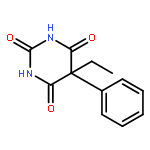
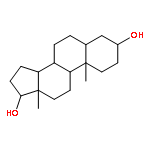
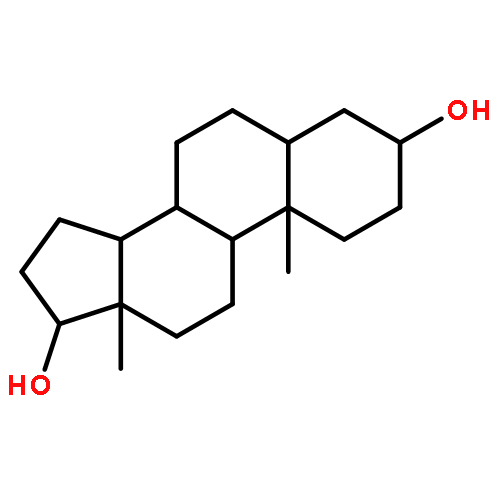
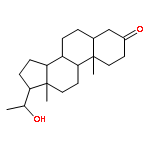
Co-reporter:Yoshiaki Kitamura, Ryo Asakura, Koki Terazawa, Aya Shibata, Masato Ikeda, Yukio Kitade
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 12(Issue 12) pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.bmcl.2017.04.023
The formation of 1,4-disubstituted 1,2,3-triazoles through copper-catalyzed azide–alkyne cycloaddition (CuAAC) in oligonucleotides bearing 1-deoxy-1-ethynyl-β-d-ribofuranose (RE) can have a positive impact on the stability of oligonucleotide duplexes and stem-loop structures.Download high-res image (52KB)Download full-size image
Co-reporter:Yuki Nagaya, Yoshiaki Kitamura, Aya Shibata, Masato Ikeda, Yukihiro Akao, Yukio Kitade
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 24(Issue 24) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.bmcl.2017.10.070
Chemically modified siRNAs containing 2-O-benzyl-1-deoxy-d-ribofuranose (RHOBn) in their 3′-overhang region were significantly more resistant towards serum nucleases than siRNAs possessing the natural nucleoside in this region. The knockdown efficacies and binding affinities of these modified siRNAs to the recombinant human Argonaute protein 2 (hAgo2) PAZ domain were comparable with that of siRNA with a thymidine dimer at the 3′-end.Download high-res image (83KB)Download full-size image
Co-reporter:Yoshiaki Kitamura, Seiya Kito, Remi Nakashima, Katsuki Tanaka, Kumi Nagaoka, Yukio Kitade
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 16) pp:3870-3874
Publication Date(Web):15 August 2016
DOI:10.1016/j.bmc.2016.06.033
RNase L is activated by 2′,5′-oligoadenylates (2-5A) at subnanomolar levels to cleave single-stranded RNA. We previously reported the hypothesis that the introduction of an 8-methyladenosine residue at the 2′-terminus of the 2-5A tetramer shifts the 2-5A binding site of RNase L. In this study, we synthesized various 5′-modified 2-5A analogs with 8-methyladenosine at the 2′-terminus. The doxifluridine-conjugated 8-methyladenosine-substituted 2-5A analog was significantly more effective as an activator of RNase L than the parent 5′-monophophorylated 2-5A tetramer and showed a tumor suppressive effect against human cervical cancer cells.
Co-reporter:Yoshiaki Kitamura, Ryuto Sakamoto, Takao Shiraishi, Haruka Oguri, Satoshi Ohno, Yukio Kitade
Tetrahedron 2016 Volume 72(27–28) pp:4016-4021
Publication Date(Web):7 July 2016
DOI:10.1016/j.tet.2016.05.029
A convenient method for the post-modification of any peptide bearing a disulfide bond via rapid ligand-free copper-catalyzed azide–alkyne cycloaddition (CuAAC) was developed. N-hydroxysuccinimide (NHS) esters and maleimides bearing an aryl acetylene residue to allow installation of a terminal alkyne moiety into peptides were efficiently synthesized. The ligation of glutathione (GSH) using the maleimides and disulfide-linked dimer of GSH (GSSG) using the NHS esters proceeded smoothly to afford the corresponding terminal alkyne-labeled glutathione derivatives, respectively. The terminal alkyne-labeled GSH and GSSG efficiently coupled with 4-fluorobenzylazide to provide the corresponding glutathione analogues in quantitative yield. In any case, side reactions including cleavage of disulfide bonds were not observed. The application of somatostatin, disulfide-linked cyclic tetradecapeptide, to this method was achieved.
Co-reporter:Tomoya Tsuzuki, Hiroshi Katagiri, Yoshiaki Kitamura, Yukio Kitade, Masato Ikeda
Tetrahedron 2016 Volume 72(Issue 43) pp:6886-6891
Publication Date(Web):27 October 2016
DOI:10.1016/j.tet.2016.09.022
We describe a new polymer-supported liquid-phase submonomer synthesis (LPSS) protocol for the construction of self-assembling arylopeptoids using polyethylene glycol monomethyl ether (MPEG) as a hydrophilic polymer support. Self-assembling properties of arylopeptoids bearing MPEG (without cleavage from the polymer support for the synthesis) in aqueous media was investigated using fluorescence probe method, dynamic light scattering, and transmission electron microscopy. We found that an arylopeptoid pentamer bearing MPEG exhibits self-assembling ability to form spherical-shaped nanostructures.
Co-reporter:Masato Ikeda, Keito Horio, Tomoya Tsuzuki, Ryo Torii, Aya Shibata, Yoshiaki Kitamura, Hiroshi Katagiri, Yukio Kitade
Tetrahedron Letters 2015 Volume 56(Issue 48) pp:6726-6729
Publication Date(Web):2 December 2015
DOI:10.1016/j.tetlet.2015.10.056
Herein, we describe a new methodology for the solid phase submonomer synthesis (SPSS) of arylopeptoids using a reductive amination reaction as the key step instead of a nucleophilic substitution reaction, which is generally used in conventional SPSS of peptoids. The new SPSS enables easy access to arylopeptoid oligomers in which phenyl side groups are directly attached to the aromatic main chain.
Co-reporter:Qin Ren, Kana Tsunaba, Yoshiaki Kitamura, Remi Nakashima, Aya Shibada, Masato Ikeda, Yukio Kitade
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 6) pp:1519-1522
Publication Date(Web):15 March 2014
DOI:10.1016/j.bmcl.2014.01.082
Positron emission tomography (PET) is a highly sensitive quantitative imaging technique for studying molecular pathways and interactions in vivo. This imaging technique plays a key role in drug discovery, pharmacokinetics, pharmacodynamics, and assessing in vivo distribution. In this study, we designed an ethynylbenzene-substituted glycol (ME) as a versatile probe for PET labeling of oligonucleotides through a click reaction.
Co-reporter:Mahmoud Kandeel;Yoshihiro Noguchi
Journal of Thermal Analysis and Calorimetry 2014 Volume 115( Issue 3) pp:2089-2097
Publication Date(Web):2014 March
DOI:10.1007/s10973-013-3319-5
Plasmodium deoxyguanylate pathways are an attractive area of investigation for future metabolic and drug discovery studies due to their unusual substrate specificities. We investigated the energetic contribution to thymidylate kinase substrate binding, and the forces underlying ligand recognition. The binding constant varied from 8 × 104 M−1 at 290 K to 6 × 104 M−1 at 310 K for dGMP, and from 16 × 104 M−1 at 290 K to 4 × 104 M−1 at 310 K for TMP. ΔCp was estimated as −1.75 kJ mol−1 K−1 for TMP and +2 kJ mol−1 K−1 for dGMP. In comparison with TMP, the binding of dGMP to PfTMK produced less favorable enthalpy change, positive or favorable entropic contribution at lower temperature, positive heat capacity change, negative \( \Updelta S_{\text{HE}}^{^\circ } \), positive ΔSother, higher total solvent-exposed surface area and more or less rigid body binding. These changes indicate unfavorable conditions for proper binding and lower conformational changes, and suboptimal structural reordering during dGMP binding.
Co-reporter:Yoshiaki Kitamura, Yuki Masegi, Shunsuke Ogawa, Remi Nakashima, Yukihiro Akao, Yoshihito Ueno, Yukio Kitade
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 15) pp:4494-4501
Publication Date(Web):1 August 2013
DOI:10.1016/j.bmc.2013.05.046
We have developed chemically modified siRNAs and miRNAs bearing urea/thiourea-bridged aromatic compounds at their 3′-end for RNAi therapy. Chemically modified RNAs possessing urea/thiourea-bridged aromatic compounds instead of naturally occurring dinucleotides at the 3′-overhang region were easily prepared in good yields and were more resistant to nucleolytic hydrolysis than unmodified RNA. siRNAs containing urea or thiourea derivatives showed the desired knockdown effect. Furthermore, modified miR-143 duplexes carrying the urea/thiourea compounds in the 3′-end of each strand were able to inhibit the growth of human bladder cancer T24 cells.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Xiong Luo, Takahiro Sugiura, Remi Nakashima, Yoshiaki Kitamura, Yukio Kitade
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 14) pp:4157-4161
Publication Date(Web):15 July 2013
DOI:10.1016/j.bmcl.2013.05.036
Short interfering RNA (siRNA) has been proven to be an utilizable tool for post-transcriptional gene silencing research. In this study, we designed and synthesized two glucosamine analogues and tried to modify the siRNA using these two glucosamine analogues at the 3′-overhang region of siRNAs to improve the nuclease resistance and to overcome some other weak points. The siRNAs modified with glucosamine analogues had almost no effect of the thermal stability and showed strong resistance to nuclease degradation. Some of them kept the same gene silencing activity level as unmodified siRNA.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Mahmoud Kandeel
Journal of Computer-Aided Molecular Design 2013 Volume 27( Issue 7) pp:605-614
Publication Date(Web):2013 July
DOI:10.1007/s10822-013-9665-3
