Co-reporter:Dr. Yong Li; Mingji Dai
Angewandte Chemie 2017 Volume 129(Issue 38) pp:11782-11785
Publication Date(Web):2017/09/11
DOI:10.1002/ange.201706845
AbstractTotal syntheses of the reported structures of the rhamnofolane diterpene natural products curcusones I and J in racemic form were achieved. The synthetic strategy features a novel tandem gold-catalyzed furan formation and furan–allene [4+3] cycloaddition to build the 5,7-fused ring system with an oxa-bridge in one step, and a stereoselective exo-Diels–Alder reaction to form the 6-membered ring. The newly developed tandem gold catalysis is quite general and can be scaled up. Our syntheses suggest that structural revisions of curcusones I and J are needed.
Co-reporter:Yong Li, Mufeng Wei, Mingji Dai
Tetrahedron 2017 Volume 73, Issue 29(Issue 29) pp:
Publication Date(Web):20 July 2017
DOI:10.1016/j.tet.2016.11.005
A concise approach to synthesize the 5-7-6 tricyclic carbon skeleton of the daphnane/tigliane diterpene natural products has been accomplished via a sequential gold-catalyzed furan formation and furan-allene [4+3] cycloaddition. This work provides new avenues for rapid and diverted synthesis of the medicinally important daphnane/tigliane diterpenes and their unnatural analogs.Download high-res image (172KB)Download full-size image
Co-reporter:Haroon Mohammad, Kwaku Kyei-Baffour, Waleed Younis, Dexter C. Davis, Hassan Eldesouky, Mohamed N. Seleem, Mingji Dai
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 11(Issue 11) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.bmc.2017.03.035
•Compounds composed of the aryl isonitrile core exhibit potent antifungal activity.•Compounds inhibit growth of Candida, Cryptococcus, and Aspergillus species.•Most potent antifungal derivatives are non-toxic to mammalian cells.Invasive fungal infections present a formidable global public health challenge due to the limited number of approved antifungal agents and the emergence of resistance to the frontline treatment options, such as fluconazole. Three fungal pathogens of significant concern are Candida, Cryptococcus, and Aspergillus given their propensity to cause opportunistic infections in immunocompromised individuals. New antifungal agents composed of unique chemical scaffolds are needed to address this public health challenge. The present study examines the structure-activity relationship of a set of aryl isonitrile compounds that possess broad-spectrum antifungal activity primarily against species of Candida and Cryptococcus. The most potent derivatives are capable of inhibiting growth of these key pathogens at concentrations as low as 0.5 µM. Remarkably, the most active compounds exhibit an excellent safety profile and are non-toxic to mammalian cells even at concentrations up to 256 µM. The present study lays the foundation for further investigation of aryl isonitrile compounds as a new class of antifungal agents.Download high-res image (49KB)Download full-size image
Co-reporter:Yong Li;Xianglin Yin
Natural Product Reports (1984-Present) 2017 vol. 34(Issue 10) pp:1185-1192
Publication Date(Web):2017/10/18
DOI:10.1039/C7NP00038C
Covering: 2000 to 2017
Macrolactones are privileged structural motifs in many functional molecules, particularly natural products and pharmaceutical molecules. They are commonly synthesized from the corresponding seco acids with various stoichiometric activating reagents to promote the formation of the macrocycle. Advances in new methods and strategies for synthesizing macrolactones have been made over the years to improve the overall synthetic efficiency and economy. This highlight focuses on the recent developments of catalytic macrolactonization methods and strategies without the use of seco acids and their applications in natural product total synthesis. In particular, catalytic C–H macrolactonization, enantioselective Rh-catalyzed redox-neutral allene-acid cyclization, catalytic carbonylative macrolactonization, and NHC-catalyzed oxidative macrolactonization are highlighted.
Co-reporter:Xianglin Yin;Haroon Mohammad;Hassan E. Eldesouky;Ahmed Abdelkhalek;Mohamed N. Seleem
Chemical Communications 2017 vol. 53(Issue 53) pp:7238-7241
Publication Date(Web):2017/06/29
DOI:10.1039/C7CC02494K
A novel and efficient palladium-catalyzed aminocarbonylative lactonization of amino propargylic alcohols has been developed to provide rapid access to various bicyclic lactones especially dihydropyrrole-fused furanones, which are novel structures and have not been explored in biological and medicinal settings. This method can also be used to access β-lactone products such as 16. Preliminary biological evaluations revealed that compounds 13h and 13s demonstrated promising activity against Clostridium difficile and compounds 13h, 13k, 13s, and 16b showed activity against several important fungal pathogens.
Co-reporter:Yu Bai, Dexter C. Davis, and Mingji Dai
The Journal of Organic Chemistry 2017 Volume 82(Issue 5) pp:
Publication Date(Web):February 7, 2017
DOI:10.1021/acs.joc.7b00009
Carbon monoxide is an important one-carbon source and can be incorporated in complex molecules via various transition-metal-catalyzed carbonylation reactions. In particular, palladium-catalyzed carbonylation reactions have found broad application in total synthesis of natural products. Examples are presented in this Synopsis to highlight recent progress in this area, including our own work in macrolide and spirocyclic molecule synthesis. In these selected cases, carbon monoxide functions as a one-carbon linchpin to facilitate building structural complexity and improving synthetic efficiency.
