Co-reporter:Artur K. Mailyan, John A. Eickhoff, Anastasiia S. Minakova, Zhenhua Gu, Ping Lu, and Armen Zakarian
Chemical Reviews 2016 Volume 116(Issue 7) pp:4441
Publication Date(Web):March 25, 2016
DOI:10.1021/acs.chemrev.5b00712
The main objective of this review is to provide a comprehensive survey of methods used for stereoselective construction of carbon–nitrogen bonds during the total synthesis of nitrogen-containing natural products that have appeared in the literature since 2000. The material is organized by specific reaction in order of decreasing number of applications in natural product synthesis. About 800 total syntheses of natural products with stereogenic carbon–nitrogen bonds described since 2000 have been reviewed.
Co-reporter:Kai Yu, Ping Lu, Jeffrey J. Jackson, Thuy-Ai D. Nguyen, Joseph Alvarado, Craig E. Stivala, Yun Ma, Kyle A. Mack, Trevor W. HaytonDavid B. Collum, Armen Zakarian
Journal of the American Chemical Society 2016 Volume 139(Issue 1) pp:527-533
Publication Date(Web):December 7, 2016
DOI:10.1021/jacs.6b11673
Lithium enolates derived from carboxylic acids are ubiquitous intermediates in organic synthesis. Asymmetric transformations with these intermediates, a central goal of organic synthesis, are typically carried out with covalently attached chiral auxiliaries. An alternative approach is to utilize chiral reagents that form discrete, well-defined aggregates with lithium enolates, providing a chiral environment conducive of asymmetric bond formation. These reagents effectively act as noncovalent, or traceless, chiral auxiliaries. Lithium amides are an obvious choice for such reagents as they are known to form mixed aggregates with lithium enolates. We demonstrate here that mixed aggregates can effect highly enantioselective transformations of lithium enolates in several classes of reactions, most notably in transformations forming tetrasubstituted and quaternary carbon centers. Easy recovery of the chiral reagent by aqueous extraction is another practical advantage of this one-step protocol. Crystallographic, spectroscopic, and computational studies of the central reactive aggregate, which provide insight into the origins of selectivity, are also reported.
Co-reporter:Artur K. Mailyan, Kyle Young, Joanna L. Chen, Bradley T. Reid, and Armen Zakarian
Organic Letters 2016 Volume 18(Issue 21) pp:5532-5535
Publication Date(Web):October 25, 2016
DOI:10.1021/acs.orglett.6b02778
A method for a directed stereoselective guanidinylation of alkenes is described. The guanidine unit can be delivered as an intact fragment by a hydroxy or carboxy group, usually with a high level of stereocontrol. After the guanidine delivery, the directing group can be cleaved under exceptionally mild conditions, typically by alcoholysis in the presence of acetic acid. Broad functional group tolerance and mild reaction conditions for the cycloguanidilation suggest applications in medicinal chemistry and natural products synthesis.
Co-reporter:Qing Xiao; Kyle Young
Journal of the American Chemical Society 2015 Volume 137(Issue 18) pp:5907-5910
Publication Date(Web):April 30, 2015
DOI:10.1021/jacs.5b03531
Muironolide A is a fascinating tetrachlorinated marine polyketide isolated from the sponge of Phorbas sp. Only 90 μg had been isolated, and the structure was established by nanoscale NMR techniques. Herein we report the total synthesis of the substance with the assigned structure of muironolide A, propose a revised structure based on NMR data, and complete the enantioselective total synthesis of muironolide A.
Co-reporter:Ping Lu; Jeffrey J. Jackson; John A. Eickhoff
Journal of the American Chemical Society 2015 Volume 137(Issue 2) pp:656-659
Publication Date(Web):January 5, 2015
DOI:10.1021/ja512213c
Michael addition is a premier synthetic method for carbon–carbon and carbon–heteroatom bond formation. Using chiral dilithium amides as traceless auxiliaries, we report the direct enantioselective Michael addition of carboxylic acids. A free carboxyl group in the product provides versatility for further functionalization, and the chiral reagent can be readily recovered by extraction with aqueous acid. The method has been applied in the enantioselective total synthesis of the purported structure of pulveraven B.