RNA interference (RNAi) is a critical cellular pathway activated by double stranded RNA and regulates the gene expression of target mRNA. During RNAi, the 3′ end of siRNA binds with the PAZ domain, followed by release and rebinding in a cyclic manner, which deemed essential for proper gene silencing. Recently, we provided the forces underlying the recognition of small interfering RNA by PAZ in a computational study based on the structure of Drosophila Argonaute 2 (Ago2) PAZ domain. We have now reanalyzed these data within the view of the new available structures from human Argonauts. While the parameters of weak binding are correlated with higher (RNAi) in the Drosophila model, a different profile is predicted with the human Ago2 PAZ domain. On the basis of the human Ago2 PAZ models, the indicators of stronger binding as the total binding energy and the free energy were associated with better RNAi efficacy. This discrepancy might be attributable to differences in the binding site topology and the difference in the conformation of the bound nucleotides.
Co-reporter:Mahmoud Kandeel;Mohamed Nabih
Journal of Thermal Analysis and Calorimetry 2013 Volume 111( Issue 3) pp:1737-1741
Publication Date(Web):2013 March
DOI:10.1007/s10973-012-2265-y
Among the pharmacokinetic parameters of chemotherapeutics, serum albumin binding is a critical factor in determining drug distribution and bioavailability. In this study, the binding properties as well as the interaction of spectinomycin with Bovine serum albumin was investigated. Spectinomycin showed stronger binding with BSA at higher temperatures, which diminishes by decreasing the temperature. The binding constant of spectinomycin with BSA varied from 3.1 × 103 M−1 at 298 K to 6.3 × 103 M−1 at 313 K. By increasing the temperature, from 298 to 313 K, the binding affinity was increased by twofolds. Thermodynamic analysis indicated changes in albumin conformation and partial loss of folding during spectinomycin-albumin binding. The mild-moderate binding affinity of spectinomycin with BSA will be important in determining the drug–drug interactions at the binding sites of BSA. The presence of stronger binding ligand e.g., chloramphenicol, tetracyclines or diclofenac will compete with spectinomycin for its binding sites, therefore, lowering its serum albumin binding. The result of this study will be helpful in understanding of the binding properties and mechanisms of interaction of spectinomycin with bovine serum albumin.
Co-reporter:Yoshiaki Kitamura, Kana Edayoshi, Yukio Kitade
Tetrahedron Letters 2012 Volume 53(Issue 51) pp:6987-6989
Publication Date(Web):19 December 2012
DOI:10.1016/j.tetlet.2012.10.053
We have developed an efficient stereoselective synthesis of 1-deoxy-1-ethynyl-β-d-ribofuranose (RE), via the β-selective cyanation of the anomeric position of 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose. Under conditions for copper(I)-catalyzed alkyne–azide 1,3-dipolar cycloaddition, the cycloaddition of RE with 4-fluorobenzylazide was accomplished within 5 min, to afford the corresponding triazole ribonucleoside in quantitative yield.
Co-reporter:Kazumi Taniho, Remi Nakashima, Mahmoud Kandeel, Yoshiaki Kitamura, Yukio Kitade
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 7) pp:2518-2521
Publication Date(Web):1 April 2012
DOI:10.1016/j.bmcl.2012.01.132
To elucidate the role of the sugar moiety in the two natural nucleotides of the 3′-overhang region of small interfering RNA (siRNA), we synthesized siRNAs that incorporated two abasic nucleosides, 1-deoxy-d-ribofuranose (RH). We improved the method for preparing an O-protected abasic nucleoside, 1-deoxy-2,3,5-tri-O-benzoyl-β-d-ribofuranose, via the reductive cleavage of the anomeric position of 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose. To incorporate RH into oligonucleotides by the standard phosphoramidite solid phase method, RH was converted into its phosphoramidite derivative and the solid support linked to a controlled pore glass resin. Chemically modified RNAs possessing RH at the 3′-overhang region were easily prepared in good yields. siRNAs containing RH showed moderate nuclease-resistance and a desirable knockdown effect.
Co-reporter:Takao Shiraishi, Yoshiaki Kitamura, Yoshihito Ueno and Yukio Kitade
Chemical Communications 2011 vol. 47(Issue 9) pp:2691-2693
Publication Date(Web):12 Jan 2011
DOI:10.1039/C0CC04979D
We synthesised aryl acetylene derivatives as versatile probes for labelling of oligonucleotides. RNA oligomers bearing an aryl acetylene molecule rapidly reacted with benzylazide derivatives under ligand-free click reaction conditions.
Co-reporter:Mahmoud Keel
Journal of Molecular Recognition 2011 Volume 24( Issue 2) pp:322-332
Publication Date(Web):
DOI:10.1002/jmr.1074
Abstract
Plasmodium deoxyguanylate pathways are an attractive area of investigation for future metabolic and drug discovery studies due to their unique substrate specificities. We investigated the energetic contribution to guanylate kinase substrate binding and the forces underlying ligand recognition. In the range from 20 to 35°C, the thermodynamic profiles displayed marked decrease in binding enthalpy, while the free energy of binding showed little changes. GMP produced a large binding heat capacity change of −356 cal mol−1 K−1, indicating considerable conformational changes upon ligand binding. Interestingly, the calculated ΔCp was −32 cal mol−1 K−1, indicating that the accessible surface area is not the central change in substrate binding, and that other entropic forces, including conformational changes, are more predominant. The thermodynamic signature for GMP is inconsistent with rigid-body association, while dGMP showed more or less rigid-body association. These binding profiles explain the poor catalytic efficiency and low affinity for dGMP compared with GMP. At low temperature, the ligands bind to the receptor site under the effect of hydrophobic forces. Interestingly, by increasing the temperature, the entropic forces gradually vanish and proceed to a nonfavorable contribution, and the interaction occurs mainly through bonding, electrostatic forces, and van der Waals interactions. Copyright © 2010 John Wiley & Sons, Ltd.
Co-reporter:Kumi Nagaoka, Yoshiaki Kitamura, Yoshihito Ueno, Yukio Kitade
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 3) pp:1186-1188
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmcl.2009.12.005
Human ribonuclease L (RNase L), an interferon-induced endoribonuclease, becomes enzymatically active after binding to 2-5A. The 5′-phosphoryl group of 2-5A is reportedly necessary for the conformational change leading to RNase L activation. However, we found that 5′-O-dephosphorylated 2-5A tetramer analogs with 8-methyladenosine at the 2′-terminus were more effective as an activator of RNase L than the parent 2-5A tetramer. Introduction of 8-methyladenosine is thought to induce a dramatic shift of 2-5A in the binding site of RNase L.The 5′-phosphoryl group of 2-5A is reportedly necessary for the conformational change leading to RNase L activation. However, we found that 5′-O-dephosphorylated 2-5A tetramer analogs with 8-methyladenosine at the 2′-terminus were more effective as an activator of RNase L than the parent 2-5A tetramer. Introduction of 8-methyladenosine is thought to induce a dramatic shift of 2-5A in the binding site of RNase L.
Co-reporter:Mahmoud Kandeel
Journal of Bioenergetics and Biomembranes 2010 Volume 42( Issue 5) pp:361-369
Publication Date(Web):2010 October
DOI:10.1007/s10863-010-9304-9
Nucleoside diphosphate kinases (NDKs) play a key role in maintaining the intracellular energy resources as well as the balance of nucleotide pools. Recently, attention has been directed to NDKs owing to its role in activating various chemotherapeutic agents. The binding affinity of different nucleotides with P. falciparum NDK was varied according to the following order ADP ~ GDP > dGDP > dADP > dTDP > CDP > dCDP > UDP. The binding of purines nucleotides was stronger than pyrimidines. Furthermore, PfNDK showed more preferences to ribonucleotides over deoxyribonucleotides. Pyrimidines showed lower negative free energy compared with that of purines. The interaction of all nucleotides showed favorable enthalpic and entropic terms. However, the enthalpic terms were the main deriving forces for purine nucleotides, while the entropic contributions were the predominant forces for pyrimidines. Interestingly, TDP showed marked affinity and more favorable enthalpic and less entropic contributions. In conclusion, the size of nucleotide was the critical factor in PfNDK ligand affinity.
Co-reporter:Yoshihito Ueno, Akihiro Kawamura, Keiji Takasu, Shinji Komatsuzaki, Takumi Kato, Satoru Kuboe, Yoshiaki Kitamura and Yukio Kitade
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 13) pp:2761-2769
Publication Date(Web):12 May 2009
DOI:10.1039/B901631G
This paper describes the synthesis and properties of a novel molecular beacon (MB) containing a benzene-phosphate backbone at its stem moiety. The fluorescence intensity of MBs was found to stabilize by the introduction of the benzene-phosphate backbone at its stem moiety. Furthermore, an MB containing the benzene-phosphate backbone was more resistant to DNase I (endonuclease) than an MB comprising natural DNA and 2′-O-methyl-RNA. These results indicate that the MB with the benzene-phosphate backbone is superior as a molecular beacon as compared to the MB composed of natural DNA and 2′-O-methyl-RNA.
Co-reporter:Yoshihito Ueno, Kayo Yoshikawa, Yoshiaki Kitamura, Yukio Kitade
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 3) pp:875-877
Publication Date(Web):1 February 2009
DOI:10.1016/j.bmcl.2008.11.107