Co-reporter:Yu Bai, Xingyu Shen, Yong Li, and Mingji Dai
Journal of the American Chemical Society 2016 Volume 138(Issue 34) pp:10838-10841
Publication Date(Web):August 10, 2016
DOI:10.1021/jacs.6b07585
Spinosyn A (1), a complex natural product featuring a unique 5,6,5,12-fused tetracyclic core structure, is the major component of spinosad, an organic insecticide and an FDA-approved agent used worldwide. Herein, we report an efficient total synthesis of (−)-spinosyn A with 15 steps in the longest linear sequence and 23 steps total from readily available compounds 14 and 23. The synthetic approach features several important catalytic transformations including a chiral amine-catalyzed intramolecular Diels–Alder reaction to afford 22 in excellent diastereoselectivity, a one-step gold-catalyzed propargylic acetate rearrangement to convert 28 to α-iodoenone 31, an unprecedented palladium-catalyzed carbonylative Heck macrolactonization to form the 5,12-fused macrolactone in one step, and a gold-catalyzed Yu glycosylation to install the challenging β-forosamine. This total synthesis is highly convergent and modular, thus offering opportunities to synthesize spinosyn analogues in order to address the emerging cross-resistance problems.
Co-reporter:Dexter C. Davis, Katherine L. Walker, Chunhua Hu, Richard N. Zare, Robert M. Waymouth, and Mingji Dai
Journal of the American Chemical Society 2016 Volume 138(Issue 33) pp:10693-10699
Publication Date(Web):July 26, 2016
DOI:10.1021/jacs.6b06573
A palladium-catalyzed cascade carbonylative spirolactonization of hydroxycyclopropanols has been developed to efficiently synthesize oxaspirolactones common to many complex natural products of important therapeutic value. The mild reaction conditions, high atom economy, broad substrate scope, and scalability of this new method were highlighted in expedient total syntheses of the Turkish tobacco natural products α-levantanolide and α-levantenolide in two and four steps, respectively. The hydroxycyclopropanol substrates are readily available in one step via a Kulinkovich reaction of the corresponding lactones. Mechanistic studies utilizing high-resolution electrospray ionization mass spectrometry (ESI-MS) identified several key intermediates in the catalytic cycle, as well as those related to catalyst decomposition and competitive pathways.
Co-reporter:Zhishi Ye and Mingji Dai
Organic Letters 2015 Volume 17(Issue 9) pp:2190-2193
Publication Date(Web):April 17, 2015
DOI:10.1021/acs.orglett.5b00828
A novel copper-catalyzed electrophilic amination of cyclopropanols with O-benzoyl-N,N-dialkylhydroxylamines to synthesize various β-aminoketones via a sequence that includes C–C bond cleavage and Csp3–N bond formation is reported. The reaction conditions are mild and tolerate a wide range of functional groups including benzoate, tosylate, expoxide, and α,β-unsaturated carbonyls, which are incompatible in the traditional amine nucleophilic conjugate addition and the Mannich reaction conditions. Preliminary mechanistic studies and a proposed catalytic cycle of this umpolung β-aminoketone synthesis process have been described as well.
Co-reporter:Yong Li, Zhishi Ye, Tabitha M. Bellman, Teng Chi, and Mingji Dai
Organic Letters 2015 Volume 17(Issue 9) pp:2186-2189
Publication Date(Web):April 17, 2015
DOI:10.1021/acs.orglett.5b00782
The first copper-catalyzed ring-opening electrophilic trifluoromethylation and trifluoromethylthiolation of cyclopropanols to form Csp3–CF3 and Csp3–SCF3 bonds have been realized. These transformations are efficient for the synthesis of β-CF3- and β-SCF3-substituted carbonyl compounds that are otherwise challenging to access. The reaction conditions are mild and tolerate a wide range of functional groups. Application to a concise synthesis of LY2409021, a glucagon receptor antagonist that is used in clinical trials for type 2 diabetes mellitus, is reported as well.
Co-reporter:Zhishi Ye, Tarsis F. Brust, Val J. Watts, and Mingji Dai
Organic Letters 2015 Volume 17(Issue 4) pp:892-895
Publication Date(Web):February 10, 2015
DOI:10.1021/ol503748t
Substituted allylic amines and their derivatives are key structural motifs of many drug molecules and natural products. A general, mild, and practical palladium-catalyzed γ-arylation of tertiary allylic amines, one of the most challenging Heck arylation substrates, has been developed. The γ-arylation products were obtained in excellent regio- and stereoselectivity. Moreover, novel and potent adenylyl cyclase inhibitors with the potential for treating neuropathic and inflammatory pain have been identified from the γ-arylation products.
Co-reporter:Zhishi Ye, Kristen E. Gettys, Xingyu Shen, and Mingji Dai
Organic Letters 2015 Volume 17(Issue 24) pp:6074-6077
Publication Date(Web):December 4, 2015
DOI:10.1021/acs.orglett.5b03096
Novel and general copper-catalyzed cyclopropanol ring opening cross-coupling reactions with difluoroalkyl bromides, perfluoroalkyl iodides, monofluoroalkyl bromides, and 2-bromo-2-alkylesters to synthesize various β-(fluoro)alkylated ketones are reported. The reactions feature mild conditions and excellent functional group compatibility and can be scaled up to gram scale. Preliminary mechanistic studies suggest the involvement of radical intermediates. The difluoroalkyl–alkyl cross-coupling products can also be readily converted to more valuable and diverse gem-difluoro-containing compounds by taking advantage of the carbonyl group resulting from cyclopropanol ring opening.
