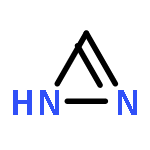
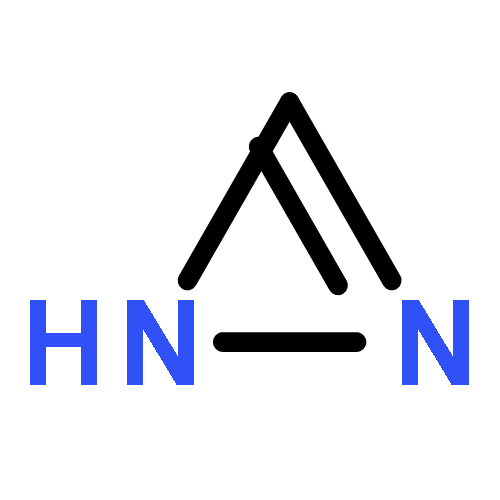
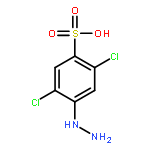
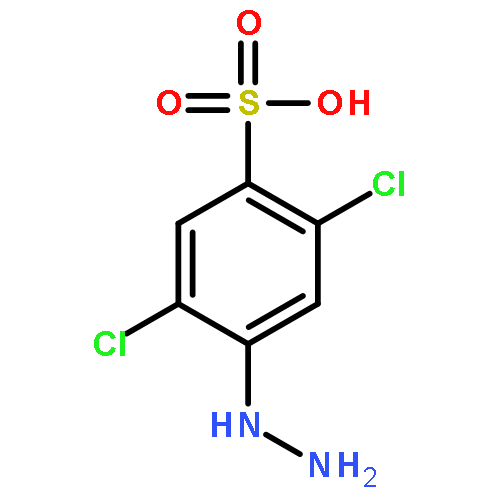
Co-reporter:David Schaller, Stefanie Hagenow, Gina Alpert, Alexandra Naß, Robert Schulz, Marcel Bermudez, Holger Stark, and Gerhard Wolber
ACS Medicinal Chemistry Letters June 8, 2017 Volume 8(Issue 6) pp:648-648
Publication Date(Web):May 4, 2017
DOI:10.1021/acsmedchemlett.7b00118
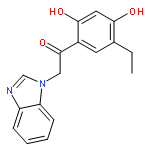
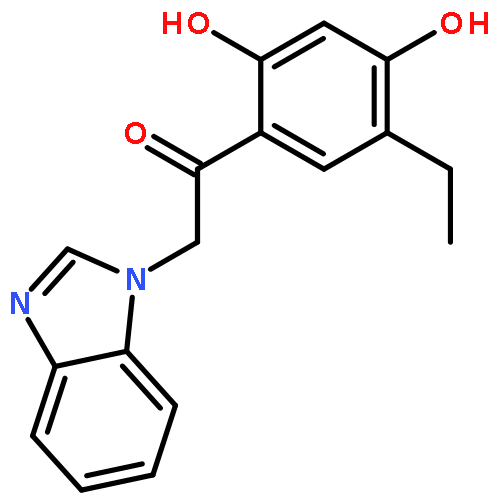
In this study, we report a ligand-centric data mining approach that guided the identification of suitable target profiles for treating obesity. The newly developed method is based on identifying target pairs for synergistic positive effects and also encompasses the exclusion of compounds showing a detrimental effect on obesity treatment (off-targets). Ligands with known activity against obesity-relevant targets were compared using fingerprint representations. Similar compounds with activities to different targets were evaluated for the mechanism of action since activation or deactivation of drug targets determines the pharmacological effect. In vitro validation of the modeling results revealed that three known modulators of melanin-concentrating hormone receptor 1 (MCHR1) show a previously unknown submicromolar affinity to the histamine H3 receptor (H3R). This synergistic activity may present a novel therapeutic option against obesity.Keywords: fingerprints; histamine H3 receptor; melanin-concentrating hormone receptor 1; Multitarget drugs; obesity;
Co-reporter:Marcel Bermudez, Andreas Bock, Fabian Krebs, Ulrike Holzgrabe, Klaus Mohr, Martin J. Lohse, and Gerhard Wolber
ACS Chemical Biology July 21, 2017 Volume 12(Issue 7) pp:1743-1743
Publication Date(Web):June 6, 2017
DOI:10.1021/acschembio.7b00275
G protein-coupled receptors transmit extracellular signals across cell membranes via different G protein classes and β-arrestins. Some pathways may be therapeutically beneficial, whereas others may be detrimental under certain pathophysiological conditions. For many GPCRs, biased agonists are available, which preferentially signal through one pathway or a subset of pathways, and harnessing biased agonism could be a potential novel therapeutic strategy. However, the incomplete mechanistic understanding of biased agonism hampers rational design of biased ligands. Using the muscarinic M2 receptor as a model system, we have analyzed the relationship between ligand-dependent conformational changes as revealed in all-atom MD simulations and the activation of specific G proteins. We find that the extent of closure of the extracellular, allosteric binding site interferes with the activation of certain G proteins. Our data allow the rational design of Gi-biased agonists at the M2 receptor and delineate a simple principle which may be translated to other GPRCs.