Unintended (off-target) transcript silencing is a critical problem associated with RNA interference (RNAi)-based therapeutic applications. This paper shows that the incorporation of appropriate alkyl linkers at the center of the sense strands can suppress the off-target effects induced by the sense strands without reducing the RNAi-inducing activity of the antisense strands.
Co-reporter:Yoshihito Ueno, Yuuji Watanabe, Aya Shibata, Kayo Yoshikawa, Takashi Takano, Michinori Kohara, Yukio Kitade
Bioorganic & Medicinal Chemistry 2009 17(5) pp: 1974-1981
Publication Date(Web):
DOI:10.1016/j.bmc.2009.01.033
Co-reporter:Takayuki Ando, Masafumi Iwata, Fazila Zulfiqar, Tatsuya Miyamoto, Masayuki Nakanishi, Yukio Kitade
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 7) pp:3809-3815
Publication Date(Web):1 April 2008
DOI:10.1016/j.bmc.2008.01.046
2-Modified aristeromycin derivatives and their related analogs were synthesized to investigate their inhibitory activity against human and Plasmodium falciparum S-adenosyl-l-homocysteine hydrolase (PfSAHH). 2-Fluoroaristeromycin showed a strong inhibitory activity against PfSAHH selectively and complete resistance to adenosine deaminase.
Co-reporter:Hiroharu Kojima, Atsushi Kozaki, Masafumi Iwata, Takayuki Ando, Yukio Kitade
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 13) pp:6575-6579
Publication Date(Web):1 July 2008
DOI:10.1016/j.bmc.2008.05.020
3′,4′-Epoxynoraristeromycin analogs were designed and synthesized. Their affinities with human and Plasmodium falciparum S-adenosyl-l-homo-cysteine hydrolase were investigated.
Co-reporter:Yoshihito Ueno, Koshi Kawada, Tomoharu Naito, Aya Shibata, Kayo Yoshikawa, Hye-Sook Kim, Yusuke Wataya, Yukio Kitade
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 16) pp:7698-7704
Publication Date(Web):15 August 2008
DOI:10.1016/j.bmc.2008.07.010
Short-interfering RNAs (siRNAs) conjugated with lipophilic groups at their 3′-termini were synthesized. The properties of the synthesized siRNAs were examined in detail, and it was found that at low concentrations, their silencing abilities were dependent on the positions of the modifications and the types of organic molecules attached. Although the modification of siRNAs with palmitic acid or oleic acid at the 3′-end slightly reduced their silencing activities, siRNAs had enough abilities to induce RNAi at 10 nM concentrations. On the other hand, the modification of siRNAs with cholesterol at the 3′-end of the passenger strand was tolerated; however, the modification at the guide strand significantly reduces its silencing activity. The siRNAs modified with the lipophilic groups did not possess ability to penetrate the plasma membranes of HT-1080 cells without the transfection reagent. However, the results described in this report will aid in designing novel siRNAs with cell membrane-permeable molecules.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Yoshihito Ueno, Takumi Inoue, Mahito Yoshida, Kayo Yoshikawa, Aya Shibata, Yoshiaki Kitamura, Yukio Kitade
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 19) pp:5194-5196
Publication Date(Web):1 October 2008
DOI:10.1016/j.bmcl.2008.08.086
We describe the synthesis and silencing activities of siRNA possessing N1-[3,5-bis(hydroxymethyl)phenyl]thymine (bt) in their 3′-overhang regions. We found that an siRNA possessing bt in the 3′-overhang region was more effective than an siRNA with natural nucleosides and the siRNA possessing 1,3-bis(hydroxymethyl)benzene (b) without a nucleobase at the 3′-overhang region in in vitro experiment using HeLa cells system. Furthermore, the RNA possessing bt at its 3′-end was more resistant to nucleolytic hydrolysis by snake venom phosphodiesterase (a 3′-exonuclease) than the RNA possessing the natural nucleoside 2′-deoxythymidine at the 3′-end. Thus, the compound bt will be a novel 3′-overhang moiety that can enhance the silencing activity and nuclease-resistant property of siRNAs.
Co-reporter:Takayuki Ando, Kenji Kojima, Praveen Chahota, Atsushi Kozaki, Nikalje D. Milind, Yukio Kitade
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 8) pp:2615-2618
Publication Date(Web):15 April 2008
DOI:10.1016/j.bmcl.2008.03.029
4′-Modified noraristeromycin (NAM) analogs, 4′-sulfo-, 4′-sulfamoy, 4′-azido and 4′-amino-NAM, were systematically synthesized. The inhibitory activities of these analogs and related compounds against Plasmodium falciparum and human S-adenosyl-l-homocysteine hydrolase were investigated.
Co-reporter:Takayuki Ando, Hisashi Shinohara, Xiong Luo, Mahmoud Kandeel, Yukio Kitade
Carbohydrate Research 2007 Volume 342(Issue 17) pp:2641-2648
Publication Date(Web):10 December 2007
DOI:10.1016/j.carres.2007.08.009
1′,2′-cis-β-Glycosyladenine nucleosides, such as β-altroside, β-mannoside, and β-idoside, were efficiently synthesized from the corresponding 1′,2′-trans-β-6-chloropurine derivatives, β-glucoside, and β-galactoside. Nucleophilic substitution of the O-trifluoromethanesulfonyl groups at the C-2′ and/or 3′ was carried out using tetrabutylammonium acetate or cesium acetate under mild conditions. Subsequent deprotection and amidation afforded the desired compounds, 1′,2′-cis-β-pyranosyladenine nucleosides.
Co-reporter:Aya Kato, Yuri Yasuda, Yoshiaki Kitamura, Mahmoud Kandeel, Yukio Kitade
Parasitology International (September 2012) Volume 61(Issue 3) pp:501-503
Publication Date(Web):1 September 2012
DOI:10.1016/j.parint.2012.03.001
During the course of our research into new anti-malaria drugs, Plasmodium falciparum thymidylate kinase (PfTMK) has emerged as an important drug target because of its unique substrate specificity. Compared with human thymidylate kinase (HsTMK), PfTMK shows broader substrate specificity, which includes both purine and pyrimidine nucleotides. PfTMK accepts both 2′-deoxyguanosine monophosphate (dGMP) and thymidine monosphosphate (TMP) as substrates. We have evaluated the inhibitory activity of seven carbocyclic thymidine analogs and report the first structure‒activity relationship for these inhibitors against PfTMK. The 2′,3′ dideoxycarbocyclic derivative of thymidine showed the most potent inhibition of the enzyme. The KidTMP and KidGMP values were 20 and 7 μM respectively. Thus, further modifications of carbocyclic thymidine analogs represent a good strategy for developing more powerful thymidylate kinase inhibitors.Graphical abstractDownload full-size imageHighlights► We designed a series of carbocyclic nucleoside analogs. ► We established the first structure–activity relationship. ► Several carbocyclic thymidine analogs were good inhibitors. ► The Ki values against PfTMK was in the low micromolar range.
Co-reporter:Yoshihiro Noguchi, Yuri Yasuda, Makoto Tashiro, Tadashi Kataoka, Yoshiaki Kitamura, Mahmoud Kandeel, Yukio Kitade
Parasitology International (August 2013) Volume 62(Issue 4) pp:368-371
Publication Date(Web):1 August 2013
DOI:10.1016/j.parint.2013.03.009
•We synthesized enantioselective 2′,3′-dideoxycarbocyclic pyrimidine nucleosides.•We screened several carbocyclic pyrimidine analogues for inhibitory activity against PfTMK.•The fluorinated derivative exhibited the most potent inhibitory activity.Plasmodium falciparum thymidylate kinase (PfTMK) is a promising antimalarial target due to its unique substrate specificity. Recently, we reported that 2′,3′-dideoxycarbocyclic thymidine showed moderate inhibitory activity and reported the related structure–activity relationship for inhibitors against PfTMK. In this study, we have designed and synthesized enantioselective 2′,3′-dideoxycarbocyclic pyrimidine nucleosides based on our previous results and screened them for inhibitory activity against PfTMK. The most potent inhibitor showed KiTMP and KidGMP values of 14 and 20 μM, respectively. The fluorinated dideoxy derivative (-)-7, exhibited lower KiTMP and higher KidGMP compared with that of the parent compound (KiTMP, KidGMP equals 20 and 7 μM, respectively). The modification of carbocyclic pyrimidine nucleosides is a promising strategy for developing powerful PfTMK inhibitors.Download full-size image
Co-reporter:Takao Shiraishi, Yoshiaki Kitamura, Yoshihito Ueno and Yukio Kitade
Chemical Communications 2011 - vol. 47(Issue 9) pp:NaN2693-2693
Publication Date(Web):2011/01/12
DOI:10.1039/C0CC04979D
We synthesised aryl acetylene derivatives as versatile probes for labelling of oligonucleotides. RNA oligomers bearing an aryl acetylene molecule rapidly reacted with benzylazide derivatives under ligand-free click reaction conditions.
Co-reporter:Yoshihito Ueno, Akihiro Kawamura, Keiji Takasu, Shinji Komatsuzaki, Takumi Kato, Satoru Kuboe, Yoshiaki Kitamura and Yukio Kitade
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 13) pp:NaN2769-2769
Publication Date(Web):2009/05/12
DOI:10.1039/B901631G
This paper describes the synthesis and properties of a novel molecular beacon (MB) containing a benzene-phosphate backbone at its stem moiety. The fluorescence intensity of MBs was found to stabilize by the introduction of the benzene-phosphate backbone at its stem moiety. Furthermore, an MB containing the benzene-phosphate backbone was more resistant to DNase I (endonuclease) than an MB comprising natural DNA and 2′-O-methyl-RNA. These results indicate that the MB with the benzene-phosphate backbone is superior as a molecular beacon as compared to the MB composed of natural DNA and 2′-O-methyl-RNA.

