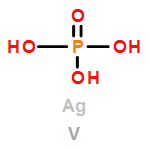
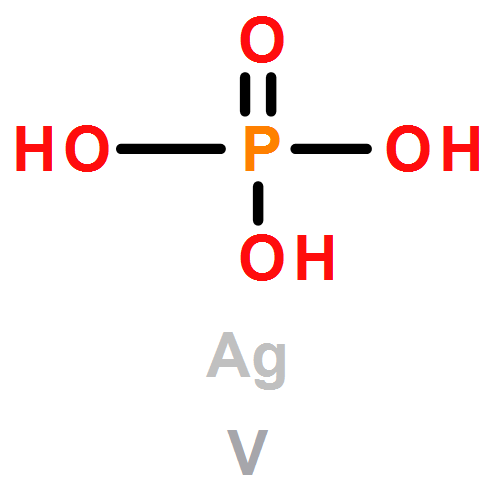
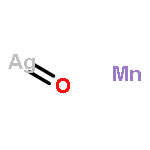
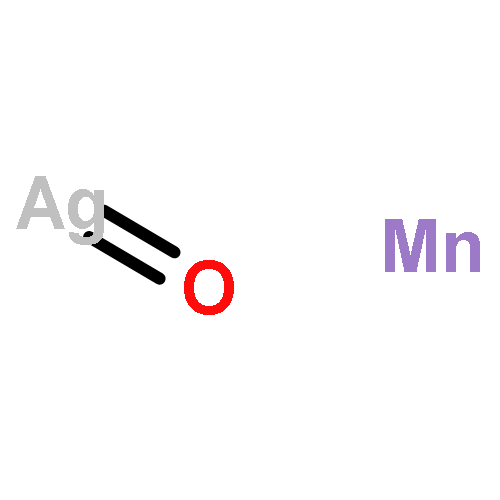
Co-reporter:David C. Bock;Christopher J. Pelliccione;Wei Zhang;Janis Timoshenko;K. W. Knehr;Alan C. West;Feng Wang;Yan Li;Anatoly I. Frenkel;Esther S. Takeuchi;Amy C. Marschilok
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 31) pp:20867-20880
Publication Date(Web):2017/08/09
DOI:10.1039/C7CP03312E
The iron oxide magnetite, Fe3O4, is a promising conversion type lithium ion battery anode material due to its high natural abundance, low cost and high theoretical capacity. While the close packing of ions in the inverse spinel structure of Fe3O4 enables high energy density, it also limits the kinetics of lithium ion diffusion in the material. Nanosizing of Fe3O4 to reduce the diffusion path length is an effective strategy for overcoming this issue and results in improved rate capability. However, the impact of nanosizing on the multiple structural transformations that occur during the electrochemical (de)lithiation reaction in Fe3O4 is poorly understood. In this study, the influence of crystallite size on the lithiation-conversion mechanisms in Fe3O4 is investigated using complementary X-ray techniques along with transmission electron microscopy (TEM) and continuum level simulations on electrodes of two different Fe3O4 crystallite sizes. In situ X-ray diffraction (XRD) measurements were utilized to track the changes to the crystalline phases during (de)lithiation. X-ray absorption spectroscopy (XAS) measurements at multiple points during the (de)lithiation processes provided local electronic and atomic structural information. Tracking the crystalline and nanocrystalline phases during the first (de)lithiation provides experimental evidence that (1) the lithiation mechanism is non-uniform and dependent on crystallite size, where increased Li+ diffusion length in larger crystals results in conversion to Fe0 metal while insertion of Li+ into spinel-Fe3O4 is still occurring, and (2) the disorder and size of the Fe metal domains formed when either material is fully lithiated impacts the homogeneity of the FeO phase formed during the subsequent delithiation.
Co-reporter:Qing Zhang;Andrea M. Bruck;David C. Bock;Jing Li;Varun Sarbada;Robert Hull;Eric A. Stach;Esther S. Takeuchi;Amy C. Marschilok
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 21) pp:14160-14169
Publication Date(Web):2017/05/31
DOI:10.1039/C7CP02239E
Li1+nV3O8 (n = 0–0.2) has been extensively investigated as a cathode material for Li ion batteries because of its superior electrochemical properties including high specific energy and good rate capability. In this paper, a synchrotron based energy dispersive X-ray diffraction (EDXRD) technique was employed to profile the phase transitions and the spatial phase distribution of a Li1.1V3O8 electrode during electrochemical (de)lithiation in situ and operando. As annealing temperature during the preparation of the Li1.1V3O8 material has a strong influence on the morphology and crystallinity, and consequently influences the electrochemical outcomes of the material, Li1.1V3O8 materials prepared at two different temperatures, 500 and 300 °C (LVO500 and LVO300), were employed in this study. The EDXRD spectra of LVO500 and LVO300 cells pre-discharged at C/18, C/40 and C/150 were recorded in situ, and phase localization and relative intensity of the peaks were compared. For cells discharged at the C/18 rate, although α and β phases were distributed uniformly within the LVO500 electrode, they were localized on two sides of the LVO300 electrode. Discharging rates of C/40 and C/150 led to homogeneous β phase formation in both LVO500 and LVO300 electrodes. Furthermore, the phase distribution as a function of position and (de)lithiation extent was mapped operando as the LVO500 cell was (de)lithiated. The operando data indicate that (1) the lithiation reaction initiated from the side of the electrode facing the Li anode and proceeded towards the side facing the steel can, (2) during discharge the phase transformation from a Li-poor to a Li-rich α phase and the formation of a β phase can proceed simultaneously in the electrode after the first formation of a β phase, and (3) the structural evolution occurring during charging is not the reverse of that during discharge and takes place homogenously throughout the electrode.
Co-reporter:Jessica L. Durham;Alexander B. Brady;Christina A. Cama;David C. Bock;Christopher J. Pelliccione;Qing Zhang;Mingyuan Ge;Yue Ru Li;Yiman Zhang;Hanfei Yan;Xiaojing Huang;Yong Chu;Esther S. Takeuchi;Amy C. Marschilok
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 33) pp:22329-22343
Publication Date(Web):2017/08/23
DOI:10.1039/C7CP04012A
The structure of pristine AgFeO2 and phase makeup of Ag0.2FeO1.6 (a one-pot composite comprised of nanocrystalline stoichiometric AgFeO2 and amorphous γ-Fe2O3 phases) was investigated using synchrotron X-ray diffraction. A new stacking-fault model was proposed for AgFeO2 powder synthesized using the co-precipitation method. The lithiation/de-lithiation mechanisms of silver ferrite, AgFeO2 and Ag0.2FeO1.6 were investigated using ex situ, in situ, and operando characterization techniques. An amorphous γ-Fe2O3 component in the Ag0.2FeO1.6 sample is quantified. Operando XRD of electrochemically reduced AgFeO2 and Ag0.2FeO1.6 composites demonstrated differences in the structural evolution of the nanocrystalline AgFeO2 component. As complimentary techniques to XRD, ex situ X-ray Absorption Spectroscopy (XAS) provided insight into the short-range structure of the (de)lithiated nanocrystalline electrodes, and a novel in situ high energy X-ray fluorescence nanoprobe (HXN) mapping measurement was applied to spatially resolve the progression of discharge. Based on the results, a redox mechanism is proposed where the full reduction of Ag+ to Ag0 and partial reduction of Fe3+ to Fe2+ occur on reduction to 1.0 V, resulting in a Li1+yFeIIIFeIIyO2 phase. The Li1+yFeIIIFeIIyO2 phase can then reversibly cycle between Fe3+ and Fe2+ oxidation states, permitting good capacity retention over 50 cycles. In the Ag0.2FeO1.6 composite, a substantial amorphous γ-Fe2O3 component is observed which discharges to rock salt LiFe2O3 and Fe0 metal phase in the 3.5–1.0 V voltage range (in parallel with the AgFeO2 mechanism), and reversibly reoxidizes to a nanocrystalline iron oxide phase.
Co-reporter:Jiefu Yin;Alexander B. Brady;Esther S. Takeuchi;Amy C. Marschilok
Chemical Communications 2017 vol. 53(Issue 26) pp:3665-3668
Publication Date(Web):2017/03/28
DOI:10.1039/C7CC00265C
MgMn2O4 nanoparticles with crystallite sizes of 11 (MMO-1) and 31 nm (MMO-2) were synthesized and their magnesium-ion battery-relevant electrochemistry was investigated. MMO-1 delivered an initial capacity of 220 mA h g−1 (678 mW h g−1). Electrolyte water content had a profound effect on cycle retention.