Co-reporter:Jamil Al-Asri, Erika Fazekas, Gábor Lehoczki, Andrej Perdih, Cornelia Görick, Matthias F. Melzig, Gyöngyi Gyémánt, Gerhard Wolber, Jérémie Mortier
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 20) pp:6725-6732
Publication Date(Web):15 October 2015
DOI:10.1016/j.bmc.2015.09.007
Starch catabolism leading to high glucose level in blood is highly problematic in chronic metabolic diseases, such as type II diabetes and obesity. α-Amylase catalyzes the hydrolysis of starch, increasing blood sugar concentration. Its inhibition represents a promising therapeutic approach to control hyperglycaemia. However, only few drug-like molecule inhibitors without sugar moieties have been discovered so far, and little information on the enzymatic mechanism is available. This work aims at the discovery of novel small α-amylase binders using a systematic in silico methodology. 3D-pharmacophore-based high throughput virtual screening of small compounds libraries was performed to identify compounds with high α-amylase affinity. Twenty-seven compounds were selected and biologically tested, revealing IC50 values in the micromolar range and ligand efficiency higher than the one of the bound form of acarbose, which is used as a reference for α-amylase inhibition.
Co-reporter:Christin Rakers, Sverre-Morten Schwerdtfeger, Jérémie Mortier, Susanne Duwe, Thorsten Wolff, Gerhard Wolber, Matthias F. Melzig
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 17) pp:4312-4317
Publication Date(Web):1 September 2014
DOI:10.1016/j.bmcl.2014.07.010
The constant risk of emerging new influenza virus strains that are resistant to established inhibitors like oseltamivir leaves influenza neuraminidase (NA) a prominent target for drug design. The inhibitory activity of several flavonoid derivatives was experimentally tested in comparison to oseltamivir for the NA expressed by the seasonal influenza virus strains A/California/7/09 (A(H1N1)pdm09), A/Perth/16/09 (A(H3N2)), and B/Brisbane/60/08. IC50 values of polyphenols confirmed moderate inhibition in the μM range. Structurally, the amount and site of glycosylation of tested flavonoids have no significant influence on their inhibitory potency. In a pharmacophore-based docking approach the structure–activity relationship was evaluated. Molecular dynamics simulations revealed highly flexible parts of the enzyme and the contribution of salt bridges to the structural stability of NA. The findings of this study elucidate the impact of flavonoids on viral neuraminidase activity and the analysis of their modes of action provide valuable information about the mechanism of NA inhibition.
