Co-reporter:Dr. Uttam Chakraborty;Dr. Serhiy Demeshko; Dr. Franc Meyer;M. Sc. Christophe Rebreyend; Dr. Bas de Bruin; Dr. Mihail Atanasov; Dr. Frank Neese;Dr. Bernd Mühldorf; Dr. Robert Wolf
Angewandte Chemie 2017 Volume 129(Issue 27) pp:8107-8112
Publication Date(Web):2017/06/26
DOI:10.1002/ange.201702454
AbstractDer 15-Valenzelektronen-Komplex [CpArFe(IiPr2Me2)] (1, CpAr=C5(C6H4-4-Et)5; IiPr2Me2=1,3-Diisopropyl-4,5-dimethylimidazolin-2-yliden) ist ausgehend von der Eisen(II)-Vorstufe [CpArFe(μ-Br)]2 in hohen Ausbeuten zugänglich. 57Fe-Mößbauer- und EPR-Daten, magnetische Messungen und Ab-initio-Ligandenfeldrechnungen deuten auf einen S=3/2-Grundzustand mit einer großen negativen Nullfeldaufspaltung hin. Infolgedessen weist 1 magnetische Anisotropie mit einer effektiven Spinumkehrbarriere von Ueff=64 cm−1 auf. Zudem katalysiert 1 die Dehydrierung von Dimethylamin-Boran unter Bildung von Tetramethyl-1,3-diaza-2,4-diboretan unter milden Bedingungen.
Co-reporter:Dr. Uttam Chakraborty;Dr. Serhiy Demeshko; Dr. Franc Meyer;M. Sc. Christophe Rebreyend; Dr. Bas de Bruin; Dr. Mihail Atanasov; Dr. Frank Neese;Dr. Bernd Mühldorf; Dr. Robert Wolf
Angewandte Chemie International Edition 2017 Volume 56(Issue 27) pp:7995-7999
Publication Date(Web):2017/06/26
DOI:10.1002/anie.201702454
AbstractThe 15 valence-electron iron(I) complex [CpArFe(IiPr2Me2)] (1, CpAr=C5(C6H4-4-Et)5; IiPr2Me2=1,3-diisopropyl-4,5-dimethylimidazolin-2-ylidene) was synthesized in high yield from the FeII precursor [CpArFe(μ-Br)]2. 57Fe Mössbauer and EPR spectroscopic data, magnetic measurements, and ab initio ligand-field calculations indicate an S= 3/2 ground state with a large negative zero-field splitting. As a consequence, 1 features magnetic anisotropy with an effective spin-reversal barrier of Ueff=64 cm−1. Moreover, 1 catalyzes the dehydrogenation of N,N-dimethylamine–borane, affording tetramethyl-1,3-diaza-2,4-diboretane under mild conditions.
Co-reporter:Robert Wolf;Markus Plois;Alexer Hepp
European Journal of Inorganic Chemistry 2017 Volume 2017(Issue 40) pp:4814-4814
Publication Date(Web):2017/11/02
DOI:10.1002/ejic.201701166
No abstract is available for this article.
Co-reporter:M. Sc. Peter Coburger;Dr. Serhiy Demeshko;M. Sc. Christian Rödl; Dr. Evamarie Hey-Hawkins; Dr. Robert Wolf
Angewandte Chemie 2017 Volume 129(Issue 50) pp:16087-16091
Publication Date(Web):2017/12/11
DOI:10.1002/ange.201709140
AbstractDie oxidative P-P-Bindungsaddition eines ortho-Carboran-substituierten 1,2-Diphosphetans an Cobalt(−I) in [K(thf)0.2][Co(η4-cod)2)] (cod=1,5-Cycloctadien) ermöglichte die Darstellung des ersten homoleptischen Cobalt(III)-Phosphanidokomplexes [K(thf)4][Co{1,2-(PtBu)2C2B10H12}2] (1). Diese Verbindung ist ein seltenes Beispiel eines verzerrt tetraedrisch koordinierten 3d6-Komplexes mit einem Low-spin-Grundzustand. Magnetische Messungen ergaben, dass 1 zwischen 2 und 270 K im Festkörper und bei 298 K in einer [D8]THF-Lösung diamagnetisch ist. Dichtefunktionalrechnungen zeigten, dass der diamagnetische Grundzustand durch die starken σ-Donor- und die moderaten π-Donoreigenschaften der Bisphosphanidoliganden verursacht wird.
Co-reporter:Stefan Pelties, Andreas W. Ehlers and Robert Wolf
Chemical Communications 2016 vol. 52(Issue 39) pp:6601-6604
Publication Date(Web):11 Apr 2016
DOI:10.1039/C6CC01572G
A new reaction mode for bicyclo[1.1.0]tetraphosphabutanes is reported. The CS and CN bonds of phenyl isothiocyanate reversibly insert into a P–P bond of [{CpNi(IMes)}2(μ-η1:η1-P4)] (1Mes, IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene), forming isomers 2a and 2b. X-ray crystallography and 31P{1H} NMR spectroscopy revealed similar bicyclo[3.1.0]heterohexane structures for these compounds.
Co-reporter:Stefan Pelties, Emma Carter, Andrea Folli, Mary F. Mahon, Damien M. Murphy, Michael K. Whittlesey, and Robert Wolf
Inorganic Chemistry 2016 Volume 55(Issue 21) pp:11006
Publication Date(Web):October 12, 2016
DOI:10.1021/acs.inorgchem.6b01540
Potassium graphite reduction of the half-sandwich Ni(II) ring-expanded diamino/diamidocarbene complexes CpNi(RE-NHC)Br gave the Ni(I) derivatives CpNi(RE-NHC) (where RE-NHC = 6-Mes (1), 7-Mes (2), 6-MesDAC (3)) in yields of 40%–50%. The electronic structures of paramagnetic 1–3 were investigated by CW X-/Q-band electron paramagnetic resonance (EPR) and Q-band 1H electron nuclear double resonance (ENDOR) spectroscopy. While small variations in the g-values were observed between the diaminocarbene complexes 1 and 2, pronounced changes in the g-values were detected between the almost isostructural species (1) and diamidocarbene species (3). These results highlight the sensitivity of the EPR g-tensor to changes in the electronic structure of the Ni(I) centers generated by incorporation of heteroatom substituents onto the backbone ring positions. Variable-temperature EPR analysis also revealed the presence of a second Ni(I) site in 3. The experimental g-values for these two Ni(I) sites detected by EPR in frozen solutions of 3 are consistent with resolution on the EPR time scale of the disordered components evident in the X-ray crystallographically determined structure and the corresponding density functional theory (DFT)-calculated g-tensor. Q-band 1H ENDOR measurements revealed a small amount of unpaired electron spin density on the Cp rings, consistent with the calculated SOMO of complexes 1–3. The magnitude of the 1H A values for 3 were also notably larger, compared to 1 and 2, again highlighting the influence of the diamidocarbene on the electronic properties of 3.