![Glycine, L-γ-glutamyl-S-[1-[[4-[1-[(4-fluorophenyl)methyl]-1H-1,2,3-triazol-4-yl]phenyl]methyl]-2,5-dioxo-3-pyrrolidinyl]-L-cysteinyl-](/data/chemimg/3722700/1401893-53-5.png)
![Glycine, L-γ-glutamyl-S-[1-[[4-[1-[(4-fluorophenyl)methyl]-1H-1,2,3-triazol-4-yl]phenyl]methyl]-2,5-dioxo-3-pyrrolidinyl]-L-cysteinyl-](/data/chemimg/3722700/1401893-53-5_b.png)
![Glycine, L-γ-glutamyl-S-[1-[(4-ethynylphenyl)methyl]-2,5-dioxo-3-pyrrolidinyl]-L-cysteinyl-](/data/chemimg/3723500/1401893-51-3.png)
![Glycine, L-γ-glutamyl-S-[1-[(4-ethynylphenyl)methyl]-2,5-dioxo-3-pyrrolidinyl]-L-cysteinyl-](/data/chemimg/3723500/1401893-51-3_b.png)
![Benzamide, N-[5-(2,5-dihydro-2,5-dioxo-1H-pyrrol-1-yl)pentyl]-4-ethynyl-](/data/chemimg/3723500/1401893-43-3.png)
![Benzamide, N-[5-(2,5-dihydro-2,5-dioxo-1H-pyrrol-1-yl)pentyl]-4-ethynyl-](/data/chemimg/3723500/1401893-43-3_b.png)
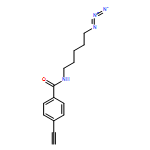
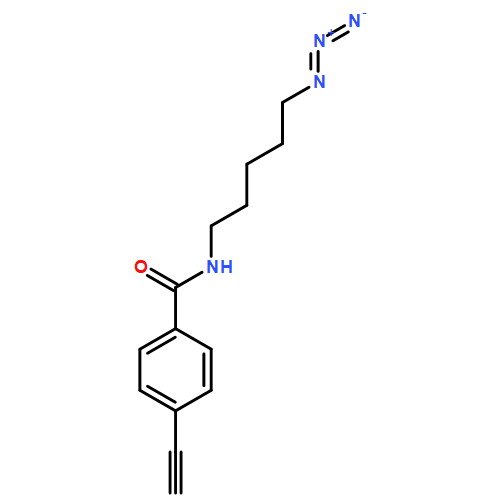
![Pentanoic acid, 5-[(4-ethynylbenzoyl)amino]-, 2,5-dioxo-1-pyrrolidinyl ester](/data/chemimg/3723500/1401893-34-2.png)
![Pentanoic acid, 5-[(4-ethynylbenzoyl)amino]-, 2,5-dioxo-1-pyrrolidinyl ester](/data/chemimg/3723500/1401893-34-2_b.png)
![Pentanoic acid, 5-[(4-ethynylbenzoyl)amino]-](/data/chemimg/3723500/1401893-28-4.png)
![Pentanoic acid, 5-[(4-ethynylbenzoyl)amino]-](/data/chemimg/3723500/1401893-28-4_b.png)
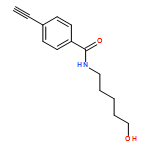
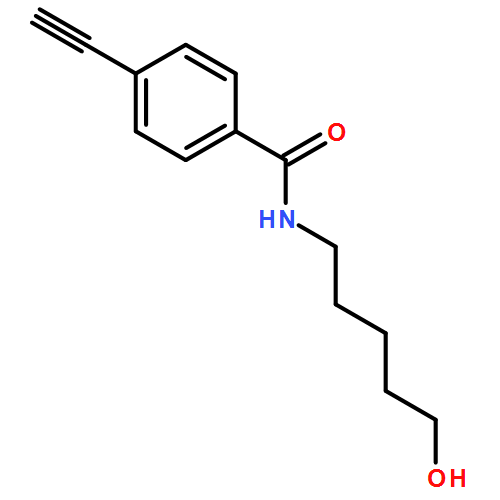