Co-reporter:Altug S. Poyraz;Jianping Huang;Christopher J. Pelliccione;Xiao Tong;Shaobo Cheng;Lijun Wu;Yimei Zhu;Amy C. Marschilok;Esther S. Takeuchi
Journal of Materials Chemistry A 2017 vol. 5(Issue 32) pp:16914-16928
Publication Date(Web):2017/08/15
DOI:10.1039/C7TA03476H
The role of tunnel cations in the electrochemistry of α-MnO2 materials has been long discussed and demands investigation as the electrochemistry of α-MnO2 materials is strongly dependent on the material specific properties (i.e. morphology, surface area, crystallite size, and chemical composition). Here, we systematically synthesized a series of α-MnO2 samples with differing K+ content but similar physicochemical and morphological properties allowing direct investigation of the role of tunnel cation (K+) on the lithium ion electrochemistry of α-MnO2 cathodes. The nanofibrous α-MnO2 materials have a chemical composition of KxMn8O16·yH2O, where 0 ≤ x ≤ 0.75 and 0.53 ≤ y ≤ 0.81. The α-MnO2 materials have similar morphology, crystallite size (17–19 nm), surface area (66–76 m2 g−1), and tunnel water content (0.53–0.81). The electrochemistry of the α-MnO2 materials was evaluated using cyclic voltammetry, galvanostatic cycling, and galvanostatic intermittent titration type tests. The α-MnO2 materials with 0 to 0.32 K+ content showed discharge curves with higher voltage, higher specific energies, and improved capacity retention compared to the 0.75 K+ containing α-MnO2 material. Fewer structural distortions were observed in lithiated samples with lower K+ content through modelling of X-ray absorption spectroscopy data indicating improved structural stability of those samples which positively impacted the electrochemistry.
Co-reporter:Altug S. Poyraz;Jianping Huang;Christopher J. Pelliccione;Xiao Tong;Shaobo Cheng;Lijun Wu;Yimei Zhu;Amy C. Marschilok;Esther S. Takeuchi
Journal of Materials Chemistry A 2017 vol. 5(Issue 32) pp:16914-16928
Publication Date(Web):2017/08/15
DOI:10.1039/C7TA03476H
The role of tunnel cations in the electrochemistry of α-MnO2 materials has been long discussed and demands investigation as the electrochemistry of α-MnO2 materials is strongly dependent on the material specific properties (i.e. morphology, surface area, crystallite size, and chemical composition). Here, we systematically synthesized a series of α-MnO2 samples with differing K+ content but similar physicochemical and morphological properties allowing direct investigation of the role of tunnel cation (K+) on the lithium ion electrochemistry of α-MnO2 cathodes. The nanofibrous α-MnO2 materials have a chemical composition of KxMn8O16·yH2O, where 0 ≤ x ≤ 0.75 and 0.53 ≤ y ≤ 0.81. The α-MnO2 materials have similar morphology, crystallite size (17–19 nm), surface area (66–76 m2 g−1), and tunnel water content (0.53–0.81). The electrochemistry of the α-MnO2 materials was evaluated using cyclic voltammetry, galvanostatic cycling, and galvanostatic intermittent titration type tests. The α-MnO2 materials with 0 to 0.32 K+ content showed discharge curves with higher voltage, higher specific energies, and improved capacity retention compared to the 0.75 K+ containing α-MnO2 material. Fewer structural distortions were observed in lithiated samples with lower K+ content through modelling of X-ray absorption spectroscopy data indicating improved structural stability of those samples which positively impacted the electrochemistry.
Co-reporter:Jianping Huang, Altug S. Poyraz, Seung-Yong Lee, Lijun Wu, Yimei Zhu, Amy C. Marschilok, Kenneth J. Takeuchi, and Esther S. Takeuchi
ACS Applied Materials & Interfaces 2017 Volume 9(Issue 5) pp:
Publication Date(Web):September 1, 2016
DOI:10.1021/acsami.6b08549
Manganese oxides are considered attractive cathode materials for rechargeable batteries due to the high abundance and environmental friendliness of manganese. In particular, cryptomelane and hollandite are desirable due to their ability to host cations within their octahedral molecular sieve (OMS-2) α-MnO2 structure. In this work, we investigate silver containing α-MnO2 structured materials (AgxMn8O16, x = 1.22, L-Ag-OMS-2 or 1.66, H-Ag-OMS-2) as host materials for Li ion and Na ion insertion/deinsertion. The results indicate a significant difference in the lithiation versus sodiation process of the OMS-2 materials. Initial reduction of Ag1.22Mn8O16 to 1.0 V delivered ∼370 mAh/g. Cycling of Ag1.22Mn8O16 between voltage ranges of 3.8–1.7 V and 3.8–1.3 V in a Na battery delivered initial capacities of 113 and 247 mAh/g, respectively. In contrast, Ag1.66Mn8O16 delivered only 15 mAh/g, ∼ 0.5 electron equivalents, to 1.7 and 1.3 V. Study of the system by electrochemical impedance spectroscopy (EIS) showed a significant decrease in charge transfer resistance from 2029 Ω to 594 Ω after 1.5 electron equivalents per Ag1.22Mn8O16 formula unit of Na ion insertion. In contrast, both Ag1.22Mn8O16 and Ag1.66Mn8O16 exhibited gradual impedance increases during lithiation. The formation of silver metal could be detected only in the sodiated material by X-ray diffraction (XRD). Thus, the impedance of Ag-OMS-2 decreases upon sodiation coincident with the formation of silver metal during the discharge process, consistent with the more favorable formation of silver metal during the sodiation process relative to the lithation process.Keywords: alpha manganese oxide; electrochemistry; silver hollandite; sodium battery; X-ray diffraction;
Co-reporter:Jessica L. Durham, Altug S. Poyraz, Esther S. Takeuchi, Amy C. Marschilok, and Kenneth J. Takeuchi
Accounts of Chemical Research 2016 Volume 49(Issue 9) pp:1864
Publication Date(Web):August 26, 2016
DOI:10.1021/acs.accounts.6b00318
Electric energy storage devices such as batteries are complex systems comprised of a variety of materials with each playing separate yet interactive roles, complicated by length scale interactions occurring from the molecular to the mesoscale. Thus, addressing specific battery issues such as functional capacity requires a comprehensive perspective initiating with atomic level concepts. For example, the electroactive materials which contribute to the functional capacity in a battery comprise approximately 30% or less of the total device mass. Thus, the design and implementation of multifunctional materials can conceptually reduce or eliminate the contribution of passive materials to the size and mass of the final system. Material multifunctionality can be achieved through appropriate material design on the atomic level resulting in bimetallic electroactive materials where one metal cation forms mesoscale conductive networks upon discharge while the other metal cations can contribute to atomic level structure and net functional secondary capacity, a device level issue. Specifically, this Account provides insight into the multimechanism electrochemical redox processes of bimetallic cathode materials based on transition metal oxides (MM′O) or phosphorus oxides (MM′PO) where M = Ag and M′ = V or Fe. One discharge process can be described as reduction-displacement where Ag+ is reduced to Ag0 and displaced from the parent structure. This reduction-displacement reaction in silver-containing bimetallic electrodes allows for the in situ formation of a conductive network, enhancing the electrochemical performance of the electrode and reducing or eliminating the need for conductive additives. A second discharge process occurs through the reduction of the second transition metal, V or Fe, where the oxidation state of the metal center is reduced and lithium cations are inserted into the structure. As both metal centers contribute to the functional capacity, determining the kinetically and thermodynamically preferred reduction processes at various states of discharge is critical to elucidating the mechanism. Specific advanced in situ and ex situ characterization techniques are conducive to gaining insight regarding the electrochemical behavior of these multifunctional materials over multiple length scales. At the material level, optical microscopy, scanning electron microscopy, and local conductivity measurement via a nanoprobe can track the discharge mechanism of an isolated single particle. At the mesoscale electrode level, in situ data from synchrotron based energy dispersive X-ray diffraction (EDXRD) within fully intact steel batteries can be used to spatially map the distribution of silver metal generated through reduction displacement as a function of discharge depth and discharge rate. As illustrated here, appropriate design of materials with multiple electrochemically active metal centers and properties tuned through strategically conceptualized materials synthesis may provide a path toward the next generation of high energy content electroactive materials and systems. Full understanding of the multiple electrochemical mechanisms can be achieved only by utilizing advanced characterization tools over multiple length scales.
Co-reporter:Wei Zhang;David C. Bock;Christopher J. Pelliccione;Yan Li;Lijun Wu;Yimei Zhu;Amy. C. Marschilok;Esther S. Takeuchi;Feng Wang
Advanced Energy Materials 2016 Volume 6( Issue 10) pp:
Publication Date(Web):
DOI:10.1002/aenm.201502471
Metal oxides, such as Fe3O4, hold promise for future battery applications due to their abundance, low cost, and opportunity for high lithium storage capacity. In order to better understand the mechanisms of multiple-electron transfer reactions leading to high capacity in Fe3O4, a comprehensive investigation on local ionic transport and ordering is made by probing site occupancies of anions (O2−) and cations (Li+, Fe3+/Fe2+) using multiple synchrotron X-ray and electron-beam techniques, in combination with ab-initio calculations. Results from this study provide the first experimental evidence that the cubic-close-packed (ccp) O-anion array in Fe3O4 is sustained throughout the lithiation and delithiation processes, thereby enabling multiple lithium intercalation and conversion reactions. Cation displacement/reordering occurs within the ccp O-anion framework, which leads to a series of phase transformations, starting from the inverse spinel phase and turning into intermediate rock-salt-like phases (LixFe3O4; 0 < x < 2), then into a cation-segregated phase (Li2O⋅FeO), and finally converting into metallic Fe and Li2O. Subsequent delithiation and lithiation processes involve interconversion between metallic Fe and FeO-like phases. These results may offer new insights into the structure-determined ionic transport and electrochemical reactions in metal oxides, and those of other compounds sharing a ccp anion framework, reminiscent of magnetite.
