Web):17 MAR 2004
DOI:10.1002/anie.200353132
Bioactive organometallic compounds: Iron-containing nucleoside analogues, which can be stereoselectively synthesized in a few steps starting from simple carbohydrates, cause tumor cells to undergo apoptosis (programmed cell death; see picture for the typical morphological characteristics of BJAB tumor cells after apoptosis).
Co-reporter:Timm Graening Dipl.-Chem.;Hans-Günther Schmalz Dr.
Angewandte Chemie 2004 Volume 116(Issue 25) pp:
Publication Date(Web):19 APR 2004
DOI:10.1002/ange.200300615
Colchicin, das Hauptalkaloid der Herbstzeitlose, ist einer der prominentesten Naturstoffe, der wie andere Tubulin-bindende Wirkstoffe (z. B. Taxol und Epothilone) über ein großes pharmazeutisches Potenzial verfügt. Die ersten Synthesen von Colchicin in den späten 50er Jahren waren Meilensteine der Naturstoffsynthese. Aber auch heute noch stellt dieses strukturell vermeintlich einfache Molekül eine Herausforderung dar. So ist es erst in den letzten Jahren gelungen, Synthesen zu entwickeln, die effizient auch neue, strukturell modifizierte Colchicin-Derivate totalsynthetisch zugänglich machen. Die vergleichende Betrachtung aller bekannten Colchicin-Synthesen in diesem Aufsatz verdeutlicht nicht nur den Fortschritt der Organischen Synthese in den letzten Jahrzehnten. Es wird vielmehr gezeigt, dass die synthetischen Fragestellungen, die dieses Molekül aufwirft, nur mit einem hohen Maß an chemischer Kreativität beantwortet werden können. Nur wenige Zielverbindungen sind auf so facettenreiche Weise synthetisiert worden wie Colchicin.
Co-reporter:Daniel Schlawe Dr.;André Majdalani;Juraj Velcicky Dr.;Erik Heßler;Thomas Wieder Priv.-Doz. Dr.;Aram Prokop Dr. Dr.;Hans-Günther Schmalz Dr.
Angewandte Chemie 2004 Volume 116(Issue 13) pp:
Publication Date(Web):17 MAR 2004
DOI:10.1002/ange.200353132
Metallorganische Verbindungen als biologische Wirkstoffe: Die stereoselektive Synthese eisenhaltiger Nucleosidanaloga gelingt in wenigen Schritten aus einfachen Kohlenhydraten. Die Produkte lösen bei Tumorzellen den programmierten Zelltod (Apoptose) aus. Das Bild zeigt die typischen morphologischen Merkmale bei apoptotischen BJAB-Tumorzellen.
Co-reporter:Timm Graening Dipl.-Chem.;Hans-Günther Schmalz Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 25) pp:
Publication Date(Web):19 APR 2004
DOI:10.1002/anie.200300615
Colchicine, the major alkaloid of the meadow saffron, is one of the most prominent natural products and, like other tubulin-binding natural products (e.g. taxol and the epothilones), exhibits great pharmaceutical potential. The first syntheses in the late 1950s were milestones in natural product synthesis. But even today this structurally supposedly simple molecule poses a challenge to synthetic chemists. Only in the last years have syntheses been developed that are efficient enough to provide novel structurally modified colchicine analogues. The comparative examination of all known colchicine total syntheses undertaken in this Review not only reveals the tremendous progress in synthetic organic methodology over the past decades, but also shows how the unique synthetic problems posed by this molecule can be solved in an exceptionally creative manner. Only a few target molecules have been synthesized in such multifaceted ways.
Co-reporter:Juraj Velcicky Dr.;Andreas Lanver Dipl.-Chem.;Johann Lex Dr.;Aram Prokop Dr.;Thomas Wieder Priv. Doz. Dr.;Hans-Günther Schmalz Dr.
Chemistry - A European Journal 2004 Volume 10(Issue 20) pp:
Publication Date(Web):2 SEP 2004
DOI:10.1002/chem.200400079
A diversity-oriented, enantioselective synthesis of new (monoprotected) carbocyclic nucleoside analogues (CNAs) with the nucleobase attached to a 3-hydroxymethyl-4-trialkylsilyloxymethylcyclopent-2-en-1-yl scaffold was developed. As a key intermediate, racemic (5SR,8RS)-8-allyloxy-2-trimethylsilyl-7-oxa-bicyclo[3.3.0]-oct-1-en-3-one was prepared from 1,1-diallyloxy-3-trimethylsilyl-2-propyne in a cobalt-mediated Pauson–Khand reaction. The enantiomerically pure material was obtained through efficient kinetic resolution (selectivity factor s≥40 at −78 °C) by means of an oxazaborolidine-catalyzed borane reduction (CBS reduction) with catecholborane. The absolute configuration of the resolved products was determined by CD spectroscopy, Mosher ester analysis, and chemical correlation. Subsequent steps involve diastereoselective ketone reduction and fully regio- and diastereoselective introduction of the nucleobase through Pd0-catalyzed allylic substitution. The generality of the method was demonstrated by preparation of CNAs in both enantiomeric series with all five natural nucleobases, as well as 5-bromouracil, 5-fluorouracil, and 6-chloropurine. Screening of the various compounds in a cytotoxicity assay with BJAB and ALL tumor cell lines revealed that some of the compounds possess pronounced antitumoral properties (LD50 values down to 9 μM, as determined by lactate dehydrogenase release after 48 h). By measuring DNA fragmentation, it could be shown that the activity results from induction of apoptosis.
Co-reporter:Timm Graening Dipl.-Chem.;Hans-Günther Schmalz Dr.
Angewandte Chemie 2003 Volume 115(Issue 23) pp:
Publication Date(Web):12 JUN 2003
DOI:10.1002/ange.200301644
Vielseitig wie die Natur: Die Pd-katalysierte enantioselektive allylische Substitution ist eine leistungsfähige Methode der modernen Synthesechemie. Insbesondere die Substitutionsreaktionen von cyclischen allylischen Substraten (siehe Bild) haben sich in letzter Zeit als wertvoll erwiesen, da sie neuartige und hocheffiziente Strategien für die Totalsynthese einer Vielzahl komplexer Naturstoffe anbieten.
Co-reporter:Timm Graening Dipl.-Chem.;Hans-Günther Schmalz Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 23) pp:
Publication Date(Web):12 JUN 2003
DOI:10.1002/anie.200301644
As versatile as Nature: Pd-catalyzed enantioselective allylic substitution is a powerful reaction in modern synthetic chemistry. The reactions of cyclic allylic substrates (see figure) have recently been proven to be of particular value as they lead to novel and highly efficient strategies in the total synthesis of various natural products.
Co-reporter:Florian Blume;Saskia Zemolka;Thorsten Fey;Remo Kranich;Hans-Günther Schmalz
Advanced Synthesis & Catalysis 2002 Volume 344(Issue 8) pp:
Publication Date(Web):25 SEP 2002
DOI:10.1002/1615-4169(200209)344:8<868::AID-ADSC868>3.0.CO;2-M
Based on a general modular synthetic scheme, a variety of chiral bidentate P/P-, P/S-, P/N-, and P/Se-ligands is accessible in an efficient divergent manner starting from phenol or naphthol derived backbone systems. A library of 20 selected ligands was tested in the Rh-catalyzed asymmetric hydroboration of styrene to give 1-phenylethanol in up to 91% ee after oxidative work-up. It was demonstrated that small variations of the ligand structures lead to pronounced, unpredictable differences in the performance of the in situ generated rhodium complexes. The modular approach should be applicable for the identification and optimization of suitable ligands for other transition metal-catalyzed transformations with comparably low effort.
Co-reporter:Timm Graening Dipl.-Chem.;Willy Friedrichsen Dr.;Johann Lex Dr.;Hans-Günther Schmalz Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 9) pp:
Publication Date(Web):2 MAY 2002
DOI:10.1002/1521-3773(20020503)41:9<1524::AID-ANIE1524>3.0.CO;2-9