Co-reporter:Uttam Chakraborty, Bernd Mühldorf, Niels J. C. van Velzen, Bas de Bruin, Sjoerd Harder, and Robert Wolf
Inorganic Chemistry 2016 Volume 55(Issue 6) pp:3075-3078
Publication Date(Web):March 7, 2016
DOI:10.1021/acs.inorgchem.5b02979
The 17 valence electron (VE) open-shell nickel gallanediyl complex [CpArNi{Ga(nacnac)}] (3, Ar = C5(C6H4-4-Et)5, nacnac = HC[C(Me)N(C6H3-2,6-iPr2)]2), having an unsupported Ni–Ga bond, was synthesized from [CpArNi(μ-Br)]2 (1) by reducing the adduct [CpArNi(μ-Br){Ga(nacnac)}] (2) or, alternatively, trapping the “CpArNiI” synthon with Ga(nacnac); spectroscopic and DFT studies showed that the single unpaired electron in 3 resides mainly at the Ni center.
Co-reporter:Uttam Chakraborty, Moritz Modl, Bernd Mühldorf, Michael Bodensteiner, Serhiy Demeshko, Niels J. C. van Velzen, Manfred Scheer, Sjoerd Harder, and Robert Wolf
Inorganic Chemistry 2016 Volume 55(Issue 6) pp:3065-3074
Publication Date(Web):March 7, 2016
DOI:10.1021/acs.inorgchem.5b02896
The preparation of new stable half-sandwich transition metal complexes, having a bulky cyclopentadienyl ligand C5(C6H4-4-Et)5 (CpAr1) or C5(C6H4-4-nBu)5 (CpAr2), is reported. The tetrahydrofuran (THF) adduct [CpAr1Fe(μ-Br)(THF)]2 (1a) was synthesized by reacting K[CpAr1] with [FeBr2(THF)2] in THF, and its molecular structure was determined by X-ray crystallography. Complex 1a easily loses its coordinated THF molecules under vacuum to form the solvent-free complex [CpAr1Fe(μ-Br)]2 (1b). The analogous complexes [CpAr1Co(μ-Br)]2 (2), [CpAr1Ni(μ-Br)]2 (3), and [CpAr2Ni(μ-Br)]2 (4) were synthesized from CoBr2 and [NiBr2(1,2-dimethoxyethane)]. The mononuclear, low-spin cobalt(III) and nickel(III) complexes [CpAr2MI2] (5, M = Co; 6, M = Ni) were prepared by reacting the radical CpAr2 with NiI2 and CoI2. The complexes were characterized by NMR and UV–vis spectroscopies and by elemental analyses. Single-crystal X-ray structure analyses revealed that the dimeric complexes 1a, 1b, and 3 have a planar M2Br2 core, whereas 2 and 4 feature a puckered M2Br2 ring.
Co-reporter:Christian M. Hoidn and Robert Wolf
Dalton Transactions 2016 vol. 45(Issue 21) pp:8875-8884
Publication Date(Web):22 Apr 2016
DOI:10.1039/C6DT00336B
A novel, versatile route to phosphorus- and carbon-substituted η5-phosphacyclohexadienyl complexes was developed. Reaction of the anionic 2,4,6-triphenylphosphinine iron complex [K([18]crown-6)(thf)2][Cp*Fe(PC5Ph3H2)] (1) with selected main group element electrophiles afforded the new complexes [Cp*Fe(2-endo-H-PC5Ph3H2)] (endo-3), [Cp*Fe(2-exo-H-PC5Ph3H2)] (exo-3), [Cp*Fe(1-Me-PC5Ph3H2)] (4), [Cp*Fe(1-Me3Si-PC5Ph3H2)] (5), [Cp*Fe(1-PPh2-PC5Ph3H2)] (6) and [Cp*Fe(2-BCat-PC5Ph3H2)] (7, BCat = 2-benzo[d][1,3,2]dioxaborol-2-yl). Initial attack of the electrophile at phosphorus was observed, leading to a P-substitued phosphinine ligand. A subsequent rearragement occured in some cases, resulting in C-substituted phosphinine complexes endo-3, exo-3 and 7. The new complexes were characterized by 1H, 31P{1H}, and 13C{1H} NMR spectroscopy, UV-vis spectroscopy and elemental analysis; their molecular structures were determined by X-ray crystallography.
Co-reporter:Christian Rödl
European Journal of Inorganic Chemistry 2016 Volume 2016( Issue 5) pp:736-742
Publication Date(Web):
DOI:10.1002/ejic.201500583
Abstract
Complexes [(C4Me4)Co(CO)2{Co(P2C2tBu2)2}] (1, C4Me4 = tetramethylcyclobutadiene) and [CpNi{Co(P2C2tBu2)2}(PPh3)] (2, Cp = cyclopentadienyl) were synthesized by transmetalating [Tl(thf)2{Co(P2C2tBu2)2}] with [(C4Me4)Co(CO)2I] and [CpNiBr(PPh3)]. Compounds 1 and 2 were fully characterized by X-ray crystallography, multinuclear NMR, UV/Vis, and IR spectroscopy, and elemental analysis. Their molecular structures show σ-coordination of one phosphorus atom of the [Co(P2C2tBu2)2]– anion to the second metal atom (cobalt or nickel). Time-dependent density functional theory (TD-DFT) calculations were applied to gain insight into the electronic structures and the nature of the electronic transitions observed in the UV/Vis spectra. Cyclic voltammetry studies revealed the similar redox behavior of 1 and 2, which are reversibly oxidized to the corresponding monocations 1+ and 2+ with retention of the dinuclear structures. The cyclic voltammograms furthermore show that 1 and 2 decompose upon reduction with cleavage of the dinuclear structures into the free [Co(P2C2tBu2)2]– anion and further uncharacterized products.
Co-reporter:Stefan Pelties and Robert Wolf
Organometallics 2016 Volume 35(Issue 16) pp:2722-2727
Publication Date(Web):August 8, 2016
DOI:10.1021/acs.organomet.6b00473
The nickel(I) complex [(η5-Cp)Ni(IPr)] (Cp = cyclopentadienyl, IPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin- 2-ylidene) reacts with phenyl isothiocyanate to afford the new heteronickelacycle [(IPr)NiSC(NPh)N(Ph)CS] (1), which contains the novel bidentate and dianionic ligand {SC(NPh)N(Ph)CS}2–. Complex 1 is also obtained from the nickel(0) compound [(IPr)Ni(styrene)2] with two equivalents of PhNCS. The γ-lactam-nickelacycle [(IPr)NiN(Ph)C(O)CH2CH(Ph) (3) is formed from [(IPr)Ni(styrene)2] and one equivalent of phenyl isocyanate. Complex 3 is a rare example of a nickel(II) complex with a T-shaped structure. Compounds 1 and 3 were characterized by X-ray crystallography, 1H and 13C NMR spectroscopy, UV–vis spectroscopy, and elemental analysis.
Co-reporter:Daniel Paul, Bernhard Beiring, Markus Plois, Nuria Ortega, Slawomir Kock, Danny Schlüns, Johannes Neugebauer, Robert Wolf, and Frank Glorius
Organometallics 2016 Volume 35(Issue 20) pp:3641-3646
Publication Date(Web):October 10, 2016
DOI:10.1021/acs.organomet.6b00702
This study describes the structural investigation of a highly versatile ruthenium-NHC (N-heterocyclic carbene) catalyst complex, which has been established for the asymmetric hydrogenation of various aromatic compounds. A complex containing an unusual doubly deprotonated NHC ligand was isolated and identified as the precatalyst to this complex. When its subsequent reactivity was monitored, two additional precatalysts, featuring partially hydrogenated naphthyl substituents, were characterized spectroscopically. Ligand hydrogenation appears to be a key activation process en route to the active catalyst.