Co-reporter:Jianping Huang, Amy C. Marschilok, Esther S. Takeuchi, and Kenneth J. Takeuchi
Chemistry of Materials 2016 Volume 28(Issue 7) pp:2191
Publication Date(Web):March 7, 2016
DOI:10.1021/acs.chemmater.6b00124
Silver vanadium phosphorus oxide, Ag2VO2PO4, is a promising cathode material for Li batteries due in part to its large capacity and high current capability. Herein, a new synthesis of Ag2VO2PO4 based on microwave heating is presented, where the reaction time is reduced by approximately 100× relative to other reported methods, and the crystallite size is controlled via synthesis temperature, showing a linear correlation of crystallite size with temperature. Notably, under galvanostatic reduction, the Ag2VO2PO4 sample with the smallest crystallite size delivers the highest capacity and shows the highest loaded voltage. Further, pulse discharge tests show a significant resistance decrease during the initial discharge coincident with the formation of Ag metal. Thus, the magnitude of the resistance decrease observed during pulse tests depends on the Ag2VO2PO4 crystallite size, with the largest resistance decrease observed for the smallest crystallite size. Additional electrochemical measurements indicate a quasi-reversible redox reaction involving Li+ insertion/deinsertion, with capacity fade due to structural changes associated with the discharge/charge process. In summary, this work demonstrates a faster synthetic approach for bimetallic polyanionic materials which also provides the opportunity for tuning of electrochemical properties through control of material physical properties such as crystallite size.
Co-reporter:Yiman Zhang, Kevin C. Kirshenbaum, Amy C. Marschilok, Esther S. Takeuchi, and Kenneth J. Takeuchi
Chemistry of Materials 2016 Volume 28(Issue 21) pp:7619
Publication Date(Web):October 12, 2016
DOI:10.1021/acs.chemmater.6b02343
Ag7Fe3(P2O7)4 is an example of an electrochemical displacement material which contains two different electrochemically active metal cations, where one cation (Ag+) forms metallic silver nanoparticles external to the crystals of Ag7Fe3(P2O7)4 via an electrochemical reduction displacement reaction, while the other cation (Fe3+) is electrochemically reduced with the retention of iron cations within the anion structural framework concomitant with lithium insertion. These contrasting redox chemistries within one pure cathode material enable high rate capability and reversibility when Ag7Fe3(P2O7)4 is employed as cathode material in a lithium ion battery (LIB). Further, pyrophosphate materials are thermally and electrically stable, desirable attributes for cathode materials in LIBs. In this paper, a bimetallic pyrophosphate material Ag7Fe3(P2O7)4 is synthesized and confirmed to be a single phase by Rietveld refinement. Electrochemistry of Ag7Fe3(P2O7)4 is reported for the first time in the context of lithium based batteries using cyclic voltammetry and galvanostatic discharge–charge cycling. The reduction displacement reaction and the lithium (de)insertion processes are investigated using ex situ X-ray absorption spectroscopy and X-ray diffraction of electrochemically reduced and oxidized Ag7Fe3(P2O7)4. Ag7Fe3(P2O7)4 exhibits good reversibility at the iron centers indicated by ∼80% capacity retention over 100 cycles following the initial formation cycle and excellent rate capability exhibited by ∼70% capacity retention upon a 4-fold increase in current.
Co-reporter:Altug S. Poyraz, Jianping Huang, Shaobo Cheng, David C. Bock, Lijun Wu, Yimei Zhu, Amy C. Marschilok, Kenneth J. Takeuchi and Esther S. Takeuchi
Green Chemistry 2016 vol. 18(Issue 11) pp:3414-3421
Publication Date(Web):29 Feb 2016
DOI:10.1039/C6GC00438E
While rechargeable lithium ion batteries (LIBs) occupy a prominent consumer presence due to their high cell potential and gravimetric energy density, there are limited opportunities for electrode recycling. Currently used or proposed cathode recycling processes are multistep procedures which involve sequences of mechanical, thermal, and chemical leaching, where only the base material is recovered and significant processing is required to generate a recycled electrode structure. Another significant issue facing lithium based batteries is capacity fade due to structural degradation of the electroactive material upon extending cycling. Herein, inspired by heterogeneous catalyst thermal regeneration strategies, we present a new facile cathode recycling process, where previously used cathodes are removed from a cell, heat treated, and then inserted into a new cell restoring the delivered capacity and cycle life. An environmentally sustainable manganese based material is employed, where binder-free self-supporting (BFSS) electrodes are prepared using a fibrous, high aspect ratio manganese oxide active material. After 200 discharge–charge cycles, the recycled BFSS electrodes display restored crystallinity and oxidation state of the manganese centers with the resulting electrochemistry (capacity and coulombic efficiency) reminiscent of freshly prepared BFSS cathodes. Notably, the BFSS electrode structure is robust with no degradation during the cell disassembly, electrode recovery, washing, and heat treatment steps; thus no post-processing is required for the recycled electrode. This work shows for the first time that a thermal regeneration method previously employed in catalyst systems can fully restore battery electrochemical performance, demonstrating a novel electrode recycling process which could open up new possibilities for energy storage devices with extended electrode lifecycles.
Co-reporter:David C. Bock, Christopher J. Pelliccione, Wei Zhang, Jiajun Wang, K. W. Knehr, Jun Wang, Feng Wang, Alan C. West, Amy C. Marschilok, Kenneth J. Takeuchi, and Esther S. Takeuchi
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 18) pp:11418
Publication Date(Web):April 20, 2016
DOI:10.1021/acsami.6b01134
Aggregation of nanosized materials in composite lithium-ion-battery electrodes can be a significant factor influencing electrochemical behavior. In this study, aggregation was controlled in magnetite, Fe3O4, composite electrodes via oleic acid capping and subsequent dispersion in a carbon black matrix. A heat treatment process was effective in the removal of the oleic acid capping agent while preserving a high degree of Fe3O4 dispersion. Electrochemical testing showed that Fe3O4 dispersion is initially beneficial in delivering a higher functional capacity, in agreement with continuum model simulations. However, increased capacity fade upon extended cycling was observed for the dispersed Fe3O4 composites relative to the aggregated Fe3O4 composites. X-ray absorption spectroscopy measurements of electrodes post cycling indicated that the dispersed Fe3O4 electrodes are more oxidized in the discharged state, consistent with reduced reversibility compared with the aggregated sample. Higher charge-transfer resistance for the dispersed sample after cycling suggests increased surface-film formation on the dispersed, high-surface-area nanocrystalline Fe3O4 compared to the aggregated materials. This study provides insight into the specific effects of aggregation on electrochemistry through a multiscale view of mechanisms for magnetite composite electrodes.Keywords: aggregate; composite; EIS; EXAFS; lithium-ion battery; magnetite; TXM
Co-reporter:Jianping Huang, Altug S. Poyraz, Kenneth J. Takeuchi, Esther S. Takeuchi and Amy C. Marschilok
Chemical Communications 2016 vol. 52(Issue 21) pp:4088-4091
Publication Date(Web):15 Feb 2016
DOI:10.1039/C6CC00025H
AgxMn8O16 (Ag-OMS-2) and KxMn8O16 (K-OMS-2) were investigated as high voltage cathode materials for Mg based batteries. Both MxMn8O16 materials delivered high initial capacities (>180 mA h g−1), and KxMn8O16 showed high cycle stability with a reversible capacity of >170 mA h g−1 after 20 cycles.
Co-reporter:Matthew M. Huie, Christina A. Cama, Paul F. Smith, Jiefu Yin, Amy C. Marschilok, Kenneth J. Takeuchi, Esther S. Takeuchi
Electrochimica Acta 2016 Volume 219() pp:267-276
Publication Date(Web):20 November 2016
DOI:10.1016/j.electacta.2016.09.107
Magnesium – ion batteries have the potential for high energy density but require new types of electrolytes for practical application. Ionic liquid (IL) electrolytes offer the opportunity for increased safety and broader voltage windows relative to traditional electrolytes. We present here a systematic study of both the conductivity and oxidative stability of hybrid electrolytes consisting of eleven ILs mixed with dipropylene glycol dimethylether (DPGDME) or acetonitrile (ACN) cosolvents and magnesium bis(trifluoromethylsulfonyl)imide (Mg(TFSI)2). Our study finds a correlation of higher conductivity of ILs with unsaturated rings and short carbon chain lengths, but by contrast, these ILs also exhibited lower oxidation voltage limits. For the cosolvent additive, although glymes have a demonstrated capability of coordination with Mg2+ ions, a decrease in conductivity compared to acetonitrile hybrid electrolytes was observed. When cycled within the appropriate voltage range, the IL-hybrid electrolytes that show the highest conductivity provide the best cathode magnesiation current densities and lowest polarization as demonstrated with a Mg0.15MnO2 and Mg0.07V2O5 cathodes.