The major alkaloid of the meadow saffron is colchicine, which displays remarkable antimitotic activity and has long been used in the treatment of acute gout and is currently being tested against a broad variety of other diseases. In a remarkable Rh-catalyzed domino transformation, the complete carbon skeleton of this important alkaloid is assembled with high efficiency (see scheme). TBS=tert-butyldimethylsilyl, TMS=trimethylsilyl.
Co-reporter:Saskia Zemolka Dipl.-Chem.;Johann Lex Dr.;Hans-Günther Schmalz Dr.
Angewandte Chemie 2002 Volume 114(Issue 14) pp:
Publication Date(Web):15 JUL 2002
DOI:10.1002/1521-3757(20020715)114:14<2635::AID-ANGE2635>3.0.CO;2-I
Eine vollständig stereoselektive Synthese von pharmakologisch relevanten trans-1,3-disubstituierten Dihydroisobenzofuranen ist durch die überraschende Selektivität der benzylischen Deprotonierung eines silylierten Tricarbonyl-Phthalan-Chrom-Komplexes möglich (siehe Schema).
Co-reporter:Timm Graening Dipl.-Chem.;Willy Friedrichsen Dr.;Johann Lex Dr.;Hans-Günther Schmalz Dr.
Angewandte Chemie 2002 Volume 114(Issue 9) pp:
Publication Date(Web):2 MAY 2002
DOI:10.1002/1521-3757(20020503)114:9<1594::AID-ANGE1594>3.0.CO;2-G
Der Wirkstoff aus der Herbstzeitlose ist Colchicin, ein starkes Mitosegift, das schon seit dem 18. Jahrhundert gegen Gelenkschmerzen und Entzündungen bei Gichtanfällen verwendet und derzeit gegen eine Vielzahl anderer Krankheiten getestet wird. In einer bemerkenswerten Rh-katalysierten Domino-Transformation gelingt der Aufbau des kompletten Kohlenstoffgerüstes dieses wichtigen Alkaloids mit hoher Effizienz (siehe Schema). TBS=tert-Butyldimethylsilyl, TMS=Trimethylsilyl.
Co-reporter:Saskia Zemolka;Johann Lex Dr.;Hans-Günther Schmalz Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 14) pp:
Publication Date(Web):15 JUL 2002
DOI:10.1002/1521-3773(20020715)41:14<2525::AID-ANIE2525>3.0.CO;2-O
A completely stereoselective synthesis of pharmacologically relevant trans-1,3-disubstituted dihydroisobenzofurans utilizes the surprising selectivity during the benzylic deprotonation of a silylated [phthalan–Cr(CO)3] complex (see scheme).
Co-reporter:Anja Pfletschinger, Wolfram Koch and Hans-Günther Schmalz
New Journal of Chemistry 2001 vol. 25(Issue 3) pp:446-450
Publication Date(Web):14 Feb 2001
DOI:10.1039/B002898N
The meta regioselectivity of the nucleophilic addition to methoxy-substituted arene-Cr(CO)3 complexes has been investigated by theoretical methods using density functional calculations employing the hybrid-DFT approach Becke3LYP and a flexible all-electron basis set. By calculating the relative energies and conformational preferences of
the competing reactive intermediates (regioisomeric primary addition products) it was, among other things, demonstrated that the preferred reaction pathway proceeds ia the most stable intermediate. Based on the theoretical results a refined mechanistic picture for a synthetically important metallorganic process was derived.
Co-reporter:Anja Pfletschinger;Wolfram Koch;Hans-Günther Schmalz
Chemistry - A European Journal 2001 Volume 7(Issue 24) pp:
Publication Date(Web):10 DEC 2001
DOI:10.1002/1521-3765(20011217)7:24<5325::AID-CHEM5325>3.0.CO;2-S
By applying the hybrid density functional method B3LYP and a flexible all-electron basis set, structures and energies of reactive intermediates derived from the 1-butyne complex of Co2(CO)6 (1) were calculated. In particular, the geometry, electronic distribution, and configurational stability of the cationic, radical, and anionic Co2(CO)6-complexed propargylic species were studied. The calculations revealed that the configurational barrier, that is, the racemization barrier for the antarafacial migration of the CHCH3 group, is low (7.6 kcal mol−1) for the radical and is similar to the experimental value for the corresponding cation (ca. 10 kcal mol−1). However, a high racemization barrier (23.7 kcal mol−1) for the anionic intermediate suggests the possibility of stereospecific reactions involving Co2(CO)6-complexed propargylic anions.
Co-reporter:Remo Kranich Dr.;Knut Eis Dr.;Oliver Geis Dipl.-Chem.;Stefan Mühle Dipl.-Chem.;Jan W. Bats Dr.;Hans-Günther Schmalz Dr.
Chemistry - A European Journal 2000 Volume 6(Issue 15) pp:
Publication Date(Web):20 JUL 2000
DOI:10.1002/1521-3765(20000804)6:15<2874::AID-CHEM2874>3.0.CO;2-1
A modular approach to a new class of structurally diverse bidentate P/N, P/P, P/S, and P/Se chelate ligands has been developed. Starting from hydroquinone, various ligands were synthesized in a divergent manner via orthogonally bis-protected bromohydroquinones as the central building block. The first donor functionality (L1) is introduced to the aromatic (hydroquinone) ligand backbone either by Pd-catalyzed cross-coupling (Suzuki coupling) with hetero-aryl bromides, by Pd-catalyzed amination, or by lithiation and subsequent treatment with electrophiles (e.g., chlorophosphanes, disulfides, diselenides, or carbamoyl chlorides). After selective deprotection, the second ligand tooth (L2) is attached by reaction of the phenolic OH functionality with a chlorophosphane, a chlorophosphite, or a related reagent. Some of the resulting chelate ligands were converted into the respective PdX2 complexes (X=Cl, I), two of which were characterized by X-ray crystallography. The methodology developed opens an access to a broad variety of new chiral and achiral transition metal complexes and is generally suited for the solid-phase synthesis of combinatorial libraries, as will be reported separately.
Co-reporter:Daniel Augner ; Oleg Krut ; Nikolay Slavov ; Dario C. Gerbino ; Hans-Georg Sahl ; Jürgen Benting ; Carl F. Nising ; Stefan Hillebrand ; Martin Krönke ;Hans-Günther Schmalz
Journal of Natural Products () pp:
Publication Date(Web):August 1, 2013
DOI:10.1021/np400301d
Pestalone (1) is a prominent marine natural product first isolated by M. Cueto et al. in 2001 from a co-fermentation of a marine fungus with a marine bacterium. For more than 10 years, 1 had been considered as a promising new antibiotic compound, the reported MIC against methicillin-resistant Staphylococcus aureus (MRSA) being 37 ng/mL. After overcoming the limited availability of 1 by total synthesis (N. Slavov et al., 2010) we performed new biological tests, which did not confirm the expected degree of antibiotic activity. The observed activity of pestalone against different MRSA strains was 3–10 μg/mL, as determined independently in two laboratories. A number of synthetic derivatives of 1 including pestalachloride A and other isoindolinones (formed from 1 by reaction with amines) did not exhibit higher activities as compared to 1 against MRSA and a series of plant pathogens.
Co-reporter:Steffen Romanski, Birgit Kraus, Miguel Guttentag, Waldemar Schlundt, Hannelore Rücker, Andreas Adler, Jörg-Martin Neudörfl, Roger Alberto, Sabine Amslinger and Hans-Günther Schmalz
Dalton Transactions 2012 - vol. 41(Issue 45) pp:NaN13875-13875
Publication Date(Web):2012/05/16
DOI:10.1039/C2DT30662J
A series of η4-acyloxycyclohexadiene–Fe(CO)3 complexes was prepared and fully characterized by spectroscopic methods including single crystal X-ray diffraction. For this purpose a new synthetic access to differently acylated 1,3- and 1,5-dienol–Fe(CO)3 complexes was developed. The enzymatically triggered CO release from these compounds was monitored (detection of CO through GC and/or by means of a myoglobin assay) and the anti-inflammatory effect of the compounds was assessed by a cellular assay based on the inhibition of NO-production by inducible NO synthase (iNOS). It was demonstrated that the properties (rate of esterase-triggered CO release, iNOS inhibition, cytotoxicity) of the complexes strongly depend on the substitution pattern of the π-ligand and the nature of the acyloxy substituent.