Co-reporter:M.Sc. Bernd Mühldorf ;Dr. Robert Wolf
Angewandte Chemie International Edition 2016 Volume 55( Issue 1) pp:427-430
Publication Date(Web):
DOI:10.1002/anie.201507170
Abstract
A mixture of the photocatalyst riboflavin tetraacetate (RFT) and the biomimetic non-heme iron complex [Fe(TPA)(MeCN)2](ClO4)2 (TPA=tris(2-pyridylmethyl)amine) efficiently catalyzes the visible-light-driven aerobic oxidation of alkyl benzenes to ketones and carboxylic acids. An RFT-catalyzed photocycle and the independent action of the iron complex as a catalyst for H2O2 disproportionation and alkyl benzene oxygenation ensure high yields and selectivities.
Co-reporter:M.Sc. Bernd Mühldorf ;Dr. Robert Wolf
Angewandte Chemie 2016 Volume 128( Issue 1) pp:437-441
Publication Date(Web):
DOI:10.1002/ange.201507170
Abstract
Eine Mischung von Riboflavintetraacetat (RFT) mit dem Nicht-Häm-Eisenkomplex [Fe(TPA)(MeCN)2](ClO4)2 (TPA=Tris(2-pyridylmethyl)amin) katalysiert effizient die aerobe Photooxidation von Alkylbenzolen zu Ketonen und Carbonsäuren. Hohe Ausbeuten und Selektivitäten werden durch den flavinkatalysierten Photoredoxzyklus und die davon unabhängige katalytische Wirkung des Eisenkomplexes in der H2O2-Disproportionierung und der Alkylbenzol-Oxygenierung erreicht.
Co-reporter:M.Sc. Thea Hering;M.Sc. Bernd Mühldorf;Dr. Robert Wolf;Dr. Burkhard König
Angewandte Chemie 2016 Volume 128( Issue 17) pp:5428-5431
Publication Date(Web):
DOI:10.1002/ange.201600783
Abstract
Chlorierte Arene sind Bestandteil vieler Pharmazeutika, Agrochemikalien und Polymere sowie wichtige Startmaterialien für übergangsmetallkatalysierte Kreuzkupplungen. Für ihre Herstellung nutzen Chemiker meist Chlorgas oder elektropositive Chlorierungsreagentien. Die Natur hingegen chloriert ausgehend von Chloridionen mithilfe von FAD-abhängigen Halogenasen und O2 als Oxidationsmittel. Wir beschreiben hier eine diesem enzymatischen Prozess nachempfundene photokatalytische oxidative Chlorierung unter Verwendung des organischen Farbstoffes Riboflavintetraacetat. Dieses chemische Verfahren ermöglicht die Umsetzung von Arenen innerhalb eines definierten Redoxbereichs und hat eine größere Substratbreite als die Aktivierung durch eine substratspezifische Enzymbindungstasche.
Co-reporter:Thea Hering;Bernd Mühldorf;Burkhard König
Angewandte Chemie International Edition 2016 Volume 55( Issue 17) pp:5342-5345
Publication Date(Web):
DOI:10.1002/anie.201600783
Abstract
Chlorine gas or electropositive chlorine reagents are used to prepare chlorinated aromatic compounds, which are found in pharmaceuticals, agrochemicals, and polymers, and serve as synthetic precursors for metal-catalyzed cross-couplings. Nature chlorinates with chloride anions, FAD-dependent halogenases, and O2 as the oxidant. A photocatalytic oxidative chlorination is described based on the organic dye riboflavin tetraacetate mimicking the enzymatic process. The chemical process allows within the suitable arene redox potential window a broader substrate scope compared to the specific activation in the enzymatic binding pocket.
Co-reporter:Uttam Chakraborty, Franziska Urban, Bernd Mühldorf, Christophe Rebreyend, Bas de Bruin, Niels van Velzen, Sjoerd Harder, and Robert Wolf
Organometallics 2016 Volume 35(Issue 11) pp:1624-1631
Publication Date(Web):May 2, 2016
DOI:10.1021/acs.organomet.6b00084
A reactive “CpArNiI” surrogate (CpAr = C5(C6H4-4-Et)5) is accessible via the reduction of the dimer [CpArNi(μ-Br)]2 with two equivalents of KC8. A trapping reaction with TEMPO afforded the new nickel(II) complex [CpArNi(η2-TEMPO)] (3), while the addition of N-heterocyclic carbenes gave the new nickel(I) radicals [CpArNi(IPr)] (4a, IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) and [CpArNi(IiPr2Me2)] (4b, IiPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene). EPR spectra supported by DFT calculations on 4a and 4b indicate that the spin density mainly resides at the nickel center. The reaction of the “CpArNi(I) source” with yellow sulfur gave the Ni2S6 complex [(CpArNi)2(μ-S6)] (5); the “subselenide” [(CpArNi)2(μ-Se2)] (6) was formed in the analogous reaction with grey selenium. All new complexes were characterized by NMR, EPR, and UV–vis spectroscopy; their molecular structures were determined by X-ray crystallography.
Co-reporter:Bernd Mühldorf and Robert Wolf
Chemical Communications 2015 vol. 51(Issue 40) pp:8425-8428
Publication Date(Web):05 Feb 2015
DOI:10.1039/C5CC00178A
The enhanced reduction potential of riboflavin tetraacetate coordinating to scandium triflate enables the challenging photocatalytic C–H oxidation of electron-deficient alkylbenzenes and benzyl alcohols.
Co-reporter:Babak Rezaei Rad, Uttam Chakraborty, Bernd Mühldorf, Julian A. W. Sklorz, Michael Bodensteiner, Christian Müller, and Robert Wolf
Organometallics 2015 Volume 34(Issue 3) pp:622-635
Publication Date(Web):January 29, 2015
DOI:10.1021/om501161y
The potassium salt [K([18]crown-6)(THF)2][Cp*Fe(η4-2,4,6-triphenylphosphinine)}] (K1, Cp* = C5Me5) can be isolated in 68% yield by reacting the anionic naphthalene complex [K([18]crown-6){Cp*Fe(η4-C10H8)}] (C10H8 = naphthalene) with 2,4,6-triphenylphosphinine. Compound K1 reacts with water to afford [K([18]-crown-6)]{Cp*Fe(η4-2,4,6-triphenyl-2,3-dihydrophosphinine 1-oxide)}] (K2) with a novel 2,3-dihydrophosphinine 1-oxide ligand. Oxidation of K1 with one equivalent of ferrocenium hexafluorophosphate yields the P–P-bonded diphosphinine complex [Cp*Fe(η5-2,4,6-triphenylphosphinine)]2 (3), while the iodide salt [Cp*Fe(η6-2,4,6-triphenylphosphinine)]I (4) can be obtained by reacting K1 with one equivalent of iodine. Reactions of 4 with LiNMe2, Cp*Li, LiBHEt3, and Ga(nacnacDipp) (nacnacDipp = HC{C(Me)N(C6H3-2,6-iPr2)}2) afford [Cp*Fe(η5-1-dimethylamino-2,4,6-triphenylphosphacyclohexadienyl)] (5), [Cp*Fe(η5-1-(η1-Cp*)-2,4,6-triphenylphosphacyclohexadienyl)] (6), [Cp*Fe(η5-1-hydro-2,4,6-triphenylphosphacyclohexadienyl)] (7), and [Cp*Fe((η5-1-{Ga(nacnacDipp)I}-2,4,6-triphenylphosphacyclohexadienyl] (8). The molecular structures of 5–8 display η5-coordinated λ3σ3-phosphinine anions. All new complexes were fully characterized by spectroscopic techniques (1H, 13C, and 31P NMR, UV–vis, and IR spectroscopy), elemental analysis, and X-ray crystallography. The electronic structures of these new phosphinine complexes were investigated theoretically at the DFT level, using molecular orbital and population analyses. The nature of the electronic transitions observed in the UV–vis spectra was analyzed using TD-DFT calculations.