![1H-1,2,3-Triazole, 1-[(4-fluorophenyl)methyl]-4-phenyl-](/data/chemimg/2342000/848950-93-6.png)
![1H-1,2,3-Triazole, 1-[(4-fluorophenyl)methyl]-4-phenyl-](/data/chemimg/2342000/848950-93-6_b.png)
![Uridine, 5'-deoxy-3'-O-[(1,1-dimethylethyl)dimethylsilyl]-5-fluoro-](http://img.cochemist.com/ccimg/500900/500882-62-2.png)
![Uridine, 5'-deoxy-3'-O-[(1,1-dimethylethyl)dimethylsilyl]-5-fluoro-](http://img.cochemist.com/ccimg/500900/500882-62-2_b.png)
![Uridine, 5'-deoxy-2'-O-[(1,1-dimethylethyl)dimethylsilyl]-5-fluoro-](http://img.cochemist.com/ccimg/500900/500882-61-1.png)
![Uridine, 5'-deoxy-2'-O-[(1,1-dimethylethyl)dimethylsilyl]-5-fluoro-](http://img.cochemist.com/ccimg/500900/500882-61-1_b.png)
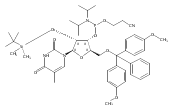


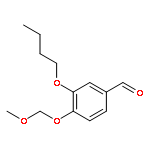
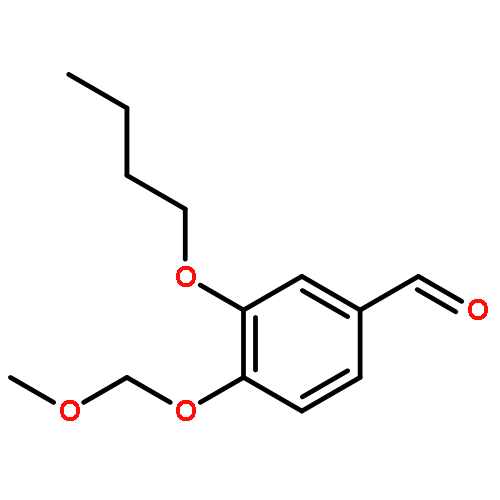