![Hexanal, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/118800/118794-69-7.png)
![Hexanal, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/118800/118794-69-7_b.png)


![Lithium, [(phenylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/116600/116511-95-6.png)
![Lithium, [(phenylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/116600/116511-95-6_b.png)













![6-[tert-butyl(dimethyl)silyl]oxyhexan-1-ol](http://img.cochemist.com/ccimg/103300/103202-59-1.png)
![6-[tert-butyl(dimethyl)silyl]oxyhexan-1-ol](http://img.cochemist.com/ccimg/103300/103202-59-1_b.png)





![(+-)-7-Amino-1,2,3,10-tetramethoxy-6,7-dihydro-5H-benzo[a]heptalen-9-on](http://img.cochemist.com/ccimg/102500/102491-73-6.png)
![(+-)-7-Amino-1,2,3,10-tetramethoxy-6,7-dihydro-5H-benzo[a]heptalen-9-on](http://img.cochemist.com/ccimg/102500/102491-73-6_b.png)
![Benzo[a]heptalen-10(5H)-one, 7-azido-6,7-dihydro-1,2,3,9-tetramethoxy-, (7S)-](http://img.cochemist.com/ccimg/101700/101675-42-7.png)
![Benzo[a]heptalen-10(5H)-one, 7-azido-6,7-dihydro-1,2,3,9-tetramethoxy-, (7S)-](http://img.cochemist.com/ccimg/101700/101675-42-7_b.png)
![Benzoic acid, 4-methoxy-, [(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/101000/100969-61-7.png)
![Benzoic acid, 4-methoxy-, [(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/101000/100969-61-7_b.png)




![Propanoic acid, [(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/99100/99068-36-7.png)
![Propanoic acid, [(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/99100/99068-36-7_b.png)




![Benzene, 1-[(1E)-3-chloro-1-propen-1-yl]-4-methoxy-](/data/chemimg/2074200/94607-41-7.png)
![Benzene, 1-[(1E)-3-chloro-1-propen-1-yl]-4-methoxy-](/data/chemimg/2074200/94607-41-7_b.png)