Co-reporter:Christina A. Cama, Christopher J. Pelliccione, Alexander B. Brady, Jing Li, Eric A. Stach, Jiajun Wang, Jun Wang, Esther S. Takeuchi, Kenneth J. Takeuchi and Amy C. Marschilok
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 25) pp:16930-16940
Publication Date(Web):06 Jun 2016
DOI:10.1039/C6CP02974D
Copper ferrite, CuFe2O4, is a promising candidate for application as a high energy electrode material in lithium based batteries. Mechanistic insight on the electrochemical reduction and oxidation processes was gained through the first X-ray absorption spectroscopic study of lithiation and delithiation of CuFe2O4. A phase pure tetragonal CuFe2O4 material was prepared and characterized using laboratory and synchrotron X-ray diffraction, Raman spectroscopy, and transmission electron microscopy. Ex situ X-ray absorption spectroscopy (XAS) measurements were used to study the battery redox processes at the Fe and Cu K-edges, using X-ray absorption near-edge structure (XANES), extended X-ray absorption fine structure (EXAFS), and transmission X-ray microscopy (TXM) spectroscopies. EXAFS analysis showed upon discharge, an initial conversion of 50% of the copper(II) to copper metal positioned outside of the spinel structure, followed by a migration of tetrahedral iron(III) cations to octahedral positions previously occupied by copper(II). Upon charging to 3.5 V, the copper metal remained in the metallic state, while iron metal oxidation to iron(III) was achieved. The results provide new mechanistic insight regarding the evolution of the local coordination environments at the iron and copper centers upon discharging and charging.
Co-reporter:Christopher J. Pelliccione, Yue Ru Li, Amy C. Marschilok, Kenneth J. Takeuchi and Esther S. Takeuchi
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 4) pp:2959-2967
Publication Date(Web):22 Dec 2015
DOI:10.1039/C5CP05926G
The combination of ex situ X-ray diffraction (XRD) and X-ray absorption spectroscopy (XAS) measurements on 2D layered copper birnessite cathode materials for lithium ion battery applications provides detailed insight into both bulk-crystalline and localized atomic structural changes resulting from electrochemically driven lithium insertion and de-insertion. Copper birnessite electrodes that had been galvanostatically discharged and charged were measured with XRD to determine the accompanying long-range crystalline structure changes, while Mn and Cu K-edge XAS measurements provided a detailed view of the Mn and Cu oxidation state changes along with variations of the local neighboring atom environments around the Mn and Cu centers. While not detectable with XRD spectra, through XAS measurements it was determined that the copper ions (Cu2+) are reduced to form amorphous nano-sized Cu metal, and can be oxidized back to Cu2+. The reversible nature of the interconversion provides a rationale to the enhanced discharge capacity of copper birnessite relative to the analogous copper-free birnessite materials. The manganese oxide octahedra comprising the 2D layers in the original copper birnessite crystal structure disperse during lithium insertion, and revert back close to their original orientation after lithium de-insertion. During electrochemical oxidation or reduction the layered birnessite structure does not collapse, even though significant local disordering around Mn and Cu centers is directly observed.
Co-reporter:Jiefu Yin, Esther S. Takeuchi, Kenneth J. Takeuchi, Amy C. Marschilok
Inorganica Chimica Acta 2016 Volume 453() pp:230-237
Publication Date(Web):1 November 2016
DOI:10.1016/j.ica.2016.08.026
•MgxMnO2 prepared via low temperature precipitation and ion exchange.•First electrochemical study of MgxMnO2 as a function of crystallite size.•Battery performance was studied in Li+, Na+, and Mg2+ based electrolytes.•Small crystallite size showed enhanced capacity and rate capability.•Mg2+ ion diffusion coefficient was ∼10× lower than that of Li+ and Na+.The synthesis and characterization of Mg-birnessite (MgxMnO2) with different crystallite sizes, prepared though low temperature precipitation and ion exchange was demonstrated. The influence of crystallite size on electrochemical performance of Mg-birnessite was studied for the first time, where material with smaller crystallite size was demonstrated to have enhanced capacity and rate capability in Li ion, Na ion, and Mg ion based electrolytes. Cation diffusion using GITT type testing demonstrated the ion diffusion coefficient of Mg2+ was ∼10× lower compared with Li+ and Na+. This work illustrates that tuning of inorganic materials properties can lead to significant enhancement of electrochemical performance in lithium, sodium as well as magnesium based batteries for materials such as Mg-birnessite and provides a deliberate approach to improve electrochemical performance.Synthesis of Mg-birnessite (MgxMnO2) with different crystallite sizes via low temperature precipitation and ion exchange was demonstrated. The influence of crystallite size on electrochemistry of Mg-birnessite was studied for the first time. Material with smaller crystallite size showed enhanced capacity and rate capability in Li+, Na+, and Mg2+ based electrolytes.
Co-reporter:Alyson Abraham, Lisa M. Housel, Christianna N. Lininger, David C. Bock, Jeffrey Jou, Feng Wang, Alan C. West, Amy C. Marschilok, Kenneth J. Takeuchi, and Esther S. Takeuchi
ACS Central Science 2016 Volume 2(Issue 6) pp:380
Publication Date(Web):May 31, 2016
DOI:10.1021/acscentsci.6b00100
Electric energy storage systems such as batteries can significantly impact society in a variety of ways, including facilitating the widespread deployment of portable electronic devices, enabling the use of renewable energy generation for local off grid situations and providing the basis of highly efficient power grids integrated with energy production, large stationary batteries, and the excess capacity from electric vehicles. A critical challenge for electric energy storage is understanding the basic science associated with the gap between the usable output of energy storage systems and their theoretical energy contents. The goal of overcoming this inefficiency is to achieve more useful work (w) and minimize the generation of waste heat (q). Minimization of inefficiency can be approached at the macro level, where bulk parameters are identified and manipulated, with optimization as an ultimate goal. However, such a strategy may not provide insight toward the complexities of electric energy storage, especially the inherent heterogeneity of ion and electron flux contributing to the local resistances at numerous interfaces found at several scale lengths within a battery. Thus, the ability to predict and ultimately tune these complex systems to specific applications, both current and future, demands not just parametrization at the bulk scale but rather specific experimentation and understanding over multiple length scales within the same battery system, from the molecular scale to the mesoscale. Herein, we provide a case study examining the insights and implications from multiscale investigations of a prospective battery material, Fe3O4.
Co-reporter:Matthew M. Huie, David C. Bock, Esther S. Takeuchi, Amy C. Marschilok, Kenneth J. Takeuchi
Coordination Chemistry Reviews 2015 Volume 287() pp:15-27
Publication Date(Web):15 March 2015
DOI:10.1016/j.ccr.2014.11.005
•Mg-ion batteries may address future large scale mobile and stationary device needs.•Recent research on cathodes for Mg-ion is reviewed.•Chemical and structural details of the cathode materials are emphasized.•Particular strategies which may lead to future research initiatives are amplified.Rechargeable magnesium-ion batteries are a promising candidate technology to address future electrical energy storage needs of large scale mobile and stationary devices, due to the high environmental abundance of magnesium metal and divalent character of magnesium ion. With the recent increase in reports discussing cathode materials for magnesium-ion batteries, it is instructive to assess recent research in order to provide inspiration for future research. This review is a summary of the different chemistries and structures of the materials developed for magnesium ion cathodes. The particular strategies which may lead to future research initiatives are amplified.
Co-reporter:Kevin C. Kirshenbaum, David C. Bock, Zhong Zhong, Amy C. Marschilok, Kenneth J. Takeuchi and Esther S. Takeuchi
Journal of Materials Chemistry A 2015 vol. 3(Issue 35) pp:18027-18035
Publication Date(Web):29 Jul 2015
DOI:10.1039/C5TA04523A
In this study, we characterize the deposition of silver metal nanoparticles formed during discharge of Li/Ag2VP2O8 cells with composite cathodes containing conductive carbon additive. Using in situ energy dispersive X-ray diffraction (EDXRD) of an intact battery, the location and distribution of silver metal nanoparticles generated upon reduction-displacement deposition within an Ag2VP2O8 cathode containing a pre-existing percolation network can be observed for the first time. This study yielded unexpected results where higher rate initial discharge generated a more effective conductive matrix. This stands in contrast to cells with cathodes with no conductive additive where a low rate initial discharge proved more effective. These results provide evidence that using conductive additives in conjunction with an in situ reduction-displacement deposition of silver metal provides a path toward the ultimate goal of complete electrical contact and full utilization of all electroactive particles.