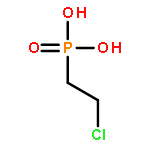
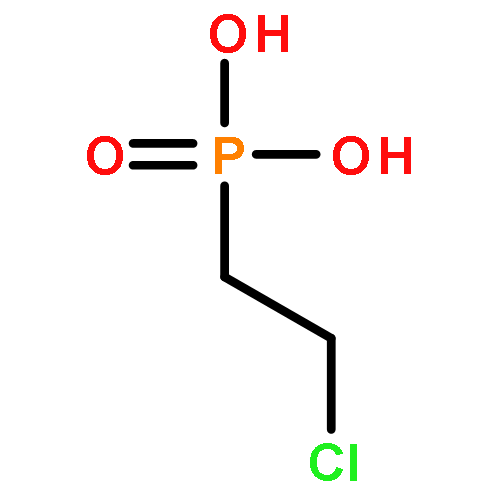






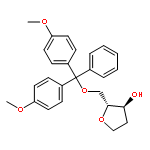
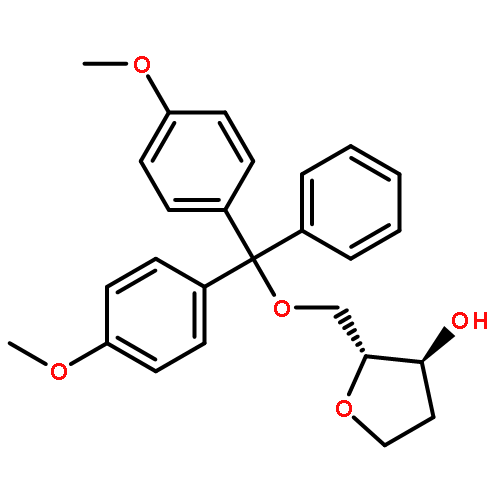
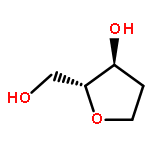
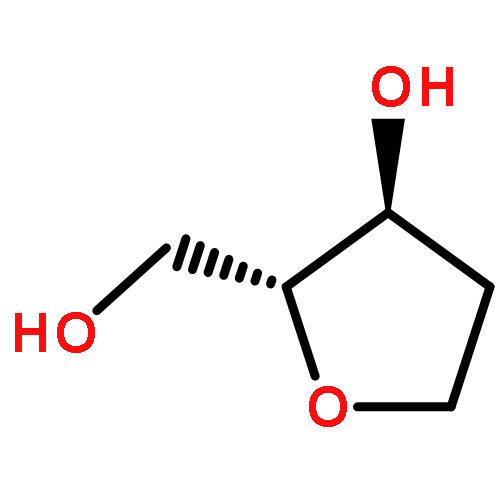