![2,6,7-Trioxabicyclo[2.2.2]octane, 1-(3-bromopropyl)-4-methyl-](http://img.cochemist.com/ccimg/89300/89276-36-8.png)
![2,6,7-Trioxabicyclo[2.2.2]octane, 1-(3-bromopropyl)-4-methyl-](http://img.cochemist.com/ccimg/89300/89276-36-8_b.png)


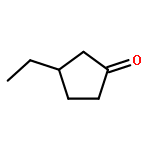
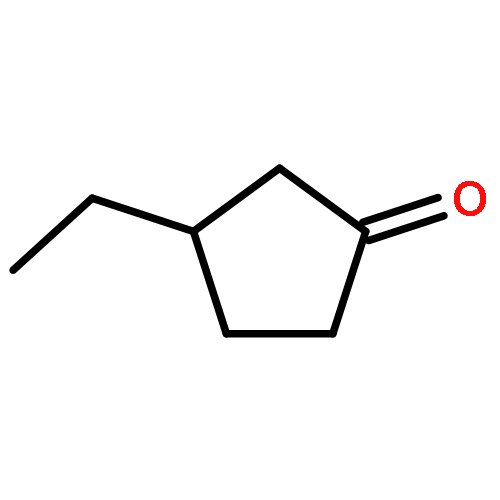
![BENZENE, 1-[(1R)-1-METHYL-2-PROPENYL]-4-(2-METHYLPROPYL)-](http://img.cochemist.com/ccimg/85800/85711-18-8.png)
![BENZENE, 1-[(1R)-1-METHYL-2-PROPENYL]-4-(2-METHYLPROPYL)-](http://img.cochemist.com/ccimg/85800/85711-18-8_b.png)
dimethyl-](http://img.cochemist.com/ccimg/85600/85514-43-8.png)
dimethyl-](http://img.cochemist.com/ccimg/85600/85514-43-8_b.png)




![Benzene,4-[(1E)-2-(3,5-dimethoxyphenyl)ethenyl]-1,2-dimethoxy-](http://img.cochemist.com/ccimg/83100/83088-26-0.png)
![Benzene,4-[(1E)-2-(3,5-dimethoxyphenyl)ethenyl]-1,2-dimethoxy-](http://img.cochemist.com/ccimg/83100/83088-26-0_b.png)
![2-Azetidinecarboxylicacid, 1-[(1,1-dimethylethyl)dimethylsilyl]-4-oxo-, (2S)-](http://img.cochemist.com/ccimg/83000/82938-50-9.png)
![2-Azetidinecarboxylicacid, 1-[(1,1-dimethylethyl)dimethylsilyl]-4-oxo-, (2S)-](http://img.cochemist.com/ccimg/83000/82938-50-9_b.png)


![2-[(6-chloro-9H-purin-9-yl)methoxy]ethyl acetate](http://img.cochemist.com/ccimg/81800/81777-47-1.png)
![2-[(6-chloro-9H-purin-9-yl)methoxy]ethyl acetate](http://img.cochemist.com/ccimg/81800/81777-47-1_b.png)




![Benzo[b]thiophene, 2-ethenyl-](http://img.cochemist.com/ccimg/78700/78646-50-1.png)
![Benzo[b]thiophene, 2-ethenyl-](http://img.cochemist.com/ccimg/78700/78646-50-1_b.png)






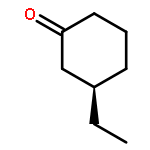
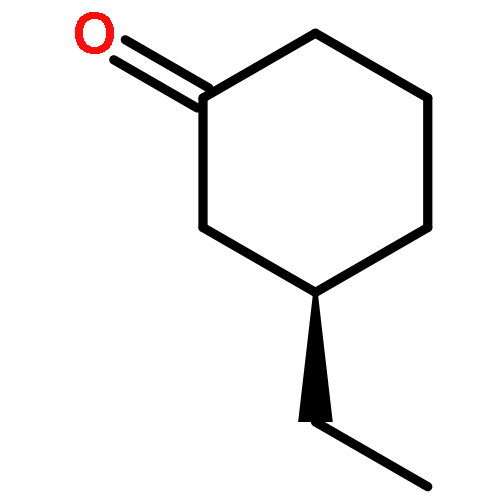


![ETHANOL, 2-[(6-CHLORO-9H-PURIN-9-YL)METHOXY]-](http://img.cochemist.com/ccimg/73500/73484-26-1.png)
![ETHANOL, 2-[(6-CHLORO-9H-PURIN-9-YL)METHOXY]-](http://img.cochemist.com/ccimg/73500/73484-26-1_b.png)




![BENZENE, 1-METHYL-4-[(1R)-1-METHYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/72800/72782-48-0.png)
![BENZENE, 1-METHYL-4-[(1R)-1-METHYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/72800/72782-48-0_b.png)










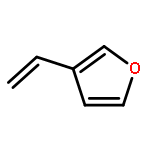

![2(1H)-Pyrimidinone, 1-(trimethylsilyl)-4-[(trimethylsilyl)amino]-](http://img.cochemist.com/ccimg/67100/67014-25-9.png)
![2(1H)-Pyrimidinone, 1-(trimethylsilyl)-4-[(trimethylsilyl)amino]-](http://img.cochemist.com/ccimg/67100/67014-25-9_b.png)








![L-Phenylalanine, N-[(phenylmethoxy)carbonyl]-, 2-propyn-1-yl ester](/data/chemimg/3313500/64286-87-9.png)
![L-Phenylalanine, N-[(phenylmethoxy)carbonyl]-, 2-propyn-1-yl ester](/data/chemimg/3313500/64286-87-9_b.png)
![2-Butyn-1-ol, 4-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/64300/64244-47-9.png)
![2-Butyn-1-ol, 4-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/64300/64244-47-9_b.png)