Co-reporter:Matthew M. Huie, Roberta A. DiLeo, Amy C. Marschilok, Kenneth J. Takeuchi, and Esther S. Takeuchi
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 22) pp:11724
Publication Date(Web):February 24, 2015
DOI:10.1021/acsami.5b00496
Batteries are multicomponent systems where the theoretical voltage and stoichiometric electron transfer are defined by the electrochemically active anode and cathode materials. While the electrolyte may not be considered in stoichiometric electron-transfer calculations, it can be a critical factor determining the deliverable energy content of a battery, depending also on the use conditions. The development of ionic liquid (IL)-based electrolytes has been a research area of recent reports by other researchers, due, in part, to opportunities for an expanded high-voltage operating window and improved safety through the reduction of flammable solvent content. The study reported here encompasses a systematic investigation of the physical properties of IL-based hybrid electrolytes including quantitative characterization of the electrolyte–separator interface via contact-angle measurements. An inverse trend in the conductivity and wetting properties was observed for a series of IL-based electrolyte candidates. Test-cell measurements were undertaken to evaluate the electrolyte performance in the presence of functioning anode and cathode materials, where several promising IL-based hybrid electrolytes with performance comparable to that of conventional carbonate electrolytes were identified. The study revealed that the contact angle influenced the performance more significantly than the conductivity because the cells containing IL–tetrafluoroborate-based electrolytes with higher conductivity but poorer wetting showed significantly decreased performance relative to the cells containing IL–bis(trifluoromethanesulfonyl)imide electrolytes with lower conductivity but improved wetting properties. This work contributes to the development of new IL battery-based electrolyte systems with the potential to improve the deliverable energy content as well as safety of lithium-ion battery systems.Keywords: contact angle; electrolyte; ionic liquid; lithium battery; separator;
Co-reporter:David C. Bock, Kevin C. Kirshenbaum, Jiajun Wang, Wei Zhang, Feng Wang, Jun Wang, Amy. C. Marschilok, Kenneth J. Takeuchi, and Esther S. Takeuchi
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 24) pp:13457
Publication Date(Web):May 29, 2015
DOI:10.1021/acsami.5b02478
When electroactive nanomaterials are fully incorporated into an electrode structure, characterization of the crystallite sizes, agglomerate sizes, and dispersion of the electroactive materials can lend insight into the complex electrochemistry associated with composite electrodes. In this study, composite magnetite electrodes were sectioned using ultramicrotome techniques, which facilitated the direct observation of crystallites and agglomerates of magnetite (Fe3O4) as well as their dispersal patterns in large representative sections of electrode, via 2D cross sectional analysis by Transmission Electron Microscopy (TEM). Further, the electrochemistry of these electrodes were recorded, and Transmission X-ray Microscopy (TXM) was used to determine the distribution of oxidation states of the reduced magnetite. Unexpectedly, while two crystallite sizes of magnetite were employed in the production of the composite electrodes, the magnetite agglomerate sizes and degrees of dispersion in the two composite electrodes were similar to each other. This observation illustrates the necessity for careful characterization of composite electrodes, in order to understand the effects of crystallite size, agglomerate size, and level of dispersion on electrochemistry.Keywords: agglomerate; crystallite size; lithium-ion battery; magnetite; transmission X-ray microscopy; ultramicrotome;
Co-reporter:David C. Bock, Ryan V. Tappero, Kenneth J. Takeuchi, Amy C. Marschilok, and Esther S. Takeuchi
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 9) pp:5429
Publication Date(Web):February 17, 2015
DOI:10.1021/am509066n
Cathode solubility in batteries can lead to decreased and unpredictable long-term battery behavior due to transition metal deposition on the negative electrode such that it no longer supports high current. Analysis of negative electrodes from cells containing vanadium oxide or phosphorus oxide based cathode systems retrieved after long-term testing was conducted. This report demonstrates the use of synchrotron based X-ray microfluorescence (XRμF) to map negative battery electrodes in conjunction with microbeam X-ray absorption spectroscopy (μXAS) to determine the oxidation states of the metal centers resident in the solid electrolyte interphase (SEI) and at the electrode surface. Based on the empirical findings, a conceptual model for the location of metal ions in the SEI and their role in impacting lithium ion mobility at the electrode surfaces is proposed.Keywords: lithium battery; silver vanadium oxide; silver vanadium phosphorus oxide; solid electrolyte interphase; X-ray microfluorescence mapping
Co-reporter:Jessica L. Durham, Kevin Kirshenbaum, Esther S. Takeuchi, Amy C. Marschilok and Kenneth J. Takeuchi
Chemical Communications 2015 vol. 51(Issue 24) pp:5120-5123
Publication Date(Web):16 Feb 2015
DOI:10.1039/C4CC10277K
A paradigm for concomitant control of crystallite size and composition of bimetallic composites via co-precipitation is introduced. Direct preparation of composites of silver ferrite and amorphous maghemite via nonstoichiometric synthesis was demonstrated. Notable impact on electrochemistry was observed, with ∼200% increase in reversible capacity for the small crystallite material.
Co-reporter:David C. Bock, Kenneth J. Takeuchi, Amy C. Marschilok and Esther S. Takeuchi
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 3) pp:2034-2042
Publication Date(Web):02 Dec 2014
DOI:10.1039/C4CP04819A
The detailed understanding of non-faradaic parasitic reactions which diminish battery calendar life is essential to the development of effective batteries for use in long life applications. The dissolution of cathode materials including manganese, cobalt and vanadium oxides in battery systems has been identified as a battery failure mechanism, yet detailed dissolution studies including kinetic analysis are absent from the literature. The results presented here provide a framework for the quantitative and kinetic analyses of the dissolution of cathode materials which will aid the broader community in more fully understanding this battery failure mechanism. In this study, the dissolution of silver vanadium oxide, representing the primary battery powering implantable cardioverter defibrillators (ICD), is compared with the dissolution of silver vanadium phosphorous oxide (AgwVxPyOz) materials which were targeted as alternatives to minimize solubility. This study contains the first kinetic analyses of silver and vanadium solution formation from Ag0.48VOPO4·1.9H2O and Ag2VP2O8, in a non-aqueous battery electrolyte. The kinetic results are compared with those of Ag2VO2PO4 and Ag2V4O11 to probe the relationships among crystal structure, stoichiometry, and solubility. For vanadium, significant dissolution was observed for Ag2V4O11 as well as for the phosphate oxide Ag0.49VOPO4·1.9H2O, which may involve structural water or the existence of multiple vanadium oxidation states. Notably, the materials from the SVPO family with the lowest vanadium solubility are Ag2VO2PO4 and Ag2VP2O8. The low concentrations and solution rates coupled with their electrochemical performance make these materials interesting alternatives to Ag2V4O11 for the ICD application.
Co-reporter:Qing Zhang, Kenneth J. Takeuchi, Esther S. Takeuchi and Amy C. Marschilok
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 35) pp:22504-22518
Publication Date(Web):03 Aug 2015
DOI:10.1039/C5CP03217B
Carbon monofluoride (CFx) has a high energy density, exceeding 2000 W h kg−1, yet its application in primary lithium batteries is limited by its power capability. Multi-walled carbon nanotubes (CNTs) are appealing additives for high-power batteries, due to their outstanding electronic transport properties, high aspect ratio necessitating low volume fraction for percolation, and high tensile strength. This perspective describes the current state of the art in lithium–carbon monofluoride (Li/CFx) batteries and highlights the opportunities for the development of high-power Li/CFx batteries via utilization of carbon nanotubes. In this report, we generated several electrode architectures using CFx/CNT combinations, and demonstrated the effectiveness of CNTs in enhancing the rate capability and energy density of Li/CFx batteries. First, we investigated the resistivity of CFx combined with CNTs and compared the CFx/CNT composites with conventional carbon additives. Second, we built CFx–CNT electrodes without metallic current collectors using CNTs as substrates, and compared their electrochemical performance with conventional CFx electrodes using aluminum foil as a current collector. Furthermore, we fabricated multi-layered CNT–CFx–CNT composite electrodes (sandwich electrodes) and studied the impact of the structure on the performance of the electrode. Our work demonstrates some of the opportunities for utilization of CNTs in CFx electrodes and the resultant implementation of CFx as a battery cathode in next-generation high-power batteries.
Co-reporter:Kevin C. Kirshenbaum, David C. Bock, Alexander B. Brady, Amy C. Marschilok, Kenneth J. Takeuchi and Esther S. Takeuchi
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 17) pp:11204-11210
Publication Date(Web):26 Mar 2015
DOI:10.1039/C5CP00961H
Previously, we reported that electrodes containing silver vanadium phosphate (Ag2VO2PO4) powder exhibit a 15000 fold increase in conductivity after discharge, concurrent with the formation of silver metal. In this study, in order to disentangle the complex nature of electrodes composed of electroactive powders, an electrochemical reduction of individual particles of Ag2VO2PO4 was conducted, to more directly probe the intrinsic materials properties of Ag2VO2PO4. Specifically, individual particle conductivity data from a nanoprobe system combined with SEM and optical imaging results revealed that the depth of discharge within an Ag2VO2PO4 particle is closely linked to the conductivity increase. Notably, the formation of silver metal may affect both inter- and intraparticle conductivity of the Ag2VO2PO4 material.