Co-reporter:Stefan Pelties, Dirk Herrmann, Bas de Bruin, František Hartl and Robert Wolf
Chemical Communications 2014 vol. 50(Issue 53) pp:7014-7016
Publication Date(Web):14 May 2014
DOI:10.1039/C4CC02601B
The reaction of the 17e nickel(I) radical [CpNi(IDipp)] (1, IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene) with P4 results in a nickel tetraphosphide [{CpNi(IDipp)}2(μ-η1:η1-P4)] with a butterfly-P42− ligand; related chalcogenides [{CpNi(IDipp)}2(μ-E2)] (E = S, Se, Te) and [{CpNi(IDipp)}2(μ-E3)] (E = S, Se) are formed with S8, Se∞ and Te∞.
Co-reporter:Babak Rezaei Rad, Dirk Herrmann, Christophe Lescop and Robert Wolf
Dalton Transactions 2014 vol. 43(Issue 11) pp:4247-4250
Publication Date(Web):03 Dec 2013
DOI:10.1039/C3DT52699B
The salt [K([18]crown-6){Cp*Fe(η4-C4py4)}] (K1, py = 2-pyridyl, Cp* = C5Me5) is accessible by the reaction of an iron(0) naphthalene precursor and bis(2-pyridyl)acetylene. Cyclic voltammetry and preparative investigations demonstrate the electron-rich nature of K1, which is reversibly oxidized to neutral [Cp*Fe(η4-C4py4)] (1) at a low potential. The first coordination studies with iron(II) and zinc(II) chloride show that all four 2-pyridyl units may be employed for metal coordination.
Co-reporter:Jennifer Malberg;Thomas Wieg;Hellmut Eckert;Michael Bodensteiner
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 10) pp:1638-1651
Publication Date(Web):
DOI:10.1002/ejic.201301173
Abstract
1,3-Diphosphacyclobutadiene complexes have previously been employed as metalloligands to a few transition metals such as tungsten, cobalt, rhodium, platinum, and gold. Here, we describe the synthesis and the structural and spectroscopic characterization of novel copper(I) and silver(I) complexes with the sandwich anions [Co(η4-P2C2R2)2]– (R = tBu or tPent). Reactions of [K(thf)3{Co(P2C2tPent2)2}] and [K(thf)2{Co(P2C2tBu2)2}] with CuCl(PPh3)/PPh3 yielded mononuclear copper(I) complexes [Cu{Co(P2C2R2)2}(PPh3)2] [R = tPent (1), tBu (2)]. The salt [Cu(PMe3)4][Co(P2C2tPent2)2] (3), which consists of a [Cu(PMe3)4]+ cation and a noncoordinated [Co(P2C2tPent2)2]– anion, was obtained when [K(thf)3{Co(P2C2tPent2)2}] was treated with CuCl(PPh3) in the presence of an excess amount of PMe3. Salt metathesis of [K(thf)3{Co(P2C2tPent2)2}] with AgCl and AgSbF6 in the presence of PMe3 afforded novel silver(I) complexes [Ag{Co(P2C2tPent2)2}(PMe3)3] (4), [Ag{Co(P2C2tPent2)2}(PMe3)2] (5), and [Ag2{Co(P2C2tPent2)2}(PMe3)5]SbF6 (6). The molecular structures of 1, 2, 5, and 6 were determined by X-ray crystallography. The solution behavior of 1–6 was analyzed by variable-temperature 1H, 31P{1H}, and 13C{1H} NMR spectroscopy in [D8]THF. 31P solution and solid-state NMR spectra, aided by two-dimensional dipolar and/or J-based correlation spectroscopy, support the proposed structures and allowed the compositions of polycrystalline precipitated products to be analyzed. Furthermore, characteristic 31P chemical shifts and indirect spin–spin coupling constants offer a clear distinction between different bonding patterns realized in this class of compounds.
Co-reporter:Dr. Jennifer Malberg;Dr. Michael Bodensteiner;MSc Daniel Paul;Dr. Thomas Wieg;Dr. Hellmut Eckert;Dr. Robert Wolf
Angewandte Chemie 2014 Volume 126( Issue 10) pp:2812-2816
Publication Date(Web):
DOI:10.1002/ange.201309493
Abstract
Molekulare Quadrate gehören zu den bekanntesten Typen von supramolekularen Architekturen. Phosphorhaltige metallorganische Baueinheiten fanden in derartigen Strukturen bisher allerdings keine Verwendung. Hier beschreiben wir die Synthese eines molekularen Quadrats aus vier Gold- und vier Cobaltatomen, das durch Selbstorganisation von anionischen 1,3-Diphosphacyclobutadien-Komplexen und Gold(I)-Kationen gebildet wird. Die röntgenkristallographische Bestimmung der Molekülstruktur der Verbindung [Au{Co(P2C2tBu2)2}]4 (1) wird durch 31P- und 13C-NMR-spektroskopische Untersuchungen im Festkörper komplementiert. DFT-Rechnungen bestätigen die Zuordnung der 31P- und 13C-NMR-Signale.
Co-reporter:M.Sc. Dominik Gärtner;Dr. Alice Welther;Dr. Babak Rezaei Rad;Dr. Robert Wolf;Dr. Axel JacobivonWangelin
Angewandte Chemie 2014 Volume 126( Issue 14) pp:3796-3800
Publication Date(Web):
DOI:10.1002/ange.201308967
Abstract
75 Jahre nach der Entdeckung der Hydroformylierung erleben Cobalt-Katalysatoren eine Renaissance in Hydrierungen. Wir haben Arenmetallate untersucht, in denen die niedervalente Metallspezies nicht durch Heteroatom-Liganden, sondern durch einfache π-Kohlenwasserstoffe stabilisiert ist. Kaliumbis(anthracen)cobaltat 1 und -ferrat 2 sind synthetische Vorstufen für quasi-“nackte” anionische Metallspezies. Eine Aggregation wird durch (labile) Koordination der in Hydrierungen vorhandenen π-Akzeptoren (Olefine, Arene, Carbonylverbindungen) effektiv unterdrückt. Kinetische Studien, NMR-Spektroskopie und Vergiftungsexperimente in Alkenhydrierungen bestätigen die Bildung eines homogenen Katalysators aus 1, der durch Ligandenaustausch gebildet und durch Koordination an Alkene stabilisiert wird. Das zugrundeliegende Katalysatorkonzept ist komplementär zur Verwendung von Komplexen mit Heteroatom-Donorliganden in reduktiven Prozessen.