Co-reporter:Manuela S. Murgueitio;Dr. Philipp Henneke;Dr. Hartmut Glossmann;Dr. Sra Santos-Sierra;Dr. Gerhard Wolber
ChemMedChem 2014 Volume 9( Issue 4) pp:813-822
Publication Date(Web):
DOI:10.1002/cmdc.201300445
Abstract
Toll-like receptors (TLRs) are critical signaling molecules with roles in various severe clinical conditions such as sepsis and rheumatoid arthritis, and have therefore been advocated as promising drug targets for the treatment of these diseases. The aim of this study was to discover small-molecule antagonists of TLR2 by computer-aided drug design. This goal poses several challenges due to the lack of available data on TLR2 modulators. To overcome these hurdles we developed a combined structure- and ligand-based virtual screening approach. First, we calculated molecular interaction fields of the TLR2 binding site to derive a structure-based 3D pharmacophore, which was then used for virtual screening. We then performed a two-step shape- and feature-based similarity search using known TLR2 ligands as query structures. A selection of virtual screening hits was biologically tested in a cell-based assay for TLR2 signaling inhibition, leading to the identification of several compounds with antagonistic activity (IC50 values) in the low-micromolar range.
Co-reporter:Jérémie Mortier, Christin Rakers, Marcel Bermudez, Manuela S. Murgueitio, ... Gerhard Wolber
Drug Discovery Today (June 2015) Volume 20(Issue 6) pp:686-702
Publication Date(Web):1 June 2015
DOI:10.1016/j.drudis.2015.01.003
•Which molecular dynamics (MD) techniques are available for drug design?•How is MD used to investigate ligand–macromolecule complexes?•How are MD studies applied to human and non-human therapeutic targets?•What are the latest advances in the field of MD?Among all tools available to design new drugs, molecular dynamics (MD) simulations have become an essential technique. Initially developed to investigate molecular models with a limited number of atoms, computers now enable investigations of large macromolecular systems with a simulation time reaching the microsecond range. The reviewed articles cover four years of research to give an overview on the actual impact of MD on the current medicinal chemistry landscape with a particular emphasis on studies of ligand–protein interactions. With a special focus on studies combining computational approaches with data gained from other techniques, this review shows how deeply embedded MD simulations are in drug design strategies and articulates what the future of this technique could be.Download high-res image (214KB)Download full-size image
Co-reporter:Manuela S. Murgueitio, Christin Rakers, Anne Frank, Gerhard Wolber
Trends in Pharmacological Sciences (February 2017) Volume 38(Issue 2) pp:155-168
Publication Date(Web):1 February 2017
DOI:10.1016/j.tips.2016.10.007
As essential proteins of the innate immune system, Toll-like receptors (TLRs) are involved in a plethora of physiological pathologies and their modulation is an ongoing quest in the field of drug discovery. Although TLRs recognize an unusually broad range of different molecular patterns, only a few small-molecule TLR modulators have been reported to date. Recent advances in crystallography and in silico techniques provide promising opportunities for TLR investigations and drug design. Here, three application areas for computational approaches are considered: (i) exploration of TLR structure and activation; (ii) understanding TLR modulation; and (iii) TLR drug discovery. By providing an overview on state-of-the-art computational methods, we highlight the value of molecular modeling in mechanistically understanding TLR function and guiding drug design.
Co-reporter:Manuela S. Murgueitio, Marcel Bermudez, Jérémie Mortier, Gerhard Wolber
Drug Discovery Today: Technologies (Autumn 2012) Volume 9(Issue 3) pp:e219-e225
Publication Date(Web):1 September 2012
DOI:10.1016/j.ddtec.2012.07.009
Despite the considerable advances in medical and pharmaceutical research during the past years, diseases caused by viruses have remained a major burden to public health. Virtual in silico screening has repeatedly proven to be useful to meet the special challenges of antiviral drug discovery. Large virtual compound libraries are filtered by different computational screening methods such as docking, ligand-based similarity searches or pharmacophore-based screening, reducing the number of candidate molecules to a smaller set of promising candidates that are then tested biologically. This rational approach makes the drug discovery process more goal-oriented and saves resources in terms of time and money. In this review we discuss how different virtual screening techniques can be applied to antiviral drug discovery, present recent success stories in this field and finally address the main differences between the methods.