![Carbamic acid, [(4-methylphenyl)sulfonyl]-, 2-propynyl ester](http://img.cochemist.com/ccimg/64000/63924-66-3.png)
![Carbamic acid, [(4-methylphenyl)sulfonyl]-, 2-propynyl ester](http://img.cochemist.com/ccimg/64000/63924-66-3_b.png)
![Phenol,4-[(1E)-2-(3,5-dimethoxyphenyl)ethenyl]-, 1-acetate](http://img.cochemist.com/ccimg/63400/63366-83-6.png)
![Phenol,4-[(1E)-2-(3,5-dimethoxyphenyl)ethenyl]-, 1-acetate](http://img.cochemist.com/ccimg/63400/63366-83-6_b.png)
![Carbamic acid, [(4-methylphenyl)sulfonyl]-, 2-butynyl ester](http://img.cochemist.com/ccimg/63200/63132-71-8.png)
![Carbamic acid, [(4-methylphenyl)sulfonyl]-, 2-butynyl ester](http://img.cochemist.com/ccimg/63200/63132-71-8_b.png)






![Benzene, [(1S)-1-methyl-2-propenyl]-](http://img.cochemist.com/ccimg/58800/58717-85-4.png)
![Benzene, [(1S)-1-methyl-2-propenyl]-](http://img.cochemist.com/ccimg/58800/58717-85-4_b.png)




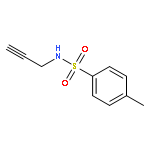



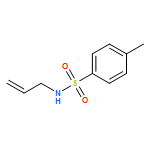
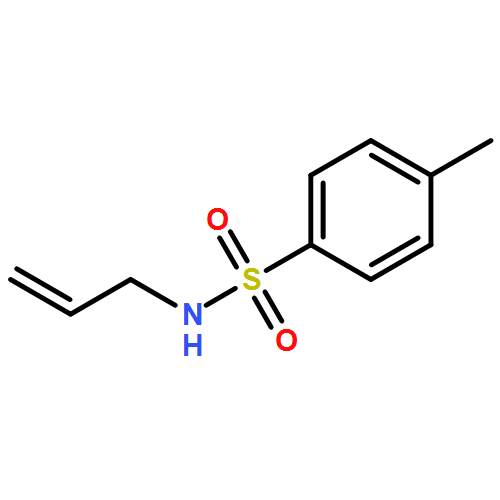
![Benzoic acid, 4-nitro-, [(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/50400/50366-20-6.png)
![Benzoic acid, 4-nitro-, [(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/50400/50366-20-6_b.png)


![2H-PYRAN, 2-[(4-BROMO-2-BUTYNYL)OXY]TETRAHYDRO-](http://img.cochemist.com/ccimg/40300/40274-28-0.png)
![2H-PYRAN, 2-[(4-BROMO-2-BUTYNYL)OXY]TETRAHYDRO-](http://img.cochemist.com/ccimg/40300/40274-28-0_b.png)

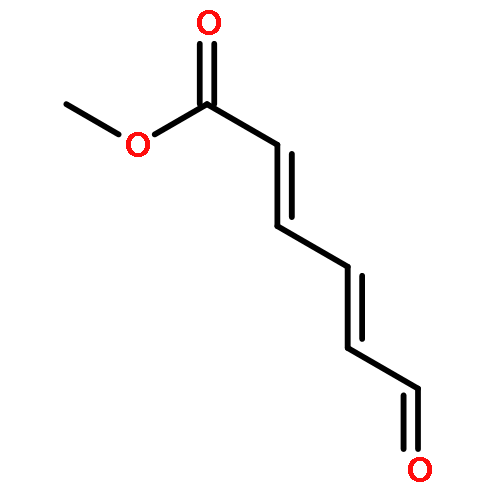
![N-(3-hydroxy-2-iodo-9,10,11-trimethoxy-6,7-dihydro-5H-dibenzo[a,c][7]annulen-5-yl)acetamide](http://img.cochemist.com/ccimg/38900/38838-27-6.png)
![N-(3-hydroxy-2-iodo-9,10,11-trimethoxy-6,7-dihydro-5H-dibenzo[a,c][7]annulen-5-yl)acetamide](http://img.cochemist.com/ccimg/38900/38838-27-6_b.png)




![Benzene, 1-methoxy-4-[(1E)-2-(4-methylphenyl)ethenyl]-](/data/chemimg/1810600/37163-68-1.png)
![Benzene, 1-methoxy-4-[(1E)-2-(4-methylphenyl)ethenyl]-](/data/chemimg/1810600/37163-68-1_b.png)



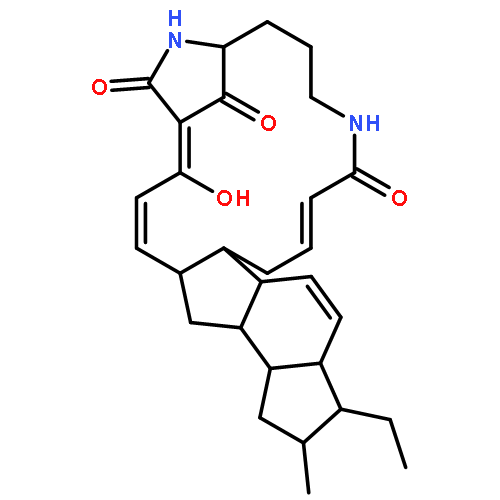








![(2R)-6-methoxy-2,5,7,8-tetramethyl-2-[(4R,8R)-4,8,12-trimethyltridecyl]-3,4-dihydro-2H-chromene](http://img.cochemist.com/ccimg/32500/32466-45-8.png)
![(2R)-6-methoxy-2,5,7,8-tetramethyl-2-[(4R,8R)-4,8,12-trimethyltridecyl]-3,4-dihydro-2H-chromene](http://img.cochemist.com/ccimg/32500/32466-45-8_b.png)










![1,2-DIMETHOXY-4-[2-(4-METHYLPHENYL)ETHENYL]BENZENE](/data/chemimg/436100/24815-56-3.png)
![1,2-DIMETHOXY-4-[2-(4-METHYLPHENYL)ETHENYL]BENZENE](/data/chemimg/436100/24815-56-3_b.png)
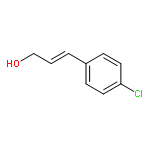
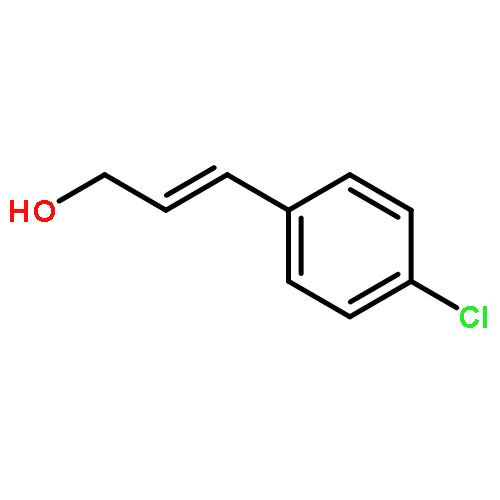
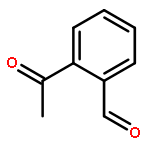