Co-reporter:Dr. Jennifer Malberg;Dr. Michael Bodensteiner;M.Sc. Daniel Paul;Dr. Thomas Wieg;Dr. Hellmut Eckert;Dr. Robert Wolf
Angewandte Chemie International Edition 2014 Volume 53( Issue 10) pp:2771-2775
Publication Date(Web):
DOI:10.1002/anie.201309493
Abstract
Molecular squares are among the most common supramolecular architectures, but phospha-organometallic complexes have not been used as building blocks for this type of structure. Herein we describe the formation of the molecular square [Au{Co(P2C2tBu2)2}]4 (1) by the self-assembly of anionic 1,3-diphosphacyclobutadiene cobalt complexes and gold(I) cations. The X-ray crystallographic determination of the molecular structure of 1 is complemented by solid-state 31P and 13C NMR investigations. High-level DFT calculations confirm the assignment of the 31P and 13C NMR resonances.
Co-reporter:M.Sc. Dominik Gärtner;Dr. Alice Welther;Dr. Babak Rezaei Rad;Dr. Robert Wolf;Dr. Axel JacobivonWangelin
Angewandte Chemie International Edition 2014 Volume 53( Issue 14) pp:3722-3726
Publication Date(Web):
DOI:10.1002/anie.201308967
Abstract
75 years after the discovery of hydroformylation, cobalt catalysts are now undergoing a renaissance in hydrogenation reactions. We have evaluated arene metalates in which the low-valent metal species is—conceptually different from heteroatom-based ligands—stabilized by π coordination to hydrocarbons. Potassium bis(anthracene)cobaltate 1 and -ferrate 2 can be viewed as synthetic precursors of quasi-“naked” anionic metal species; their aggregation is effectively impeded by (labile) coordination to the various π acceptors present in the hydrogenation reactions of unsaturated molecules (alkenes, arenes, carbonyl compounds). Kinetic studies, NMR spectroscopy, and poisoning studies of alkene hydrogenations support the formation of a homogeneous catalyst derived from 1 which is stabilized by the coordination of alkenes. This catalyst concept complements the use of complexes with heteroatom donor ligands for reductive processes.
Co-reporter:Markus Plois;Waldemar Hujo;Stefan Grimme
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 17) pp:3039-3048
Publication Date(Web):
DOI:10.1002/ejic.201300233
Abstract
The utility of the fac-[RuH3(PR3)3]– anion (R = Ph, C6H4-4-Me) for the preparation of new oligonuclear transition metal polyhydrides has been examined. The lithium salts [Li(thf)x{Ru(μ-H)3(PPh3)3}] {1: R = Ph, x = 3; 1′: R = C6H4-4-Me (Tol), x = 2.5} react with [Cp*RuCl]4 (Cp* = C5Me5), ZnCl2, and CuCl(SMe2) to form new oligonuclear polyhydrido complexes. The compounds [Cp*Ru(μ-H)3Ru(PR3)3] (2: R = Ph, 2′: R = Tol), [Zn{Ru(μ-H)3(PPh3)3}2] (3), and [Cu2{Ru(μ-H)3(PPh3)3}2] (4) were synthesized and characterized by multinuclear NMR and IR spectroscopy, and microanalysis. The molecular structures were determined by X-ray crystallography. Density functional theory calculations at the PBE-D3/def2-TZVP level support the proposed structures of the new polyhydride complexes. The impact of intramolecular London dispersion interactions on the optimized geometries is discussed.
Co-reporter:Markus Plois;Waldemar Hujo;Dr. Stefan Grimme;Christian Schwickert;Dr. Eckhard Bill;Dr. Bas deBruin;Dr. Rainer Pöttgen;Dr. Robert Wolf
Angewandte Chemie International Edition 2013 Volume 52( Issue 4) pp:1314-1318
Publication Date(Web):
DOI:10.1002/anie.201205209
Co-reporter:Jennifer Malberg, Elizabeth Lupton, Eva-Maria Schnöckelborg, Bas de Bruin, Jörg Sutter, Karsten Meyer, František Hartl, and Robert Wolf
Organometallics 2013 Volume 32(Issue 20) pp:6040-6052
Publication Date(Web):September 10, 2013
DOI:10.1021/om4005862
The dissymmetrical naphthalene-bridged complexes [Cp′Fe(μ-C10H8)FeCp*] (3; Cp* = η5-C5Me5, Cp′ = η5-C5H2-1,2,4-tBu3) and [Cp′Fe(μ-C10H8)RuCp*] (4) were synthesized via a one-pot procedure from FeCl2(thf)1.5, Cp′K, KC10H8, and [Cp*FeCl(tmeda)] (tmeda = N,N,N′,N′-tetramethylethylenediamine) or [Cp*RuCl]4, respectively. The symmetrically substituted iron ruthenium complex [Cp*Fe(μ-C10H8)RuCp*] (5) bearing two Cp* ligands was prepared as a reference compound. Compounds 3–5 are diamagnetic and display similar molecular structures, where the metal atoms are coordinated to opposite sides of the bridging naphthalene molecule. Cyclic voltammetry and UV/vis spectroelectrochemistry studies revealed that neutral 3–5 can be oxidized to monocations 3+–5+ and dications 32+–52+. The chemical oxidation of 3 and 4 with [Cp2Fe]PF6 afforded the paramagnetic hexafluorophosphate salts [Cp′Fe(μ-C10H8)FeCp*]PF6 ([3]PF6) and [Cp′Fe(μ-C10H8)RuCp*]PF6 ([4]PF6), which were characterized by various spectroscopic techniques, including EPR and 57Fe Mössbauer spectroscopy. The molecular structure of [4]PF6 was determined by X-ray crystallography. DFT calculations support the structural and spectroscopic data and determine the compositions of frontier molecular orbitals in the investigated complexes. The effects of substituting Cp* with Cp′ and Fe with Ru on the electronic structures and the structural and spectroscopic properties are analyzed.
Co-reporter:Florian D. Henne, Eva-Maria Schnöckelborg, Kai-Oliver Feldmann, Jörg Grunenberg, Robert Wolf, and Jan J. Weigand
Organometallics 2013 Volume 32(Issue 22) pp:6674-6680
Publication Date(Web):April 9, 2013
DOI:10.1021/om4002268
A bridging chloride anion between two electrophilic phosphorus centers was observed for the first time in the molecular structure of the novel P–P bonded cation [LDipp2P2Cl3]+ (2+; LDipp = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene). Cation 2+ is prepared by the reduction of the monophosphorus species [LDippPCl2]+ (1+) with sodium. DFT calculations, AIM and mechanochemical (compliance constants) analyses are used to examine the bonding situation of this unusual species. The monochloro-substituted cation [LDipp2P2Cl]+ (3+) was likewise observed as a reduction product of 1+.