![NSC 23766;N6-[2-[[4-(DIETHYLAMINO)-1-METHYLBUTYL]AMINO]-6-METHYL-4-PYRIMIDINYL]-2-METHYL-4,6-QUINOLINEDIAMINETRIHYDROCHLORIDE](http://img.cochemist.com/ccimg/733800/733767-34-5.png)
![NSC 23766;N6-[2-[[4-(DIETHYLAMINO)-1-METHYLBUTYL]AMINO]-6-METHYL-4-PYRIMIDINYL]-2-METHYL-4,6-QUINOLINEDIAMINETRIHYDROCHLORIDE](http://img.cochemist.com/ccimg/733800/733767-34-5_b.png)
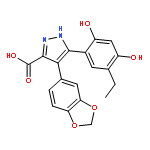
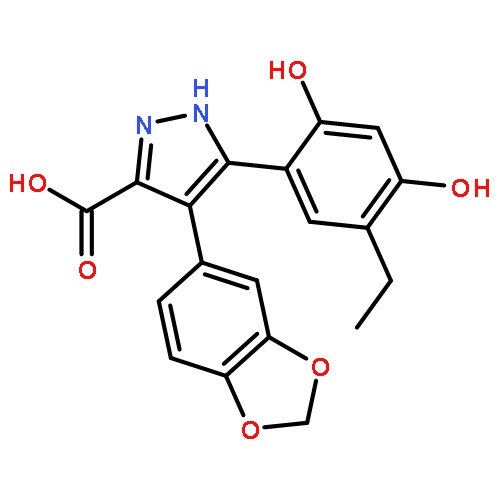
![5-Benzoxazolol, 7-ethenyl-2-[3-fluoro-4-(sulfooxy)phenyl]-](http://img.cochemist.com/ccimg/877200/877172-75-3.png)
![5-Benzoxazolol, 7-ethenyl-2-[3-fluoro-4-(sulfooxy)phenyl]-](http://img.cochemist.com/ccimg/877200/877172-75-3_b.png)
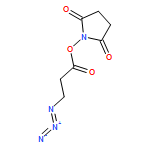
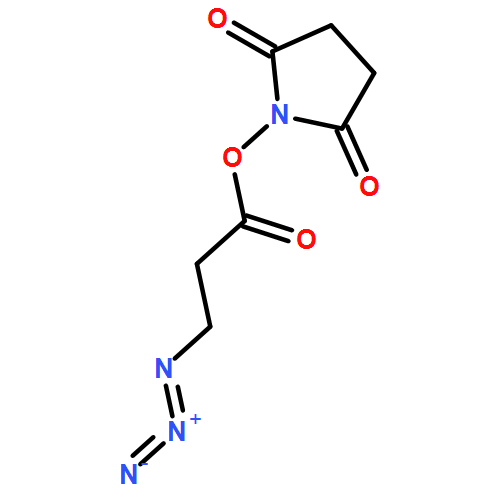




![Phenol,4-[[4-(4-chlorophenyl)-2-thiazolyl]amino]-](http://img.cochemist.com/ccimg/312700/312636-16-1.png)
![Phenol,4-[[4-(4-chlorophenyl)-2-thiazolyl]amino]-](http://img.cochemist.com/ccimg/312700/312636-16-1_b.png)


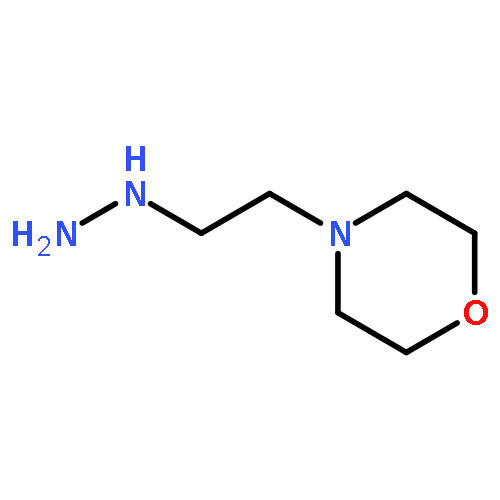
![(2,3-DIHYDRO-BENZO[1,4]DIOXIN-6-YL)-HYDRAZINE](http://img.cochemist.com/ccimg/299200/299165-45-0.png)
![(2,3-DIHYDRO-BENZO[1,4]DIOXIN-6-YL)-HYDRAZINE](http://img.cochemist.com/ccimg/299200/299165-45-0_b.png)


![6-Hydrazinyl-1,3,4-trimethyl-1H-pyrazolo[3,4-b]pyridine](http://img.cochemist.com/ccimg/175300/175202-00-3.png)