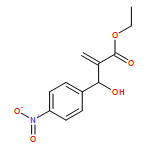
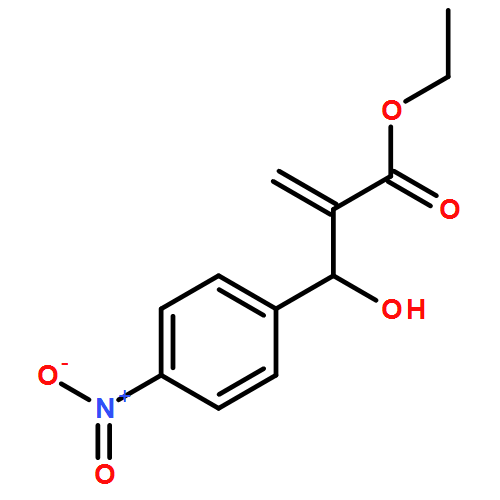

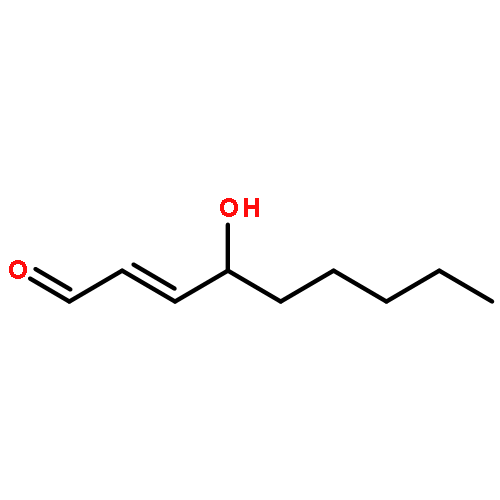


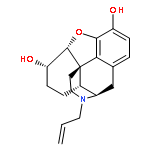
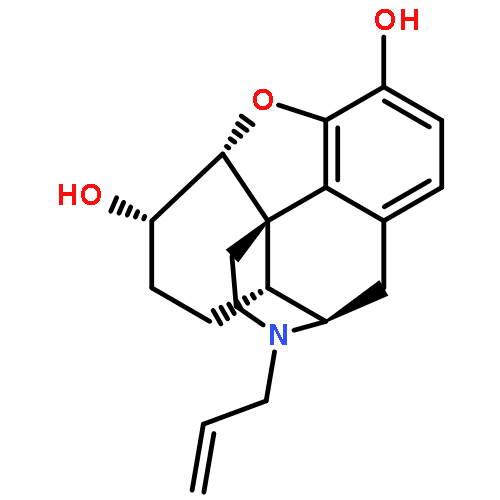
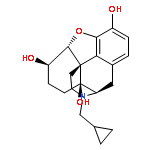
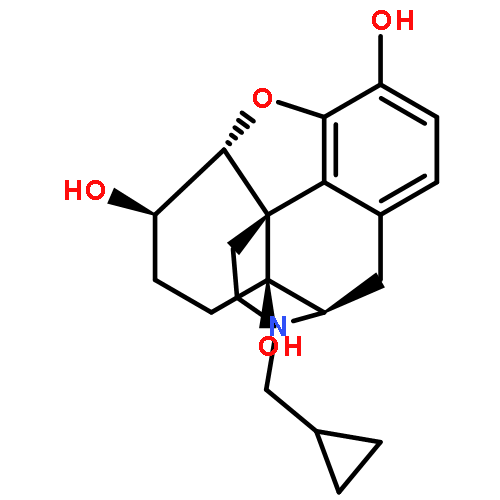







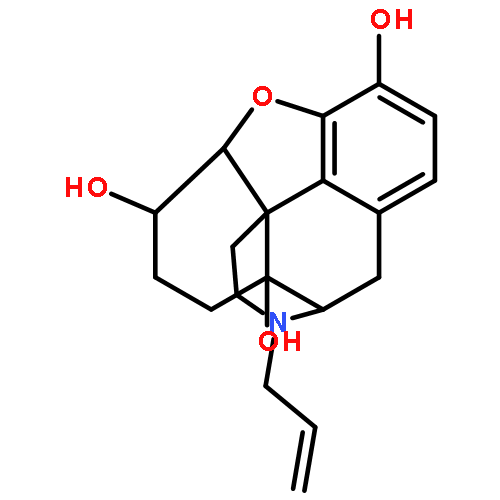


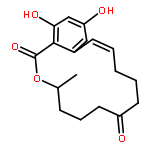
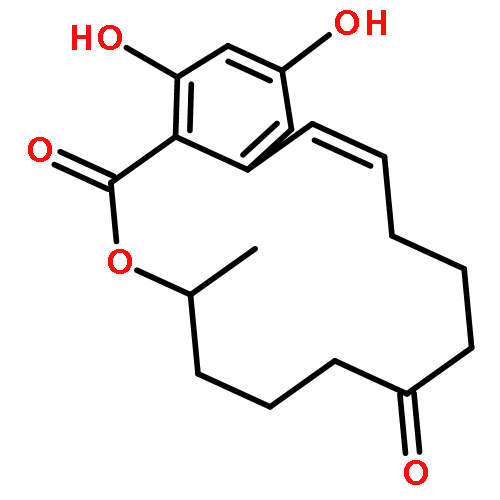
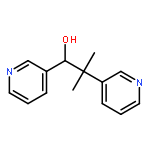