Co-reporter:Dexter C. Davis, Haroon Mohammad, Kwaku Kyei-Baffour, Waleed Younis, Cassidy Noel Creemer, Mohamed N. Seleem, Mingji Dai
European Journal of Medicinal Chemistry 2015 101() pp: 384-390
Publication Date(Web):
DOI:10.1016/j.ejmech.2015.06.031
Co-reporter:Yang Yang, Yu Bai, Siyuan Sun, and Mingji Dai
Organic Letters 2014 Volume 16(Issue 23) pp:6216-6219
Publication Date(Web):November 20, 2014
DOI:10.1021/ol503150c
Inspired by their potential biosynthesis, we have developed divergent total syntheses of seven monoterpene indole alkaloids including mersicarpine, leuconodines B and D, leuconoxine, melodinine E, leuconolam, and rhazinilam, and one unnatural analogue with an unprecedented structural skeleton. The key steps involve a Witkop–Winterfeldt oxidative indole cleavage followed by transannular cyclization. The transannular cyclization product was then converted to the corresponding structural skeletons by pairing its functional groups into different reaction modes.
Co-reporter:Wandi Zhang, Christopher W. Haskins, Yang Yang and Mingji Dai
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 45) pp:9109-9112
Publication Date(Web):07 Oct 2014
DOI:10.1039/C4OB01825G
Palladium-catalyzed synthesis of nitriles from amides has been described. Two similar, but complementary reaction conditions have been identified to convert various amides including α,β,γ,δ-unsaturated amides, cinnamides, aromatic amides and alkyl amides to the corresponding nitriles in good to excellent yield.
Co-reporter:Dr. Yang Yang;Christopher W. Haskins;Wi Zhang;Pui Leng Low ;Dr. Mingji Dai
Angewandte Chemie 2014 Volume 126( Issue 15) pp:4003-4006
Publication Date(Web):
DOI:10.1002/ange.201400416
Abstract
Divergent and concise total syntheses of two lycopodium alkaloids, lyconadins A and C have been developed. The synthesis of lyconadin A, having potent neurotrophic activity, features an efficient one-pot ketal removal and formal aza-[4+2] cyclization to form the cagelike core structure. A tandem ketal removal/Mannich reaction was developed to build the tricyclic structure of lyconadin C. Both lyconadins A and C were synthesized from a pivotal intermediate.
Co-reporter:Yu Bai;Dexter C. Davis ;Dr. Mingji Dai
Angewandte Chemie 2014 Volume 126( Issue 25) pp:6637-6640
Publication Date(Web):
DOI:10.1002/ange.201403006
Abstract
A novel Pd-catalyzed cascade alkoxycarbonylative macrolactonization to construct tetrahydropyran/tetrahydrofuran-containing bridged macrolactones in one step from alkendiols is described. Products with various ring sizes and substituents were obtained. Challenging macrolactones involving tertiary alcohols were synthesized smoothly as well. Mechanistically, experimental evidence to support a trans-oxypalladation step has been provided. The method was applied to the synthesis of potent anticancer compound 9-demethylneopeltolide.
Co-reporter:Dr. Yang Yang;Christopher W. Haskins;Wi Zhang;Pui Leng Low ;Dr. Mingji Dai
Angewandte Chemie International Edition 2014 Volume 53( Issue 15) pp:3922-3925
Publication Date(Web):
DOI:10.1002/anie.201400416
Abstract
Divergent and concise total syntheses of two lycopodium alkaloids, lyconadins A and C have been developed. The synthesis of lyconadin A, having potent neurotrophic activity, features an efficient one-pot ketal removal and formal aza-[4+2] cyclization to form the cagelike core structure. A tandem ketal removal/Mannich reaction was developed to build the tricyclic structure of lyconadin C. Both lyconadins A and C were synthesized from a pivotal intermediate.
Co-reporter:Yu Bai;Dexter C. Davis ;Dr. Mingji Dai
Angewandte Chemie International Edition 2014 Volume 53( Issue 25) pp:6519-6522
Publication Date(Web):
DOI:10.1002/anie.201403006
Abstract
A novel Pd-catalyzed cascade alkoxycarbonylative macrolactonization to construct tetrahydropyran/tetrahydrofuran-containing bridged macrolactones in one step from alkendiols is described. Products with various ring sizes and substituents were obtained. Challenging macrolactones involving tertiary alcohols were synthesized smoothly as well. Mechanistically, experimental evidence to support a trans-oxypalladation step has been provided. The method was applied to the synthesis of potent anticancer compound 9-demethylneopeltolide.
Co-reporter:Wandi Zhang, Christopher W. Haskins, Yang Yang and Mingji Dai
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 45) pp:NaN9112-9112
Publication Date(Web):2014/10/07
DOI:10.1039/C4OB01825G
Palladium-catalyzed synthesis of nitriles from amides has been described. Two similar, but complementary reaction conditions have been identified to convert various amides including α,β,γ,δ-unsaturated amides, cinnamides, aromatic amides and alkyl amides to the corresponding nitriles in good to excellent yield.
Co-reporter:Xianglin Yin, Haroon Mohammad, Hassan E. Eldesouky, Ahmed Abdelkhalek, Mohamed N. Seleem and Mingji Dai
Chemical Communications 2017 - vol. 53(Issue 53) pp:NaN7241-7241
Publication Date(Web):2017/05/11
DOI:10.1039/C7CC02494K
A novel and efficient palladium-catalyzed aminocarbonylative lactonization of amino propargylic alcohols has been developed to provide rapid access to various bicyclic lactones especially dihydropyrrole-fused furanones, which are novel structures and have not been explored in biological and medicinal settings. This method can also be used to access β-lactone products such as 16. Preliminary biological evaluations revealed that compounds 13h and 13s demonstrated promising activity against Clostridium difficile and compounds 13h, 13k, 13s, and 16b showed activity against several important fungal pathogens.