Co-reporter:Jennifer Malberg;Thomas Wieg;Dr. Hellmut Eckert;Dr. Michael Bodensteiner;Dr. Robert Wolf
Chemistry - A European Journal 2013 Volume 19( Issue 7) pp:2356-2369
Publication Date(Web):
DOI:10.1002/chem.201203606
Abstract
The synthesis and structural characterization of the first coordination compounds of bis(diphosphacyclobutadiene) cobaltate anions [M(P2C2R2)2]− is described. Reactions of the new potassium salts [K(thf)3{Co(η4-P2C2tPent2)2}] (1) and [K(thf)4{Co(η4-P2C2Ad2)2}] (2) with [AuCl(tht)] (tht=tetrahydrothiophene), [AuCl(PPh3)] and Ag[SbF6] afforded the complexes [Au{Co(P2C2tPent2)2}(PMe3)2] (3), [Au{Co(P2C2Ad2)2}]x (4), [Ag{Co(P2C2Ad2)2}]x (5), [Au(PMe3)4][Au{Co(P2C2Ad2)2}2] (6), [K([18]crown-6)(thf)2][Au{Co(P2C2Ad2)2}2] (7), and [K([18]crown-6)(thf)2][M{Co(P2C2Ad2)2}2] (8: M=Au 9: M=Ag) in moderate yields. The molecular structures of 2 and 3, and 6–9 were elucidated by X-ray crystallography. Complexes 4–9 were thoroughly characterized by 31P and 13C solid state NMR spectroscopy. The complexes [Au{Co(P2C2Ad2)2}]x (4) and [Ag{Co(P2C2Ad2)2}]x (5) exist as coordination polymers in the solid state. The linking mode between the monomeric units in the polymers is deduced. The soluble complexes 1–3, 6, and 7 were studied by multinuclear 1H-, 31P{1H}-, and 13C{1H} NMR spectroscopy in solution. Variable temperature NMR measurements of 3 and 6 in deuterated THF reveal the formation of equilibria between the ionic species [Au(PMe3)4]+, [Au(PMe3)2]+, [Co(P2C2R2)2]−, and [Au{Co(P2C2R2)2}2]− (R=tPent and Ad).
Co-reporter:Dipl.-Chem. Markus Plois;M.Sc. Waldemar Hujo;Dr. Stefan Grimme;Dipl.-Chem. Christian Schwickert;Dr. Eckhard Bill;Dr. Bas deBruin;Dr. Rainer Pöttgen;Dr. Robert Wolf
Angewandte Chemie 2013 Volume 125( Issue 4) pp:1352-1357
Publication Date(Web):
DOI:10.1002/ange.201205209
Co-reporter:Eva-Maria Schnöckelborg, Marat M. Khusniyarov, Bas de Bruin, František Hartl, Thorsten Langer, Matthias Eul, Stephen Schulz, Rainer Pöttgen, and Robert Wolf
Inorganic Chemistry 2012 Volume 51(Issue 12) pp:6719-6730
Publication Date(Web):May 25, 2012
DOI:10.1021/ic300366m
Naphthalene and anthracene transition metalates are potent reagents, but their electronic structures have remained poorly explored. A study of four Cp*-substituted iron complexes (Cp* = pentamethylcyclopentadienyl) now gives rare insight into the bonding features of such species. The highly oxygen- and water-sensitive compounds [K(18-crown-6){Cp*Fe(η4-C10H8)}] (K1), [K(18-crown-6){Cp*Fe(η4-C14H10)}] (K2), [Cp*Fe(η4-C10H8)] (1), and [Cp*Fe(η4-C14H10)] (2) were synthesized and characterized by NMR, UV–vis, and 57Fe Mössbauer spectroscopy. The paramagnetic complexes 1 and 2 were additionally characterized by electron paramagnetic resonance (EPR) spectroscopy and magnetic susceptibility measurements. The molecular structures of complexes K1, K2, and 2 were determined by single-crystal X-ray crystallography. Cyclic voltammetry of 1 and 2 and spectroelectrochemical experiments revealed the redox properties of these complexes, which are reversibly reduced to the monoanions [Cp*Fe(η4-C10H8)]− (1–) and [Cp*Fe(η4-C14H10)]− (2–) and reversibly oxidized to the cations [Cp*Fe(η6-C10H8)]+ (1+) and [Cp*Fe(η6-C14H10)]+ (2+). Reduced orbital charges and spin densities of the naphthalene complexes 1–/0/+ and the anthracene derivatives 2–/0/+ were obtained by density functional theory (DFT) methods. Analysis of these data suggests that the electronic structures of the anions 1– and 2– are best represented by low-spin FeII ions coordinated by anionic Cp* and dianionic naphthalene and anthracene ligands. The electronic structures of the neutral complexes 1 and 2 may be described by a superposition of two resonance configurations which, on the one hand, involve a low-spin FeI ion coordinated by the neutral naphthalene or anthracene ligand L, and, on the other hand, a low-spin FeII ion coordinated to a ligand radical L•–. Our study thus reveals the redox noninnocent character of the naphthalene and anthracene ligands, which effectively stabilize the iron atoms in a low formal, but significantly higher spectroscopic oxidation state.
Co-reporter:Eva-Maria Schnöckelborg;Franti&x161;ek Hartl;Thorsten Langer;Rainer Pöttgen
European Journal of Inorganic Chemistry 2012 Volume 2012( Issue 10) pp:1632-1638
Publication Date(Web):
DOI:10.1002/ejic.201200001
Abstract
Although there has been much interest in the chemistry of bimetallic transition metal complexes, compounds with naphthalene or anthracene as bridging ligands are still rare. In this article, we describe the synthesis of the homodinuclear iron complexes [Cp*Fe(μ-η4:η4-L)FeCp*] (1: L = C10H8, 2: L = C14H10; Cp* = η5-C5Me5). The complexes were characterized by 1H and 13C{1H} NMR, UV/Vis, and 57Fe Mössbauer spectroscopy, and their molecular structures were determined by X-ray crystallography. Both complexes are diamagnetic as a result of the strong magnetic coupling of the 17e FeI centers mediated by the polyarene bridge. An analysisof the redox behavior of 1 and 2 by cyclic voltammetry andUV/Vis spectroelectrochemistry shows that the complexes can be oxidized reversibly in two well-separated one-electron steps to the monocation [Cp*Fe(μ-L)FeCp*]+ and the dication [Cp*Fe(μ-L)FeCp*]2+. The reduction to the monoanion [Cp*Fe(μ-L)FeCp*]– was also observed.
Co-reporter:Markus Plois, Thomas Wiegand, and Robert Wolf
Organometallics 2012 Volume 31(Issue 24) pp:8469-8477
Publication Date(Web):November 1, 2012
DOI:10.1021/om300705x
New anionic ruthenium(II) aluminohydrides were synthesized from [Cp*RuCl]4, LiAlH4, and lithium alkyls. The reaction of [Cp*RuCl]4 with 8 equiv of LiAlH4 afforded the lithium salt [Li(DME)3][Cp*2Ru2(μ-H)(μ-AlH4)2] (1) in 65% isolated yield. Reactions of [Cp*RuCl]4, LiAlH4, and RLi (R = Me, Et, iPr, nBu) in THF yielded the new compounds [Li5(THF)2Cp*6Ru6Al4Me4H19] (2) and [Li2(THF)2Cp*RuAl2R2H7)]2 (3–5, R = Et, iPr, tBu). The compounds were characterized by single-crystal X-ray crystallography, multinuclear liquid and solid-state NMR spectroscopy, IR spectroscopy, and microanalysis. The molecular structures of 1–5 display novel anionic ruthenium(II) aluminate frameworks. In the case of 2–5, these anions associate into unusual cage structures via Li–H interactions in the solid state. Variable-temperature 1H and 7Li NMR studies on the fluxional complex 5 in deuterated toluene show that the cage structure of this compound is retained at low temperature in solution.