Co-reporter:Craig E. Stivala, Evelyne Benoit, Rómulo Aráoz, Denis Servent, Alexei Novikov, Jordi Molgó and Armen Zakarian
Natural Product Reports 2015 vol. 32(Issue 3) pp:411-435
Publication Date(Web):22 Oct 2014
DOI:10.1039/C4NP00089G
Covering: up to August 2014
From a small group of exotic compounds isolated only two decades ago, Cyclic Imine (CI) toxins have become a major class of marine toxins with global distribution. Their distinct chemical structure, biological mechanism of action, and intricate chemistry ensures that CI toxins will continue to be the subject of fascinating fundamental studies in the broad fields of chemistry, chemical biology, and toxicology. The worldwide occurrence of potent CI toxins in marine environments, their accumulation in shellfish, and chemical stability are important considerations in assessing risk factors for human health. This review article aims to provide an account of chemistry, biology, and toxicology of CI toxins from their discovery to the present day.
Co-reporter:Kyle Young;Qing Xiao
European Journal of Organic Chemistry 2015 Volume 2015( Issue 11) pp:2337-2341
Publication Date(Web):
DOI:10.1002/ejoc.201500174
Abstract
A lanthanide-catalyzed intramolecular Diels–Alder (IMDA) reaction en route to the synthesis of muironolide A is described, along with detailed crystallographic structural studies of lanthanide complexes between β-oxo amides and terpyridine. The X-ray crystallographic studies are first examples of nonsymmetric heteroleptic complexes of lanthanides with β-oxo carbonyl compounds, which reveal several structural properties that could be exploited in the development of catalytic reactions with lanthanides.
Co-reporter:Ping Lu, Aaron T. Herrmann, and Armen Zakarian
The Journal of Organic Chemistry 2015 Volume 80(Issue 15) pp:7581-7589
Publication Date(Web):July 6, 2015
DOI:10.1021/acs.joc.5b01177
A stereodivergent approach to the central thiolane subunit of Nuphar sesquiterpene thioalkaloids has been developed. This approach features a rhodium-catalyzed Stevens-type rearrangement in conjunction with an enzyme resolution reaction. Further elaboration into a polycyclic ring system via alcohol oxidation and ring-closing metathesis is also described.
Co-reporter:Hiroyuki Kobayashi, John A. Eickhoff, and Armen Zakarian
The Journal of Organic Chemistry 2015 Volume 80(Issue 20) pp:9989-9999
Publication Date(Web):September 17, 2015
DOI:10.1021/acs.joc.5b01558
Facile synthesis of a variety of α-heterosubstituted ketones under mild conditions was achieved by copper-mediated cross-coupling of thioesters with functionalized organostannanes. Application of this coupling methodology provided a concise pathway for the conversion of carboxylic acids to 2-aminoimidazoles, 2-aminothiazoles, and 2-aminooxazoles via thioesters in practical yields.
Co-reporter:Jeffrey J. Jackson;Dr. Hiroyuki Kobayashi;Sophia D. Steffens;Dr. Armen Zakarian
Angewandte Chemie 2015 Volume 127( Issue 34) pp:10109-10113
Publication Date(Web):
DOI:10.1002/ange.201504113
Abstract
The asymmetric synthesis of dragmacidin D (1) was completed in 10 steps. Its sole stereocenter was set by using direct asymmetric alkylation enabled by a C2-symmetric tetramine and lithium N-(trimethylsilyl)-tert-butylamide as the enolization reagent. A central Larock indole synthesis was employed in a convergent assembly of the heterocyclic subunits. The stereochemical evidence from this work strongly supports the predicted S configuration at the 6′′′ position, which is consistent with other members of the dragmacidin family of natural products.
Co-reporter:Jeffrey J. Jackson;Dr. Hiroyuki Kobayashi;Sophia D. Steffens;Dr. Armen Zakarian
Angewandte Chemie International Edition 2015 Volume 54( Issue 34) pp:9971-9975
Publication Date(Web):
DOI:10.1002/anie.201504113
Abstract
The asymmetric synthesis of dragmacidin D (1) was completed in 10 steps. Its sole stereocenter was set by using direct asymmetric alkylation enabled by a C2-symmetric tetramine and lithium N-(trimethylsilyl)-tert-butylamide as the enolization reagent. A central Larock indole synthesis was employed in a convergent assembly of the heterocyclic subunits. The stereochemical evidence from this work strongly supports the predicted S configuration at the 6′′′ position, which is consistent with other members of the dragmacidin family of natural products.