![6-Hydrazinyl-1,3,4-trimethyl-1H-pyrazolo[3,4-b]pyridine](http://img.cochemist.com/ccimg/175300/175202-00-3_b.png)


![Benzamide,N-[[[4-[(5-bromo-2-pyrimidinyl)oxy]-3-methylphenyl]amino]carbonyl]-2-(dimethylamino)-](http://img.cochemist.com/ccimg/134800/134742-19-1.png)
![Benzamide,N-[[[4-[(5-bromo-2-pyrimidinyl)oxy]-3-methylphenyl]amino]carbonyl]-2-(dimethylamino)-](http://img.cochemist.com/ccimg/134800/134742-19-1_b.png)


![L-Lysine,S-[2,3-bis[(1-oxohexadecyl)oxy]propyl]-N-(1-oxohexadecyl)-L-cysteinyl-L-seryl-L-lysyl-L-lysyl-L-lysyl-](http://img.cochemist.com/ccimg/112300/112208-00-1.png)
![L-Lysine,S-[2,3-bis[(1-oxohexadecyl)oxy]propyl]-N-(1-oxohexadecyl)-L-cysteinyl-L-seryl-L-lysyl-L-lysyl-L-lysyl-](http://img.cochemist.com/ccimg/112300/112208-00-1_b.png)
![Benzo[d][1,3]dioxol-5-ylhydrazine](http://img.cochemist.com/ccimg/62700/62646-09-7.png)
![Benzo[d][1,3]dioxol-5-ylhydrazine](http://img.cochemist.com/ccimg/62700/62646-09-7_b.png)
![(2R,3S,4S,5S,6R)-6-[(2S,3S,4R,5S,6S)-3-acetamido-2-[(2R,3S,4R,5S,6R)-6-[(3S,4R,5S,6S)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)tetrahydropyran-4-yl]oxy-2-carboxy-4,5-dihydroxy-tetrahydropyran-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)tetrahydropyran-4-yl]oxy-](http://img.cochemist.com/ccimg/57300/57282-61-8.png)