Co-reporter:Lijun Wu, Feng Xu, Yimei Zhu, Alexander B. Brady, Jianping Huang, Jessica L. Durham, Eric Dooryhee, Amy C. Marschilok, Esther S. Takeuchi, and Kenneth J. Takeuchi
ACS Nano 2015 Volume 9(Issue 8) pp:8430
Publication Date(Web):July 16, 2015
DOI:10.1021/acsnano.5b03274
Hollandites (OMS-2) are an intriguing class of sorbents, catalysts, and energy storage materials with a tunnel structure permitting one-dimensional insertion and deinsertion of ions and small molecules along the c direction. A 7-fold increase in delivered capacity for Li/AgxMn8O16 electrochemical cells (160 versus 23 mAh/g) observed upon a seemingly small change in silver content (x ∼1.1 (L-Ag-OMS-2) and 1.6 (H-Ag-OMS-2)) led us to characterize the structure and defects of the silver hollandite material. Herein, Ag hollandite nanorods are studied through the combined use of local (atomic imaging, electron diffraction, electron energy-loss spectroscopy) and bulk (synchrotron based X-ray diffraction, thermogravimetric analysis) techniques. Selected area diffraction and high resolution transmission electron microscopy show a structure consistent with that refined by XRD; however, the Ag occupancy varies significantly even within neighboring channels. Both local and bulk measurements indicate a greater quantity of oxygen vacancies in L-Ag-OMS-2, resulting in lower average Mn valence relative to H-Ag-OMS-2. Electron energy loss spectroscopy shows a lower Mn oxidation state on the surface relative to the interior of the nanorods, where the average Mn valence is approximately Mn3.7+ for H-Ag-OMS-2 and Mn3.5+ for L-Ag-OMS-2 nanorods, respectively. The higher delivered capacity of L-Ag-OMS-2 may be related to more oxygen vacancies compared to H-Ag-OMS-2. Thus, the oxygen vacancies and MnO6 octahedra distortion are assumed to open the MnO6 octahedra walls, facilitating Li diffusion in the ab plane. These results indicate crystallite size and surface defects are significant factors affecting battery performance.Keywords: electron energy loss spectroscopy; lithium battery; octahedral molecular sieve; oxygen defects; silver hollandite; transmission electron microscopy;
Co-reporter:Kevin Kirshenbaum;Chia-Ying Lee;Amy C. Marschilok;Zhong Zhong;David C. Bock;Esther S. Takeuchi
Science 2015 Volume 347(Issue 6218) pp:149-154
Publication Date(Web):09 Jan 2015
DOI:10.1126/science.1257289
Watching the silver lining inside
Some types of batteries contain both a transition metal reducible metal, such as the cathode material Ag2VP2O8. During operation, both Ag and V ions are reduced, and the Ag atoms can form wires to enhance the internal conductivity. Kirshenbaum et al. probe the discharge of a battery at different rates and track the formation of Ag atoms using in situ energy-dispersive x-ray diffraction (see the Perspective by Dudney and Li). They show how the discharge rate affects whether the Ag or V is preferentially reduced and also the distribution of the Ag atoms, and then correlate this to the loss of battery capacity at higher discharge rates.
Science, this issue p. 149; see also p. 131
Co-reporter:Kevin C. Kirshenbaum, David C. Bock, Zhong Zhong, Amy C. Marschilok, Kenneth J. Takeuchi and Esther S. Takeuchi
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 19) pp:9138-9147
Publication Date(Web):07 Apr 2014
DOI:10.1039/C4CP01220H
In situ, in operando characterization of electrochemical cells can provide insight into the complex discharge chemistries of batteries which may not be available with destructive methods. In this study, in situ energy-dispersive X-ray spectroscopy (EDXRD) measurements are conducted for the first time on active lithium/silver vanadium diphosphate, Li/Ag2VP2O8, electrochemical cells at several depths of discharge allowing depth profiling analysis of the reduction process. This technique enables non-destructive, in operando imaging of the reduction process within a battery electrode over a millimeter scale interrogation area with micron scale resolution. The discharge of Ag2VP2O8 progresses via a reduction displacement reaction forming conductive silver metal as a discharge product, a high Z material which can be readily detected by diffraction-based methods. The high energy X-ray capabilities of NSLS beamline X17B1 allowed spatially resolved detection of the reduction products forming conductive pathways providing insight into the discharge mechanism of Ag2VP2O8.
Co-reporter:David C. Bock, Amy C. Marschilok, Kenneth J. Takeuchi, Esther S. Takeuchi
Journal of Power Sources 2013 Volume 231() pp:219-225
Publication Date(Web):1 June 2013
DOI:10.1016/j.jpowsour.2013.01.012
Silver vanadium oxide (Ag2V4O11, SVO) has enjoyed widespread commercial success over the past 30 years as a cathode material for implantable cardiac defibrillator (ICD) batteries. Recently, silver vanadium phosphorous oxide (Ag2VO2PO4, SVPO) has been studied as possibly combining the desirable thermal stability aspects of LiFePO4 with the electrical conductivity of SVO. Further, due to the noted insoluble nature of most phosphate salts, a lower material solubility of SVPO relative to SVO is anticipated. Thus, the first vanadium dissolution studies of SVPO in battery electrolyte solutions are described herein. The equilibrium solubility of SVPO was ∼5 times less than SVO, with a rate constant of dissolution ∼3.5 times less than that of SVO. The vanadium dissolution in SVO and SVPO can be adequately described with a diffusion-layer model, as supported by the Noyes–Whitney equation. Cells prepared with vanadium-treated anodes displayed higher AC impedance and DC resistance relative to control anodes. These data support the premise that SVPO cells are likely to exhibit reduced cathode solubility and thus are less affected by increased cell resistance due to cathode solubility compared to SVO based cells.Highlights► The first vanadium dissolution studies of SVPO in battery electrolyte solutions are described. ► The equilibrium solubility of SVPO was ∼5 times less than that of silver vanadium oxide, SVO. ► These techniques establish a paradigm for other systems where solubility may be an issue.
Co-reporter:Roberta A. Di Leo, Amy C. Marschilok, Kenneth J. Takeuchi, Esther S. Takeuchi
Electrochimica Acta 2013 Volume 109() pp:27-32
Publication Date(Web):30 October 2013
DOI:10.1016/j.electacta.2013.07.041
Physical and electrochemical properties of mixtures of ionic liquids based on saturated ring systems with carbonate based solvents were investigated. The conductivity and electrochemical stability of two series of ionic liquids based on piperidinium and pyrrolidinium cations with tetrafluoroborate and bis(trifluorosulfonylimide) anions were evaluated. The effects of the ionic liquid cation, substituent chain length of the cation function group, and the anion type on conductivity and electrochemical stability as determined by cyclic voltammetry were studied. The conductivity was influenced by the substituent chain length of the ionic liquid cation and the solvent carbonate type, where higher conductivities were observed with shorter substituent chains and EC versus PC. The saturated ring ionic liquid–carbonate mixtures may show particular promise for implementation as battery electrolytes due to notable high voltage stabilities, where stability >5.5 V was maintained in the presence of lithium salt. This study should promote development of future safe, high voltage lithium ion battery systems.
Co-reporter:Melissa C. Menard, Amy C. Marschilok, Kenneth J. Takeuchi, Esther S. Takeuchi
Electrochimica Acta 2013 Volume 94() pp:320-326
Publication Date(Web):1 April 2013
DOI:10.1016/j.electacta.2013.02.012
We have investigated magnetite (Fe3O4) as an electroactive battery electrode material, where a linear relationship was observed between Fe3O4 crystallite size and capacity, with a negative slope. In order to better understand this novel relationship, we report here the Rietveld refinement and X-ray absorption spectroscopy (XAS) investigation of nanosized Fe3O4 as a function of crystallite size (7–26 nm). Rietveld refinement established that the Fe3O4 samples were phase pure, while the extended X-ray absorption fine structure (EXAFS) and X-ray absorption near edge structure (XANES) provided insight into the local geometries and electronic structure of the iron centers, including oxidation state assignment. From our current and recent studies, we suggest that the surface of the Fe3O4 crystallites is rich in Fe3+, thus as the Fe3O4 crystallite size decreases, the electrochemical capacity increases, due to a net enrichment of Fe3O4 in Fe3+.
Co-reporter:David C. Bock, Kenneth J. Takeuchi, Amy C. Marschilok and Esther S. Takeuchi
Dalton Transactions 2013 vol. 42(Issue 38) pp:13981-13989
Publication Date(Web):08 Aug 2013
DOI:10.1039/C3DT51544C
Material design strategies for energy storage applications can be considered in two major categories: (1) control of structure and composition and (2) material dimensional control such as the implementation of nanomaterials. Characterization of electrochemical properties determines energy content and possible viability for potential application. Equally critical yet more challenging is quantifying the non-Faradaic parasitic reactions of the active materials and the relationship to battery life. Understanding the significant factors associated with battery lifetimes for the implantable cardioverter defibrillator (ICD) is critical for the development of new ICD batteries. In situ dissolution of the cathode material has been identified as a major factor in premature end of life for ICD batteries. This study contains the kinetic analyses of silver and vanadium dissolution from the benchmark silver vanadium oxide (SVO) material and two silver vanadium phosphorous oxide (SVPO-H and SVPO-R) materials with differing physical properties in a non-aqueous ICD battery electrolyte. A comparison of the kinetic and mechanistic results for SVO, SVPO-H and SVPO-R provides insight for future material design approaches.