![Benzene, [(1R)-1-ethyl-2-propen-1-yl]-](/data/chemimg/1781200/20068-20-6.png)
![Benzene, [(1R)-1-ethyl-2-propen-1-yl]-](/data/chemimg/1781200/20068-20-6_b.png)










![Benzene, 1,2-dimethoxy-4-[2-(4-methoxyphenyl)ethenyl]-, (E)-](http://img.cochemist.com/ccimg/18600/18513-95-6.png)
![Benzene, 1,2-dimethoxy-4-[2-(4-methoxyphenyl)ethenyl]-, (E)-](http://img.cochemist.com/ccimg/18600/18513-95-6_b.png)







![Benzamide, N-(trimethylsilyl)-N-[9-(trimethylsilyl)-9H-purin-6-yl]-](http://img.cochemist.com/ccimg/18100/18055-47-5.png)
![Benzamide, N-(trimethylsilyl)-N-[9-(trimethylsilyl)-9H-purin-6-yl]-](http://img.cochemist.com/ccimg/18100/18055-47-5_b.png)


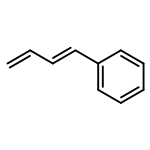
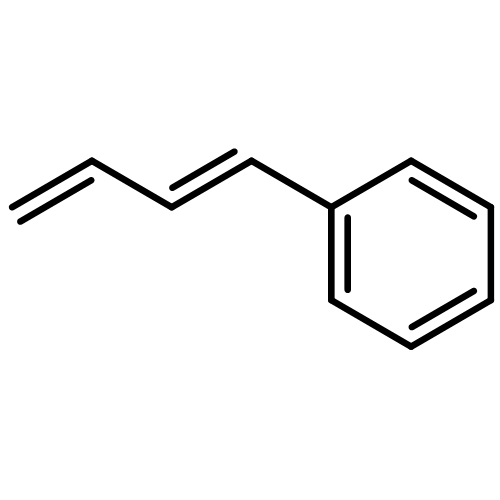








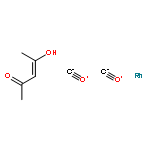
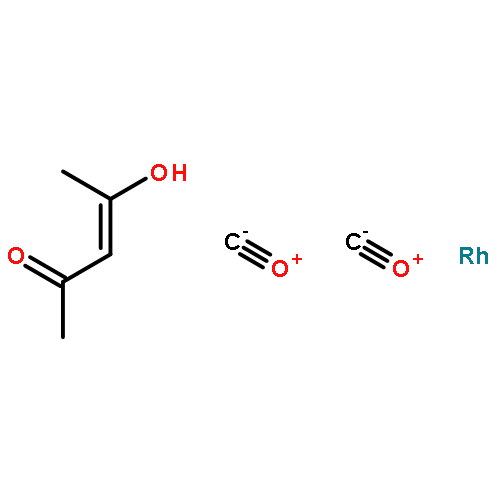






![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0.png)
![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0_b.png)
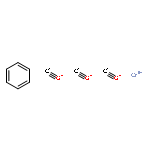
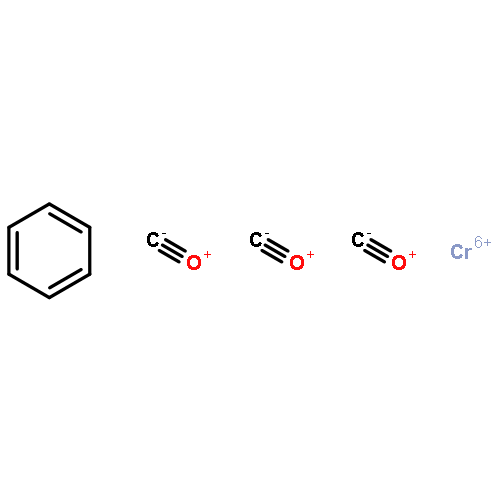


![L-Serine, 1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl-, methyl ester](http://img.cochemist.com/ccimg/7600/7535-76-4.png)
![L-Serine, 1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl-, methyl ester](http://img.cochemist.com/ccimg/7600/7535-76-4_b.png)
















![Bicyclo[4.1.0]heptan-2-one](http://img.cochemist.com/ccimg/5800/5771-58-4.png)
![Bicyclo[4.1.0]heptan-2-one](http://img.cochemist.com/ccimg/5800/5771-58-4_b.png)




![Morpholine, 4-[(triphenylphosphoranylidene)acetyl]-](http://img.cochemist.com/ccimg/4900/4885-64-7.png)
![Morpholine, 4-[(triphenylphosphoranylidene)acetyl]-](http://img.cochemist.com/ccimg/4900/4885-64-7_b.png)
![Bicyclo[3.1.0]hexan-2-one](http://img.cochemist.com/ccimg/4200/4160-49-0.png)
![Bicyclo[3.1.0]hexan-2-one](http://img.cochemist.com/ccimg/4200/4160-49-0_b.png)







![Benzoic acid,2-[(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/3300/3232-37-9.png)
![Benzoic acid,2-[(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/3300/3232-37-9_b.png)
![L-SERINE, 1-[(PHENYLMETHOXY)CARBONYL]-L-PROLYL-, METHYL ESTER](http://img.cochemist.com/ccimg/2500/2481-26-7.png)
![L-SERINE, 1-[(PHENYLMETHOXY)CARBONYL]-L-PROLYL-, METHYL ESTER](http://img.cochemist.com/ccimg/2500/2481-26-7_b.png)