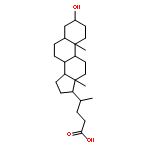







![Glycine, N-[(3a,5b,7a)-3,7-dihydroxy-24-oxocholan-24-yl]-](http://img.cochemist.com/ccimg/700/640-79-9.png)
![Glycine, N-[(3a,5b,7a)-3,7-dihydroxy-24-oxocholan-24-yl]-](http://img.cochemist.com/ccimg/700/640-79-9_b.png)
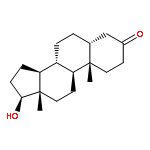
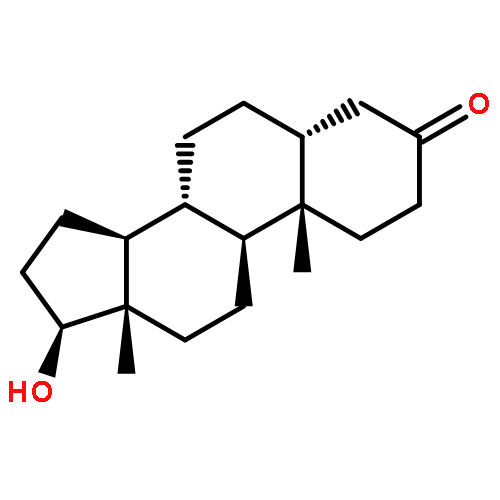

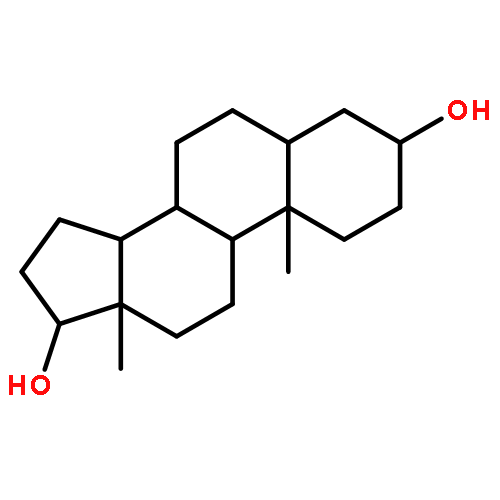
![Ethanesulfonic acid,2-[[(5b)-3,7,12,24-tetraoxocholan-24-yl]amino]-(9CI)](http://img.cochemist.com/ccimg/600/517-37-3.png)
![Ethanesulfonic acid,2-[[(5b)-3,7,12,24-tetraoxocholan-24-yl]amino]-(9CI)](http://img.cochemist.com/ccimg/600/517-37-3_b.png)
![Ethanesulfonic acid,2-[[(3a,5b)-3-hydroxy-24-oxocholan-24-yl]amino]-](http://img.cochemist.com/ccimg/600/516-90-5.png)
![Ethanesulfonic acid,2-[[(3a,5b)-3-hydroxy-24-oxocholan-24-yl]amino]-](http://img.cochemist.com/ccimg/600/516-90-5_b.png)
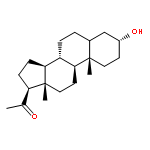
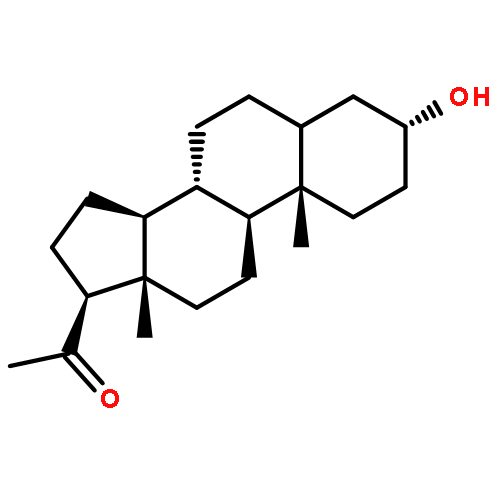
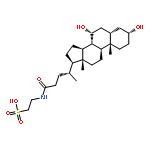

![Glycine, N-[(3a,5b)-3-hydroxy-24-oxocholan-24-yl]-](http://img.cochemist.com/ccimg/500/474-74-8.png)
![Glycine, N-[(3a,5b)-3-hydroxy-24-oxocholan-24-yl]-](http://img.cochemist.com/ccimg/500/474-74-8_b.png)



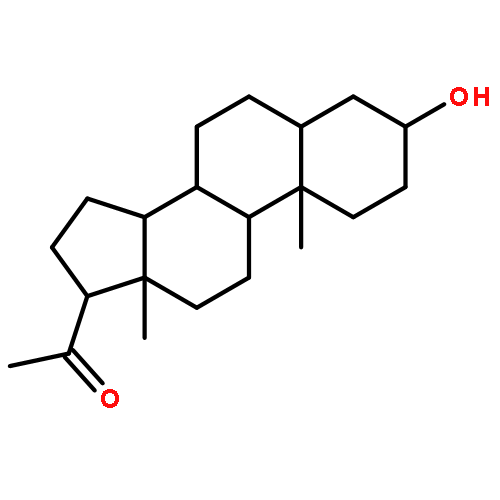


![2-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonic Acid](http://img.cochemist.com/ccimg/100/81-24-3.png)
![2-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonic Acid](http://img.cochemist.com/ccimg/100/81-24-3_b.png)
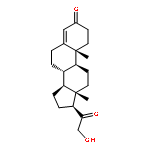
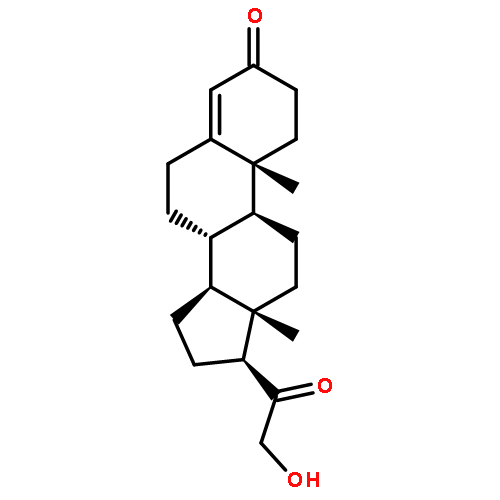




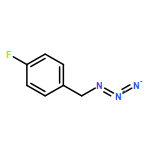



![10H-Benzo[4,5]cyclohepta[1,2-b]thiophen-10-one, 4,9-dihydro-4-(1-methyl-4-piperidinylidene)-](http://img.cochemist.com/ccimg/34600/34580-13-7.png)
![10H-Benzo[4,5]cyclohepta[1,2-b]thiophen-10-one, 4,9-dihydro-4-(1-methyl-4-piperidinylidene)-](http://img.cochemist.com/ccimg/34600/34580-13-7_b.png)