![Propanedioic acid, 2-[5-(phenylmethoxy)-2,3-pentadien-1-yl]-2-(2-propyn-1-yl)-, 1,3-dimethyl ester](/data/chemimg/3722400/2047811-92-5.png)
![Propanedioic acid, 2-[5-(phenylmethoxy)-2,3-pentadien-1-yl]-2-(2-propyn-1-yl)-, 1,3-dimethyl ester](/data/chemimg/3722400/2047811-92-5_b.png)
![Propanedioic acid, 2-[5-(phenylmethoxy)-2,3-pentadien-1-yl]-, 1,3-dimethyl ester](/data/chemimg/3722300/2047811-91-4.png)
![Propanedioic acid, 2-[5-(phenylmethoxy)-2,3-pentadien-1-yl]-, 1,3-dimethyl ester](/data/chemimg/3722300/2047811-91-4_b.png)













![2-METHYL-2-PROPANYL [(3R,6S)-6-(2,3-DIFLUOROPHENYL)-2-OXO-3-AZEPANYL]CARBAMATE](http://img.cochemist.com/ccimg/1313100/1313028-09-9.png)
![2-METHYL-2-PROPANYL [(3R,6S)-6-(2,3-DIFLUOROPHENYL)-2-OXO-3-AZEPANYL]CARBAMATE](http://img.cochemist.com/ccimg/1313100/1313028-09-9_b.png)