Co-reporter:Melissa C. Menard, Kenneth J. Takeuchi, Amy C. Marschilok and Esther S. Takeuchi
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 42) pp:18539-18548
Publication Date(Web):30 Sep 2013
DOI:10.1039/C3CP52870G
Magnetite (Fe3O4) is an abundant, low cost, environmentally benign material with potential application in batteries. Recently, low temperature coprecipitation methods have enabled preparation of a series of nanocrystalline magnetite samples with a range of crystallite sizes. Electrochemical cells based on Li/Fe3O4 show a linear increase in capacity with decreasing crystallite size at voltages ≥1.2 V where a 2× capacity improvement relative to commercial (26.2 nm) magnetite is observed. In this report, a combination of X-ray powder diffraction (XRD) and X-ray absorption spectroscopy (XAS) is used to measure magnetite structural changes occurring upon electrochemical reduction, with parent Fe3O4 crystallite size as a variable. Notably, XAS provides evidence of metallic iron formation at high levels of electrochemical reduction.
Co-reporter:Kenneth J. Takeuchi, Shali Z. Yau, Melissa C. Menard, Amy C. Marschilok, and Esther S. Takeuchi
ACS Applied Materials & Interfaces 2012 Volume 4(Issue 10) pp:5547
Publication Date(Web):October 9, 2012
DOI:10.1021/am301443g
Synthetic control of the silver content in silver hollandite, AgxMn8O16, where the silver content ranges from 1.0 ≤ x ≤ 1.8 is demonstrated. This level of compositional control was enabled by the development of a lower temperature reflux based synthesis compared to the more commonly reported hydrothermal approach. Notably, the synthetic variance of the silver content was accompanied by a concomitant variance in crystallite size as well as surface area and particle size. To verify the retention of the hollandite structure, the first Rietveld analysis of silver hollandite was conducted on samples of varying composition. The impacts of silver content, crystallite size, surface area, and particle size on electrochemical reversibility were examined under cyclic voltammetry and battery testing.Keywords: battery; compositional control; crystallite size control; electrochemistry; reflux synthesis; Rietveld analysis; silver hollandite;
Co-reporter:Jiefu Yin, Alexander B. Brady, Esther S. Takeuchi, Amy C. Marschilok and Kenneth J. Takeuchi
Chemical Communications 2017 - vol. 53(Issue 26) pp:NaN3668-3668
Publication Date(Web):2017/03/06
DOI:10.1039/C7CC00265C
MgMn2O4 nanoparticles with crystallite sizes of 11 (MMO-1) and 31 nm (MMO-2) were synthesized and their magnesium-ion battery-relevant electrochemistry was investigated. MMO-1 delivered an initial capacity of 220 mA h g−1 (678 mW h g−1). Electrolyte water content had a profound effect on cycle retention.
Co-reporter:Kevin C. Kirshenbaum, David C. Bock, Zhong Zhong, Amy C. Marschilok, Kenneth J. Takeuchi and Esther S. Takeuchi
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 19) pp:NaN9147-9147
Publication Date(Web):2014/04/07
DOI:10.1039/C4CP01220H
In situ, in operando characterization of electrochemical cells can provide insight into the complex discharge chemistries of batteries which may not be available with destructive methods. In this study, in situ energy-dispersive X-ray spectroscopy (EDXRD) measurements are conducted for the first time on active lithium/silver vanadium diphosphate, Li/Ag2VP2O8, electrochemical cells at several depths of discharge allowing depth profiling analysis of the reduction process. This technique enables non-destructive, in operando imaging of the reduction process within a battery electrode over a millimeter scale interrogation area with micron scale resolution. The discharge of Ag2VP2O8 progresses via a reduction displacement reaction forming conductive silver metal as a discharge product, a high Z material which can be readily detected by diffraction-based methods. The high energy X-ray capabilities of NSLS beamline X17B1 allowed spatially resolved detection of the reduction products forming conductive pathways providing insight into the discharge mechanism of Ag2VP2O8.
Co-reporter:Melissa C. Menard, Kenneth J. Takeuchi, Amy C. Marschilok and Esther S. Takeuchi
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 42) pp:NaN18548-18548
Publication Date(Web):2013/09/30
DOI:10.1039/C3CP52870G
Magnetite (Fe3O4) is an abundant, low cost, environmentally benign material with potential application in batteries. Recently, low temperature coprecipitation methods have enabled preparation of a series of nanocrystalline magnetite samples with a range of crystallite sizes. Electrochemical cells based on Li/Fe3O4 show a linear increase in capacity with decreasing crystallite size at voltages ≥1.2 V where a 2× capacity improvement relative to commercial (26.2 nm) magnetite is observed. In this report, a combination of X-ray powder diffraction (XRD) and X-ray absorption spectroscopy (XAS) is used to measure magnetite structural changes occurring upon electrochemical reduction, with parent Fe3O4 crystallite size as a variable. Notably, XAS provides evidence of metallic iron formation at high levels of electrochemical reduction.
Co-reporter:Qing Zhang, Kenneth J. Takeuchi, Esther S. Takeuchi and Amy C. Marschilok
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 35) pp:NaN22518-22518
Publication Date(Web):2015/08/03
DOI:10.1039/C5CP03217B
Carbon monofluoride (CFx) has a high energy density, exceeding 2000 W h kg−1, yet its application in primary lithium batteries is limited by its power capability. Multi-walled carbon nanotubes (CNTs) are appealing additives for high-power batteries, due to their outstanding electronic transport properties, high aspect ratio necessitating low volume fraction for percolation, and high tensile strength. This perspective describes the current state of the art in lithium–carbon monofluoride (Li/CFx) batteries and highlights the opportunities for the development of high-power Li/CFx batteries via utilization of carbon nanotubes. In this report, we generated several electrode architectures using CFx/CNT combinations, and demonstrated the effectiveness of CNTs in enhancing the rate capability and energy density of Li/CFx batteries. First, we investigated the resistivity of CFx combined with CNTs and compared the CFx/CNT composites with conventional carbon additives. Second, we built CFx–CNT electrodes without metallic current collectors using CNTs as substrates, and compared their electrochemical performance with conventional CFx electrodes using aluminum foil as a current collector. Furthermore, we fabricated multi-layered CNT–CFx–CNT composite electrodes (sandwich electrodes) and studied the impact of the structure on the performance of the electrode. Our work demonstrates some of the opportunities for utilization of CNTs in CFx electrodes and the resultant implementation of CFx as a battery cathode in next-generation high-power batteries.
Co-reporter:Kevin C. Kirshenbaum, David C. Bock, Alexander B. Brady, Amy C. Marschilok, Kenneth J. Takeuchi and Esther S. Takeuchi
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 17) pp:NaN11210-11210
Publication Date(Web):2015/03/26
DOI:10.1039/C5CP00961H
Previously, we reported that electrodes containing silver vanadium phosphate (Ag2VO2PO4) powder exhibit a 15000 fold increase in conductivity after discharge, concurrent with the formation of silver metal. In this study, in order to disentangle the complex nature of electrodes composed of electroactive powders, an electrochemical reduction of individual particles of Ag2VO2PO4 was conducted, to more directly probe the intrinsic materials properties of Ag2VO2PO4. Specifically, individual particle conductivity data from a nanoprobe system combined with SEM and optical imaging results revealed that the depth of discharge within an Ag2VO2PO4 particle is closely linked to the conductivity increase. Notably, the formation of silver metal may affect both inter- and intraparticle conductivity of the Ag2VO2PO4 material.
Co-reporter:Christina A. Cama, Christopher J. Pelliccione, Alexander B. Brady, Jing Li, Eric A. Stach, Jiajun Wang, Jun Wang, Esther S. Takeuchi, Kenneth J. Takeuchi and Amy C. Marschilok
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 25) pp:NaN16940-16940
Publication Date(Web):2016/06/06
DOI:10.1039/C6CP02974D
Copper ferrite, CuFe2O4, is a promising candidate for application as a high energy electrode material in lithium based batteries. Mechanistic insight on the electrochemical reduction and oxidation processes was gained through the first X-ray absorption spectroscopic study of lithiation and delithiation of CuFe2O4. A phase pure tetragonal CuFe2O4 material was prepared and characterized using laboratory and synchrotron X-ray diffraction, Raman spectroscopy, and transmission electron microscopy. Ex situ X-ray absorption spectroscopy (XAS) measurements were used to study the battery redox processes at the Fe and Cu K-edges, using X-ray absorption near-edge structure (XANES), extended X-ray absorption fine structure (EXAFS), and transmission X-ray microscopy (TXM) spectroscopies. EXAFS analysis showed upon discharge, an initial conversion of 50% of the copper(II) to copper metal positioned outside of the spinel structure, followed by a migration of tetrahedral iron(III) cations to octahedral positions previously occupied by copper(II). Upon charging to 3.5 V, the copper metal remained in the metallic state, while iron metal oxidation to iron(III) was achieved. The results provide new mechanistic insight regarding the evolution of the local coordination environments at the iron and copper centers upon discharging and charging.