![5H-Dibenzo[a,c]cycloheptene-3-carboxylicacid, 5-(acetylamino)-6,7-dihydro-9,10,11-trimethoxy-, methyl ester, (5S)-](http://img.cochemist.com/ccimg/700/641-28-1.png)
![5H-Dibenzo[a,c]cycloheptene-3-carboxylicacid, 5-(acetylamino)-6,7-dihydro-9,10,11-trimethoxy-, methyl ester, (5S)-](http://img.cochemist.com/ccimg/700/641-28-1_b.png)


![[(7S)-1,2,3,9-tetramethoxy-10-oxo-5,6,7,8,9,10-hexahydrobenzo[a]heptalen-7-yl]carbamic acid](http://img.cochemist.com/ccimg/600/518-12-7.png)
![[(7S)-1,2,3,9-tetramethoxy-10-oxo-5,6,7,8,9,10-hexahydrobenzo[a]heptalen-7-yl]carbamic acid](http://img.cochemist.com/ccimg/600/518-12-7_b.png)
![4-Pyridinecarboxylicacid, 2-[(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/500/495-84-1.png)
![4-Pyridinecarboxylicacid, 2-[(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/500/495-84-1_b.png)
![Acetamide,N-[(7S)-5,6,7,9-tetrahydro-10-hydroxy-1,2,3-trimethoxy-9-oxobenzo[a]heptalen-7-yl]-](http://img.cochemist.com/ccimg/500/477-27-0.png)
![Acetamide,N-[(7S)-5,6,7,9-tetrahydro-10-hydroxy-1,2,3-trimethoxy-9-oxobenzo[a]heptalen-7-yl]-](http://img.cochemist.com/ccimg/500/477-27-0_b.png)






![4H-Cyclohepta[b]furan, 2-butyl-5,6,7,8-tetrahydro-5-methoxy-](http://img.cochemist.com/ccimg/917100/917087-54-8.png)
![4H-Cyclohepta[b]furan, 2-butyl-5,6,7,8-tetrahydro-5-methoxy-](http://img.cochemist.com/ccimg/917100/917087-54-8_b.png)
![4H-CYCLOHEPTA[B]FURAN, 5,6,7,8-TETRAHYDRO-5-METHOXY-2-PHENYL-](http://img.cochemist.com/ccimg/917100/917087-44-6.png)
![4H-CYCLOHEPTA[B]FURAN, 5,6,7,8-TETRAHYDRO-5-METHOXY-2-PHENYL-](http://img.cochemist.com/ccimg/917100/917087-44-6_b.png)
![Bicyclo[4.1.0]heptan-2-one, 1-(1-hexyn-1-yl)-](http://img.cochemist.com/ccimg/917100/917087-37-7.png)
![Bicyclo[4.1.0]heptan-2-one, 1-(1-hexyn-1-yl)-](http://img.cochemist.com/ccimg/917100/917087-37-7_b.png)



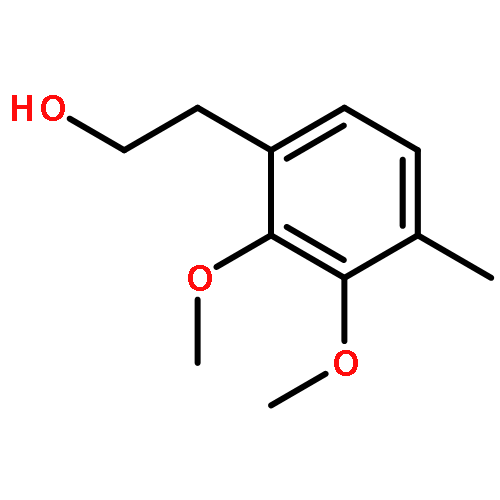


![N-Benzyl-N-methyldinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin-4-amine](http://img.cochemist.com/ccimg/490100/490023-37-5.png)
![N-Benzyl-N-methyldinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin-4-amine](http://img.cochemist.com/ccimg/490100/490023-37-5_b.png)




![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2_b.png)
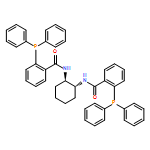
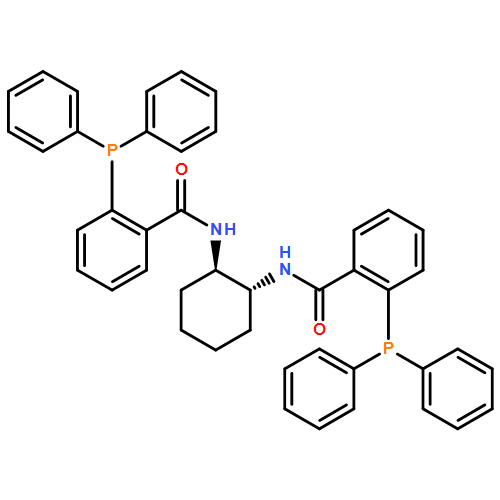


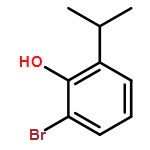
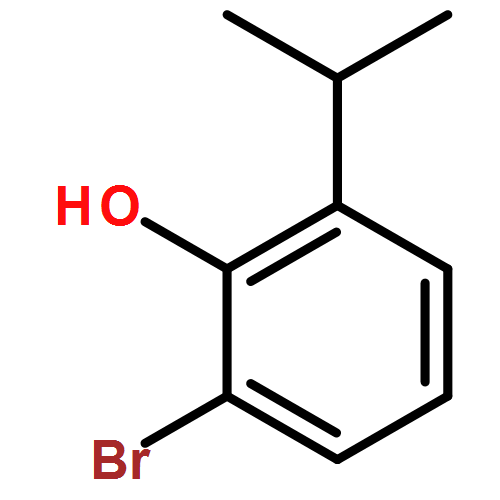
![1H,3H-Pyrrolo[1,2-c][1,3,2]oxazaborole,1-butyltetrahydro-3,3-diphenyl-, (S)-](http://img.cochemist.com/ccimg/129200/129145-37-5.png)
![1H,3H-Pyrrolo[1,2-c][1,3,2]oxazaborole,1-butyltetrahydro-3,3-diphenyl-, (S)-](http://img.cochemist.com/ccimg/129200/129145-37-5_b.png)


