![Phosphonic acid, [(2-isocyanophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/375400/375372-32-0.png)
![Phosphonic acid, [(2-isocyanophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/375400/375372-32-0_b.png)

![Silane, (1,1-dimethylethyl)[(10-iododecyl)oxy]dimethyl-](http://img.cochemist.com/ccimg/201800/201799-41-9.png)
![Silane, (1,1-dimethylethyl)[(10-iododecyl)oxy]dimethyl-](http://img.cochemist.com/ccimg/201800/201799-41-9_b.png)


![Benzenemethanamine, N-(phenylmethyl)-N-[(2E)-3-phenyl-2-propen-1-yl]-](/data/chemimg/109900/147452-43-5.png)
![Benzenemethanamine, N-(phenylmethyl)-N-[(2E)-3-phenyl-2-propen-1-yl]-](/data/chemimg/109900/147452-43-5_b.png)







![1-Propanone, 3-[bis(phenylmethyl)amino]-1-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/105200/105101-05-1.png)
![1-Propanone, 3-[bis(phenylmethyl)amino]-1-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/105200/105101-05-1_b.png)
![Silane, (1,1-dimethylethyl)[(6-iodohexyl)oxy]dimethyl-](http://img.cochemist.com/ccimg/103500/103483-32-5.png)
![Silane, (1,1-dimethylethyl)[(6-iodohexyl)oxy]dimethyl-](http://img.cochemist.com/ccimg/103500/103483-32-5_b.png)










![Pentanoic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/87800/87729-38-2.png)
![Pentanoic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/87800/87729-38-2_b.png)
![Methanesulfonic acid, 1,1,1-trifluoro-, 1,7,7-trimethylbicyclo[2.2.1]hept-2-en-2-yl ester](/data/chemimg/2010400/86544-72-1.png)
![Methanesulfonic acid, 1,1,1-trifluoro-, 1,7,7-trimethylbicyclo[2.2.1]hept-2-en-2-yl ester](/data/chemimg/2010400/86544-72-1_b.png)
![Silane, (1,1-dimethylethyl)[(5-iodopentyl)oxy]dimethyl-](/data/chemimg/1995200/85514-45-0.png)
![Silane, (1,1-dimethylethyl)[(5-iodopentyl)oxy]dimethyl-](/data/chemimg/1995200/85514-45-0_b.png)


![SILANE, (1,1-DIMETHYLETHYL)[(7-IODOHEPTYL)OXY]DIMETHYL-](http://img.cochemist.com/ccimg/78600/78535-68-9.png)
![SILANE, (1,1-DIMETHYLETHYL)[(7-IODOHEPTYL)OXY]DIMETHYL-](http://img.cochemist.com/ccimg/78600/78535-68-9_b.png)


