Co-reporter:David C. Bock, Kenneth J. Takeuchi, Amy C. Marschilok and Esther S. Takeuchi
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 3) pp:NaN2042-2042
Publication Date(Web):2014/12/02
DOI:10.1039/C4CP04819A
The detailed understanding of non-faradaic parasitic reactions which diminish battery calendar life is essential to the development of effective batteries for use in long life applications. The dissolution of cathode materials including manganese, cobalt and vanadium oxides in battery systems has been identified as a battery failure mechanism, yet detailed dissolution studies including kinetic analysis are absent from the literature. The results presented here provide a framework for the quantitative and kinetic analyses of the dissolution of cathode materials which will aid the broader community in more fully understanding this battery failure mechanism. In this study, the dissolution of silver vanadium oxide, representing the primary battery powering implantable cardioverter defibrillators (ICD), is compared with the dissolution of silver vanadium phosphorous oxide (AgwVxPyOz) materials which were targeted as alternatives to minimize solubility. This study contains the first kinetic analyses of silver and vanadium solution formation from Ag0.48VOPO4·1.9H2O and Ag2VP2O8, in a non-aqueous battery electrolyte. The kinetic results are compared with those of Ag2VO2PO4 and Ag2V4O11 to probe the relationships among crystal structure, stoichiometry, and solubility. For vanadium, significant dissolution was observed for Ag2V4O11 as well as for the phosphate oxide Ag0.49VOPO4·1.9H2O, which may involve structural water or the existence of multiple vanadium oxidation states. Notably, the materials from the SVPO family with the lowest vanadium solubility are Ag2VO2PO4 and Ag2VP2O8. The low concentrations and solution rates coupled with their electrochemical performance make these materials interesting alternatives to Ag2V4O11 for the ICD application.
Co-reporter:Christopher J. Pelliccione, Yue Ru Li, Amy C. Marschilok, Kenneth J. Takeuchi and Esther S. Takeuchi
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 4) pp:NaN2967-2967
Publication Date(Web):2015/12/22
DOI:10.1039/C5CP05926G
The combination of ex situ X-ray diffraction (XRD) and X-ray absorption spectroscopy (XAS) measurements on 2D layered copper birnessite cathode materials for lithium ion battery applications provides detailed insight into both bulk-crystalline and localized atomic structural changes resulting from electrochemically driven lithium insertion and de-insertion. Copper birnessite electrodes that had been galvanostatically discharged and charged were measured with XRD to determine the accompanying long-range crystalline structure changes, while Mn and Cu K-edge XAS measurements provided a detailed view of the Mn and Cu oxidation state changes along with variations of the local neighboring atom environments around the Mn and Cu centers. While not detectable with XRD spectra, through XAS measurements it was determined that the copper ions (Cu2+) are reduced to form amorphous nano-sized Cu metal, and can be oxidized back to Cu2+. The reversible nature of the interconversion provides a rationale to the enhanced discharge capacity of copper birnessite relative to the analogous copper-free birnessite materials. The manganese oxide octahedra comprising the 2D layers in the original copper birnessite crystal structure disperse during lithium insertion, and revert back close to their original orientation after lithium de-insertion. During electrochemical oxidation or reduction the layered birnessite structure does not collapse, even though significant local disordering around Mn and Cu centers is directly observed.
Co-reporter:Kevin C. Kirshenbaum, David C. Bock, Zhong Zhong, Amy C. Marschilok, Kenneth J. Takeuchi and Esther S. Takeuchi
Journal of Materials Chemistry A 2015 - vol. 3(Issue 35) pp:NaN18035-18035
Publication Date(Web):2015/07/29
DOI:10.1039/C5TA04523A
In this study, we characterize the deposition of silver metal nanoparticles formed during discharge of Li/Ag2VP2O8 cells with composite cathodes containing conductive carbon additive. Using in situ energy dispersive X-ray diffraction (EDXRD) of an intact battery, the location and distribution of silver metal nanoparticles generated upon reduction-displacement deposition within an Ag2VP2O8 cathode containing a pre-existing percolation network can be observed for the first time. This study yielded unexpected results where higher rate initial discharge generated a more effective conductive matrix. This stands in contrast to cells with cathodes with no conductive additive where a low rate initial discharge proved more effective. These results provide evidence that using conductive additives in conjunction with an in situ reduction-displacement deposition of silver metal provides a path toward the ultimate goal of complete electrical contact and full utilization of all electroactive particles.
Co-reporter:David C. Bock, Kenneth J. Takeuchi, Amy C. Marschilok and Esther S. Takeuchi
Dalton Transactions 2013 - vol. 42(Issue 38) pp:NaN13989-13989
Publication Date(Web):2013/08/08
DOI:10.1039/C3DT51544C
Material design strategies for energy storage applications can be considered in two major categories: (1) control of structure and composition and (2) material dimensional control such as the implementation of nanomaterials. Characterization of electrochemical properties determines energy content and possible viability for potential application. Equally critical yet more challenging is quantifying the non-Faradaic parasitic reactions of the active materials and the relationship to battery life. Understanding the significant factors associated with battery lifetimes for the implantable cardioverter defibrillator (ICD) is critical for the development of new ICD batteries. In situ dissolution of the cathode material has been identified as a major factor in premature end of life for ICD batteries. This study contains the kinetic analyses of silver and vanadium dissolution from the benchmark silver vanadium oxide (SVO) material and two silver vanadium phosphorous oxide (SVPO-H and SVPO-R) materials with differing physical properties in a non-aqueous ICD battery electrolyte. A comparison of the kinetic and mechanistic results for SVO, SVPO-H and SVPO-R provides insight for future material design approaches.
Co-reporter:Jessica L. Durham, Kevin Kirshenbaum, Esther S. Takeuchi, Amy C. Marschilok and Kenneth J. Takeuchi
Chemical Communications 2015 - vol. 51(Issue 24) pp:NaN5123-5123
Publication Date(Web):2015/02/16
DOI:10.1039/C4CC10277K
A paradigm for concomitant control of crystallite size and composition of bimetallic composites via co-precipitation is introduced. Direct preparation of composites of silver ferrite and amorphous maghemite via nonstoichiometric synthesis was demonstrated. Notable impact on electrochemistry was observed, with ∼200% increase in reversible capacity for the small crystallite material.
Co-reporter:Jianping Huang, Altug S. Poyraz, Kenneth J. Takeuchi, Esther S. Takeuchi and Amy C. Marschilok
Chemical Communications 2016 - vol. 52(Issue 21) pp:NaN4091-4091
Publication Date(Web):2016/02/15
DOI:10.1039/C6CC00025H
AgxMn8O16 (Ag-OMS-2) and KxMn8O16 (K-OMS-2) were investigated as high voltage cathode materials for Mg based batteries. Both MxMn8O16 materials delivered high initial capacities (>180 mA h g−1), and KxMn8O16 showed high cycle stability with a reversible capacity of >170 mA h g−1 after 20 cycles.
Co-reporter:Qing Zhang, Andrea M. Bruck, David C. Bock, Jing Li, Varun Sarbada, Robert Hull, Eric A. Stach, Kenneth J. Takeuchi, Esther S. Takeuchi and Amy C. Marschilok
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 21) pp:NaN14169-14169
Publication Date(Web):2017/05/03
DOI:10.1039/C7CP02239E
Li1+nV3O8 (n = 0–0.2) has been extensively investigated as a cathode material for Li ion batteries because of its superior electrochemical properties including high specific energy and good rate capability. In this paper, a synchrotron based energy dispersive X-ray diffraction (EDXRD) technique was employed to profile the phase transitions and the spatial phase distribution of a Li1.1V3O8 electrode during electrochemical (de)lithiation in situ and operando. As annealing temperature during the preparation of the Li1.1V3O8 material has a strong influence on the morphology and crystallinity, and consequently influences the electrochemical outcomes of the material, Li1.1V3O8 materials prepared at two different temperatures, 500 and 300 °C (LVO500 and LVO300), were employed in this study. The EDXRD spectra of LVO500 and LVO300 cells pre-discharged at C/18, C/40 and C/150 were recorded in situ, and phase localization and relative intensity of the peaks were compared. For cells discharged at the C/18 rate, although α and β phases were distributed uniformly within the LVO500 electrode, they were localized on two sides of the LVO300 electrode. Discharging rates of C/40 and C/150 led to homogeneous β phase formation in both LVO500 and LVO300 electrodes. Furthermore, the phase distribution as a function of position and (de)lithiation extent was mapped operando as the LVO500 cell was (de)lithiated. The operando data indicate that (1) the lithiation reaction initiated from the side of the electrode facing the Li anode and proceeded towards the side facing the steel can, (2) during discharge the phase transformation from a Li-poor to a Li-rich α phase and the formation of a β phase can proceed simultaneously in the electrode after the first formation of a β phase, and (3) the structural evolution occurring during charging is not the reverse of that during discharge and takes place homogenously throughout the electrode.
.jpg)




















































![N-PROPYL-3-METHYLPYRIDINIUM BIS(TRIFLUOROMETHYLSULFONYL)IMIDE, 99% [PMPIM]](http://img.cochemist.com/ccimg/817600/817575-06-7.png)
![N-PROPYL-3-METHYLPYRIDINIUM BIS(TRIFLUOROMETHYLSULFONYL)IMIDE, 99% [PMPIM]](http://img.cochemist.com/ccimg/817600/817575-06-7_b.png)