Co-reporter:Bernd Mühldorf and Robert Wolf
Chemical Communications 2015 - vol. 51(Issue 40) pp:NaN8428-8428
Publication Date(Web):2015/02/05
DOI:10.1039/C5CC00178A
The enhanced reduction potential of riboflavin tetraacetate coordinating to scandium triflate enables the challenging photocatalytic C–H oxidation of electron-deficient alkylbenzenes and benzyl alcohols.
Co-reporter:Babak Rezaei Rad, Dirk Herrmann, Christophe Lescop and Robert Wolf
Dalton Transactions 2014 - vol. 43(Issue 11) pp:NaN4250-4250
Publication Date(Web):2013/12/03
DOI:10.1039/C3DT52699B
The salt [K([18]crown-6){Cp*Fe(η4-C4py4)}] (K1, py = 2-pyridyl, Cp* = C5Me5) is accessible by the reaction of an iron(0) naphthalene precursor and bis(2-pyridyl)acetylene. Cyclic voltammetry and preparative investigations demonstrate the electron-rich nature of K1, which is reversibly oxidized to neutral [Cp*Fe(η4-C4py4)] (1) at a low potential. The first coordination studies with iron(II) and zinc(II) chloride show that all four 2-pyridyl units may be employed for metal coordination.
Co-reporter:Christian M. Hoidn and Robert Wolf
Dalton Transactions 2016 - vol. 45(Issue 21) pp:NaN8884-8884
Publication Date(Web):2016/04/22
DOI:10.1039/C6DT00336B
A novel, versatile route to phosphorus- and carbon-substituted η5-phosphacyclohexadienyl complexes was developed. Reaction of the anionic 2,4,6-triphenylphosphinine iron complex [K([18]crown-6)(thf)2][Cp*Fe(PC5Ph3H2)] (1) with selected main group element electrophiles afforded the new complexes [Cp*Fe(2-endo-H-PC5Ph3H2)] (endo-3), [Cp*Fe(2-exo-H-PC5Ph3H2)] (exo-3), [Cp*Fe(1-Me-PC5Ph3H2)] (4), [Cp*Fe(1-Me3Si-PC5Ph3H2)] (5), [Cp*Fe(1-PPh2-PC5Ph3H2)] (6) and [Cp*Fe(2-BCat-PC5Ph3H2)] (7, BCat = 2-benzo[d][1,3,2]dioxaborol-2-yl). Initial attack of the electrophile at phosphorus was observed, leading to a P-substitued phosphinine ligand. A subsequent rearragement occured in some cases, resulting in C-substituted phosphinine complexes endo-3, exo-3 and 7. The new complexes were characterized by 1H, 31P{1H}, and 13C{1H} NMR spectroscopy, UV-vis spectroscopy and elemental analysis; their molecular structures were determined by X-ray crystallography.
Co-reporter:Stefan Pelties, Dirk Herrmann, Bas de Bruin, František Hartl and Robert Wolf
Chemical Communications 2014 - vol. 50(Issue 53) pp:NaN7016-7016
Publication Date(Web):2014/05/14
DOI:10.1039/C4CC02601B
The reaction of the 17e nickel(I) radical [CpNi(IDipp)] (1, IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene) with P4 results in a nickel tetraphosphide [{CpNi(IDipp)}2(μ-η1:η1-P4)] with a butterfly-P42− ligand; related chalcogenides [{CpNi(IDipp)}2(μ-E2)] (E = S, Se, Te) and [{CpNi(IDipp)}2(μ-E3)] (E = S, Se) are formed with S8, Se∞ and Te∞.
Co-reporter:Stefan Pelties, Andreas W. Ehlers and Robert Wolf
Chemical Communications 2016 - vol. 52(Issue 39) pp:NaN6604-6604
Publication Date(Web):2016/04/11
DOI:10.1039/C6CC01572G
A new reaction mode for bicyclo[1.1.0]tetraphosphabutanes is reported. The CS and CN bonds of phenyl isothiocyanate reversibly insert into a P–P bond of [{CpNi(IMes)}2(μ-η1:η1-P4)] (1Mes, IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene), forming isomers 2a and 2b. X-ray crystallography and 31P{1H} NMR spectroscopy revealed similar bicyclo[3.1.0]heterohexane structures for these compounds.







![Magnesium, bromo[4-(ethoxycarbonyl)phenyl]-](http://img.cochemist.com/ccimg/364400/364358-99-6.png)
![Magnesium, bromo[4-(ethoxycarbonyl)phenyl]-](http://img.cochemist.com/ccimg/364400/364358-99-6_b.png)