![(2R,3S,4S,5S,6R)-6-[(2S,3S,4R,5S,6S)-3-acetamido-2-[(2R,3S,4R,5S,6R)-6-[(3S,4R,5S,6S)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)tetrahydropyran-4-yl]oxy-2-carboxy-4,5-dihydroxy-tetrahydropyran-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)tetrahydropyran-4-yl]oxy-](http://img.cochemist.com/ccimg/57300/57282-61-8_b.png)
![Tetrazolo[1,5-b]pyridazin-6(5H)-one, hydrazone](http://img.cochemist.com/ccimg/52500/52476-90-1.png)
![Tetrazolo[1,5-b]pyridazin-6(5H)-one, hydrazone](http://img.cochemist.com/ccimg/52500/52476-90-1_b.png)




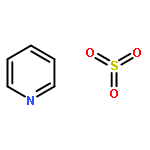
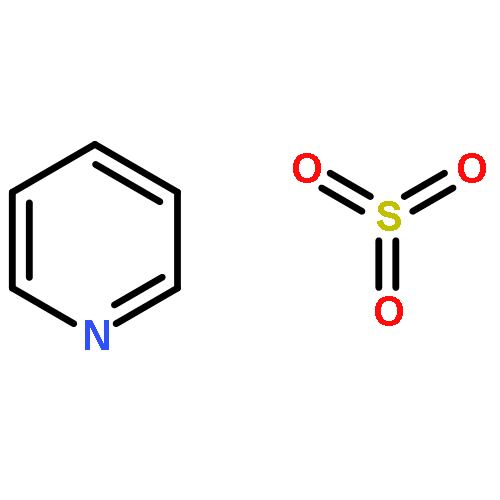




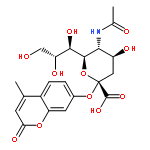
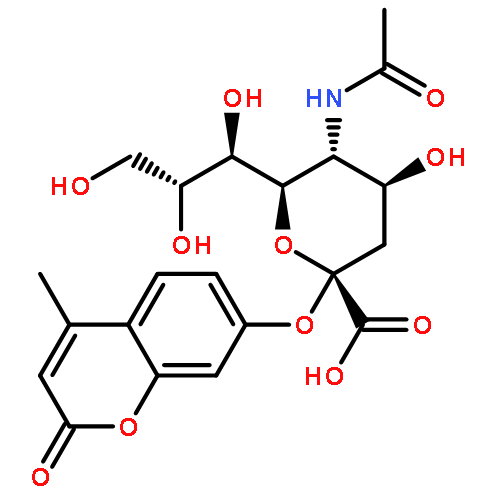
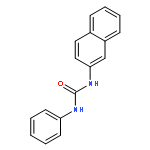
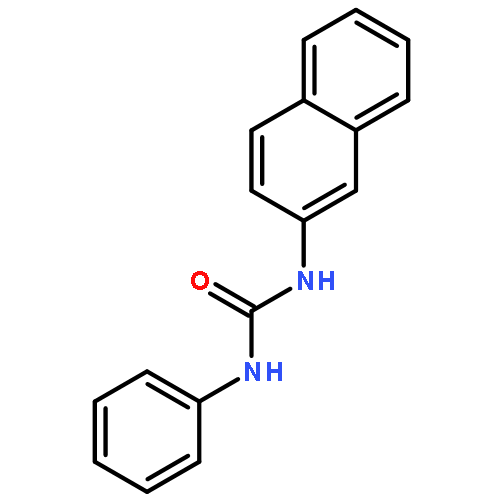

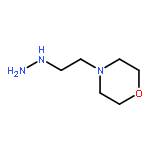
![L-Glutamic acid,N-[4-[[(2-amino-3,4,7,8-tetrahydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-](http://img.cochemist.com/ccimg/4100/4033-27-6.png)
![L-Glutamic acid,N-[4-[[(2-amino-3,4,7,8-tetrahydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-](http://img.cochemist.com/ccimg/4100/4033-27-6_b.png)

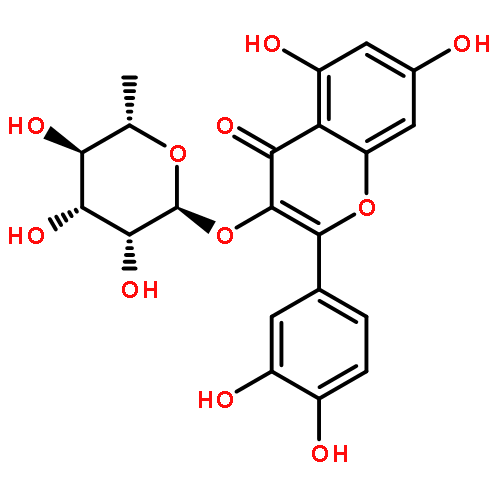
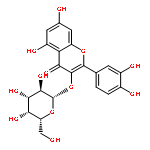
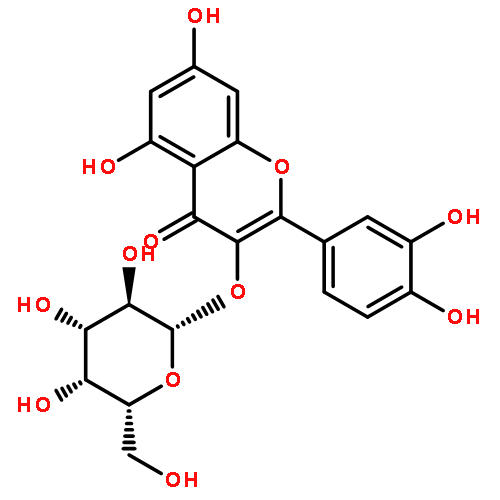
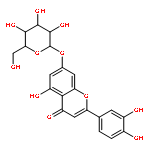
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9.png)
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9_b.png)