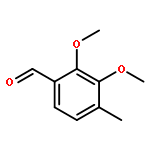







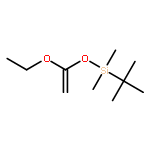
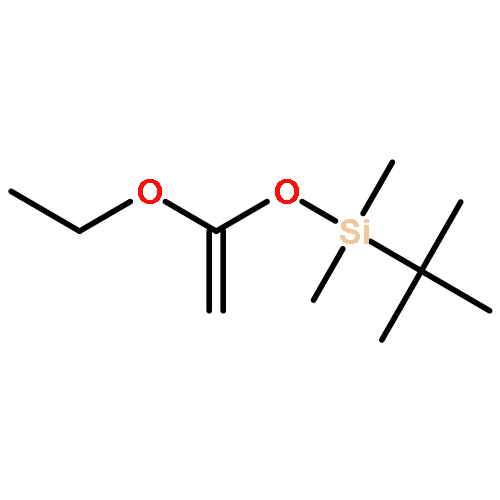
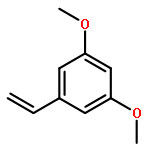
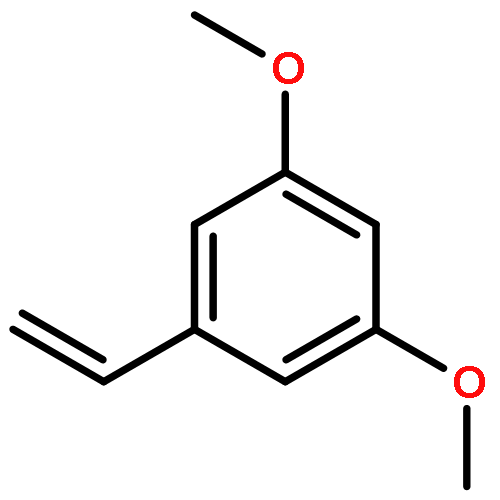


![Benzene, [(1R)-1-methyl-2-propenyl]-](http://img.cochemist.com/ccimg/36700/36617-88-6.png)
![Benzene, [(1R)-1-methyl-2-propenyl]-](http://img.cochemist.com/ccimg/36700/36617-88-6_b.png)




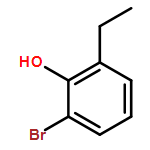
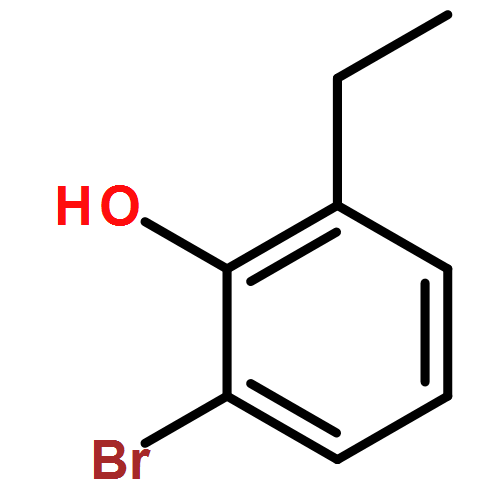


![N-(Trimethylsilyl)-2-[(trimethylsilyl)oxy]pyrimidin-4-amine](http://img.cochemist.com/ccimg/18100/18037-10-0.png)
![N-(Trimethylsilyl)-2-[(trimethylsilyl)oxy]pyrimidin-4-amine](http://img.cochemist.com/ccimg/18100/18037-10-0_b.png)

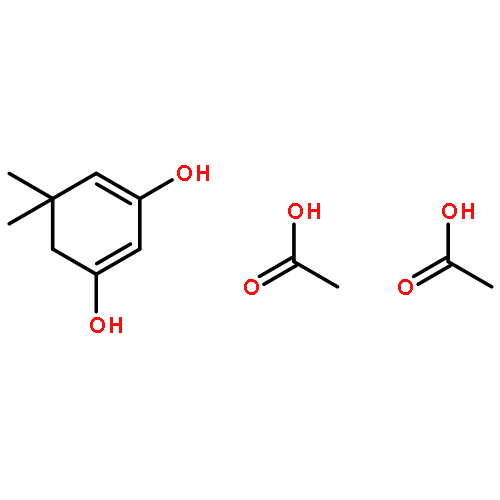

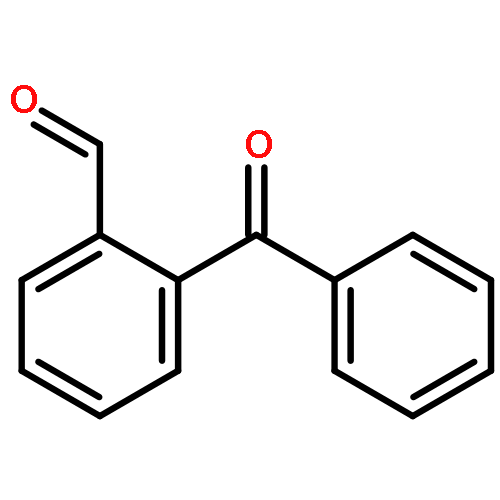










![2-Anthracenecarboxylic acid,1-[2-(5-carboxy-4-hydroxy-1-oxopentyl)-6-hydroxybenzoyl]-9,10-dihydro-3,8-dihydroxy-9,10-dioxo-](http://img.cochemist.com/ccimg/253000/252961-22-1.png)
![2-Anthracenecarboxylic acid,1-[2-(5-carboxy-4-hydroxy-1-oxopentyl)-6-hydroxybenzoyl]-9,10-dihydro-3,8-dihydroxy-9,10-dioxo-](http://img.cochemist.com/ccimg/253000/252961-22-1_b.png)






![BENZO[A]HEPTALEN-9(5H)-ONE,7-AMINO-6,7-DIHYDRO-1,2,3,10-TETRAMETHOXY-, (7S)-](http://img.cochemist.com/ccimg/3500/3476-50-4.png)
![BENZO[A]HEPTALEN-9(5H)-ONE,7-AMINO-6,7-DIHYDRO-1,2,3,10-TETRAMETHOXY-, (7S)-](http://img.cochemist.com/ccimg/3500/3476-50-4_b.png)