![[1,1'-Bicyclopropyl]-1-ol](http://img.cochemist.com/ccimg/54300/54251-80-8.png)
![[1,1'-Bicyclopropyl]-1-ol](http://img.cochemist.com/ccimg/54300/54251-80-8_b.png)

methanone](http://img.cochemist.com/ccimg/52800/52742-32-2.png)
methanone](http://img.cochemist.com/ccimg/52800/52742-32-2_b.png)

![(4-Fluoro-benzyl)-[2-(1H-indol-3-yl)-ethyl]-amine](http://img.cochemist.com/ccimg/51900/51841-40-8.png)
![(4-Fluoro-benzyl)-[2-(1H-indol-3-yl)-ethyl]-amine](http://img.cochemist.com/ccimg/51900/51841-40-8_b.png)








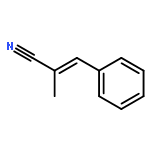
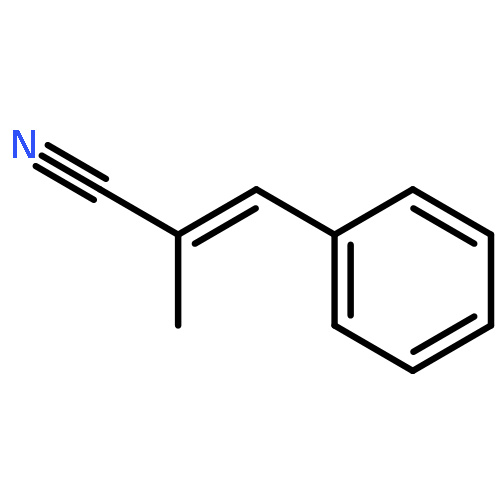
![Benzonitrile, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/38200/38175-96-1.png)
![Benzonitrile, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/38200/38175-96-1_b.png)







![Phosphonic acid, [(2-cyanophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/24000/23973-65-1.png)
![Phosphonic acid, [(2-cyanophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/24000/23973-65-1_b.png)











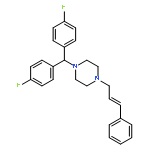
![Piperazine,1-(diphenylmethyl)-4-[(2E)-3-phenyl-2-propen-1-yl]-](http://img.cochemist.com/ccimg/16700/16699-20-0.png)
![Piperazine,1-(diphenylmethyl)-4-[(2E)-3-phenyl-2-propen-1-yl]-](http://img.cochemist.com/ccimg/16700/16699-20-0_b.png)





















![PHOSPHONIC ACID, [(2-NITROPHENYL)METHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/5000/4914-12-9.png)
![PHOSPHONIC ACID, [(2-NITROPHENYL)METHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/5000/4914-12-9_b.png)
![1,6-Dioxaspiro[4.5]decan-2-one](http://img.cochemist.com/ccimg/4800/4744-44-9.png)
![1,6-Dioxaspiro[4.5]decan-2-one](http://img.cochemist.com/ccimg/4800/4744-44-9_b.png)




![(4aS,4bR,6aS,8S,10aS,10bS,12aS)-8-hydroxy-10a,12a-dimethylhexadecahydro-2H-naphtho[2,1-f]chromen-2-one](http://img.cochemist.com/ccimg/2100/2061-71-4.png)
![(4aS,4bR,6aS,8S,10aS,10bS,12aS)-8-hydroxy-10a,12a-dimethylhexadecahydro-2H-naphtho[2,1-f]chromen-2-one](http://img.cochemist.com/ccimg/2100/2061-71-4_b.png)




















![1H-1,4-Diazepine, hexahydro-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/872200/872103-28-1.png)
![1H-1,4-Diazepine, hexahydro-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/872200/872103-28-1_b.png)







![2-CHLORO-5,6-DIHYDROCYCLOPENTA[B]PYRIDIN-7-ONE](http://img.cochemist.com/ccimg/109300/109240-30-4.png)
![2-CHLORO-5,6-DIHYDROCYCLOPENTA[B]PYRIDIN-7-ONE](http://img.cochemist.com/ccimg/109300/109240-30-4_b.png)