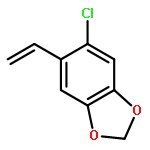
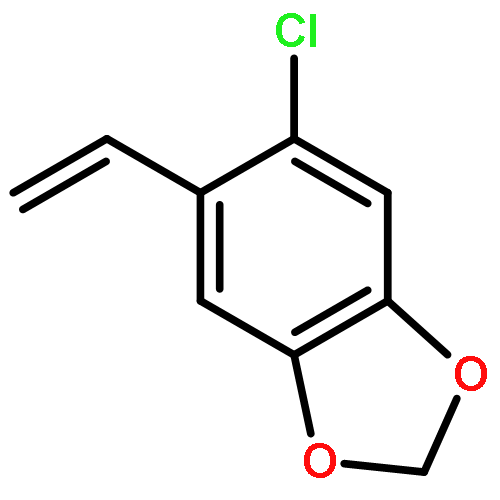
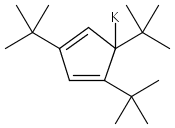



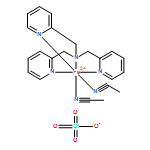


![Benzoic acid, 4-[(phenylmethyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/123900/123876-56-2.png)
![Benzoic acid, 4-[(phenylmethyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/123900/123876-56-2_b.png)
![Ruthenium, tetra-m3-chlorotetrakis[(1,2,3,4,5-h)-1,2,3,4,5-pentamethyl-2,4-cyclopentadien-1-yl]tetra-](http://img.cochemist.com/ccimg/113900/113860-07-4.png)
![Ruthenium, tetra-m3-chlorotetrakis[(1,2,3,4,5-h)-1,2,3,4,5-pentamethyl-2,4-cyclopentadien-1-yl]tetra-](http://img.cochemist.com/ccimg/113900/113860-07-4_b.png)
![1-chloro-4-[3-(4-chlorophenyl)but-1-enyl]benzene](http://img.cochemist.com/ccimg/108800/108708-08-3.png)
![1-chloro-4-[3-(4-chlorophenyl)but-1-enyl]benzene](http://img.cochemist.com/ccimg/108800/108708-08-3_b.png)
![Phosphine,(tricyclo[3.3.1.13,7]dec-1-ylmethylidyne)-](http://img.cochemist.com/ccimg/101100/101055-70-3.png)
![Phosphine,(tricyclo[3.3.1.13,7]dec-1-ylmethylidyne)-](http://img.cochemist.com/ccimg/101100/101055-70-3_b.png)
![[1,1'-Biphenyl]-4-ol, 4'-ethenyl-](http://img.cochemist.com/ccimg/93300/93249-93-5.png)
![[1,1'-Biphenyl]-4-ol, 4'-ethenyl-](http://img.cochemist.com/ccimg/93300/93249-93-5_b.png)












![Benzene, 1-methyl-3-[(1Z)-2-phenylethenyl]-](/data/chemimg/1779000/19466-29-6.png)
![Benzene, 1-methyl-3-[(1Z)-2-phenylethenyl]-](/data/chemimg/1779000/19466-29-6_b.png)








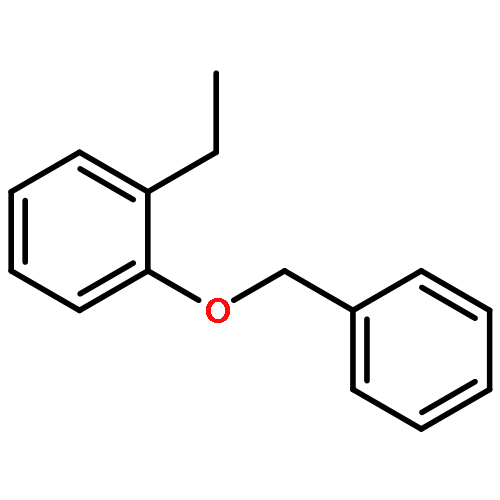
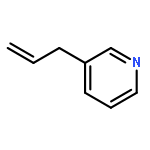
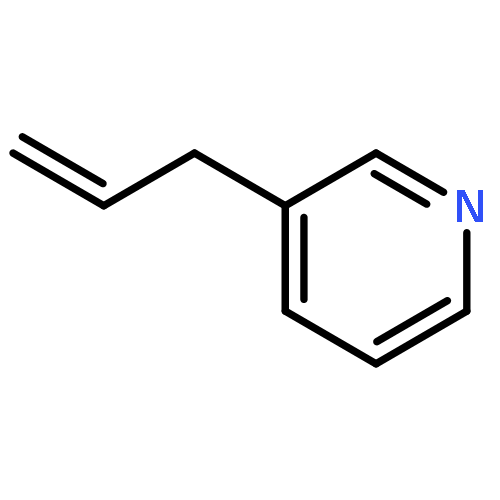
![TRICYCLO[8.2.2.2<SUP>4,7</SUP>]HEXADECA-1(12),4,6,10,13,15-HEXAENE-5,11-DIYLBIS[BIS(3,5-DIMETHYLPHENYL)PHOSPHINE]](http://img.cochemist.com/ccimg/7300/7289-53-4.png)
![TRICYCLO[8.2.2.2<SUP>4,7</SUP>]HEXADECA-1(12),4,6,10,13,15-HEXAENE-5,11-DIYLBIS[BIS(3,5-DIMETHYLPHENYL)PHOSPHINE]](http://img.cochemist.com/ccimg/7300/7289-53-4_b.png)
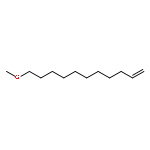
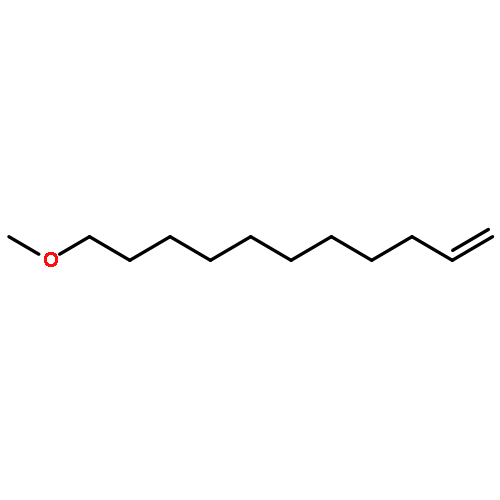
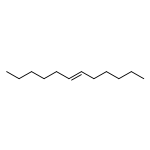
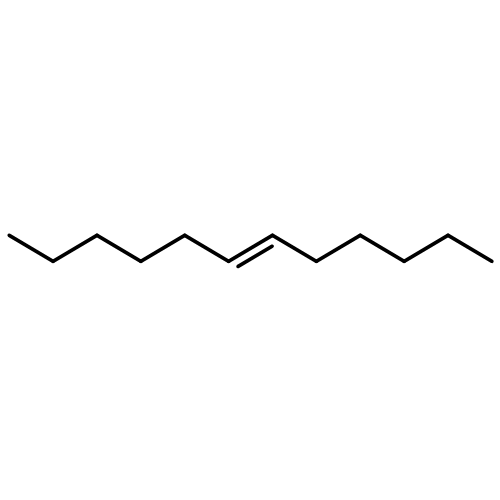


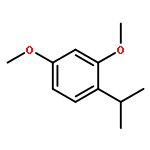

















![Benzene, 1-chloro-4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/1700/1657-50-7.png)
![Benzene, 1-chloro-4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/1700/1657-50-7_b.png)









![Benzoic acid, 4-[(E)-(phenylmethylene)amino]-, ethyl ester](http://img.cochemist.com/ccimg/870700/870621-09-3.png)
![Benzoic acid, 4-[(E)-(phenylmethylene)amino]-, ethyl ester](http://img.cochemist.com/ccimg/870700/870621-09-3_b.png)




![N-[(3-methylphenyl)methyl]aniline](http://img.cochemist.com/ccimg/75400/75366-13-1.png)
![N-[(3-methylphenyl)methyl]aniline](http://img.cochemist.com/ccimg/75400/75366-13-1_b.png)
![Benzoic acid, 4-[(phenylmethyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/61500/61439-53-0.png)
![Benzoic acid, 4-[(phenylmethyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/61500/61439-53-0_b.png)


![Benzene, 1-methyl-4-[[(2E)-3-phenyl-2-propen-1-yl]oxy]-](/data/chemimg/1815800/38276-67-4.png)
![Benzene, 1-methyl-4-[[(2E)-3-phenyl-2-propen-1-yl]oxy]-](/data/chemimg/1815800/38276-67-4_b.png)






![Benzenamine, N-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-96-3.png)
![Benzenamine, N-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-96-3_b.png)




![Dibenzo[a,e]cyclooctene](http://img.cochemist.com/ccimg/300/262-89-5.png)
![Dibenzo[a,e]cyclooctene](http://img.cochemist.com/ccimg/300/262-89-5_b.png)