Co-reporter:Ping Lu ; Artur Mailyan ; Zhenhua Gu ; David M. Guptill ; Hengbin Wang ; Huw M. L. Davies
Journal of the American Chemical Society 2014 Volume 136(Issue 51) pp:17738-17749
Publication Date(Web):November 19, 2014
DOI:10.1021/ja510573v
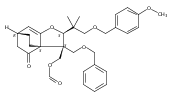
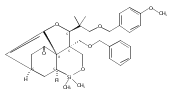
The evolution of a program directed at the enantioselective total synthesis of maoecrystal V, a highly modified ent-kauranoid, is described. An early stage chiral auxiliary-directed asymmetric C–H functionalization for the construction of a key benzofuran intermediate enabled the first asymmetric synthesis of the natural enantiomer of maoecrystal V, confirming the assigned stereochemistry. A divergent course of the central intramolecular Diels–Alder reaction, which is dependent on the nature of the dienophile, initially led to the development of an unanticipated and previously unknown isomer of maoecrystal V, which we named maoecrystal ZG. In light of the reported selective and potent cytotoxic activity of maoecrystal V, the cytotoxic properties of maoecrystal ZG were also investigated.
Co-reporter:Phillip J. Mabe and Armen Zakarian
Organic Letters 2014 Volume 16(Issue 2) pp:516-519
Publication Date(Web):December 31, 2013
DOI:10.1021/ol403398u
A mild method for α-hydroxylation of N-acyl oxazolidinones by asymmetric radical addition of the 2,2,6,6-tetramethylpiperidine N-oxy (TEMPO) radical to titanium enolates was developed. The high diastereoselectivity and broad scope of the reaction show synthetic utility for the α-hydroxylation of substrates that are not tolerant to strongly basic conditions.
Co-reporter:Joseph Alvarado, Aaron T. Herrmann, and Armen Zakarian
The Journal of Organic Chemistry 2014 Volume 79(Issue 13) pp:6206-6220
Publication Date(Web):June 13, 2014
DOI:10.1021/jo500957d
A direct α-fluorination of N-acyloxazolidinones based on the unique reactivity of group IVa metal enolates has been developed. The reaction is an experimentally simple, low-cost, quick, and energy-efficient alternative for asymmetric α-fluorination of N-acyloxazolidinones. Preliminary studies have shown compatibility with alkyl, alkenyl, and alkynyl, aromatic, and several heteroaromatic substituents. High diastereoselectivities have been achieved with most substrates tested, and the reaction is typically complete within 1 h at ambient temperature.
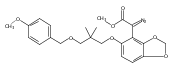
Co-reporter:Ping Lu ; Zhenhua Gu
Journal of the American Chemical Society 2013 Volume 135(Issue 39) pp:14552-14555
Publication Date(Web):September 19, 2013
DOI:10.1021/ja408231t
A total synthesis of the unusual ent-kaurane maoecrystal V is described. The synthesis strategy features a counterintuitive early disconnection of the lactone subunit to a polycyclic enol ether intermediate in order to preserve the central tetrahydrofuran ring until the beginning stages of the synthesis. This strategy enables an application of C–H functionalization at the early phase of the synthesis during the construction of a dihydrobenzofuran intermediate.
Co-reporter:Yun Ma ; Craig E. Stivala ; Ashley M. Wright ; Trevor Hayton ; Jun Liang ; Ivan Keresztes ; Emil Lobkovsky ; David B. Collum
Journal of the American Chemical Society 2013 Volume 135(Issue 45) pp:16853-16864
Publication Date(Web):May 8, 2013
DOI:10.1021/ja403076u
A combination of X-ray crystallography, 6Li, 15N, and 13C NMR spectroscopies, and density functional theory computations affords insight into the structures and reactivities of intervening aggregates underlying highly selective asymmetric alkylations of carboxylic acid dianions (enediolates) mediated by the dilithium salt of a C2-symmetric chiral tetraamine. Crystallography shows a trilithiated n-butyllithium–dilithiated amide that has dimerized to a hexalithiated form. Spectroscopic studies implicate the non-dimerized trilithiated mixed aggregate. Reaction of the dilithiated amide with the dilithium enediolate derived from phenylacetic acid affords a tetralithio aggregate comprised of the two dianions in solution and the dimerized octalithio form in the solid state. Computational studies shed light on the details of the solution structures and afford a highly predictive stereochemical model.
Co-reporter:Qing Xiao, Kyle Young, and Armen Zakarian
Organic Letters 2013 Volume 15(Issue 13) pp:3314-3317
Publication Date(Web):June 13, 2013
DOI:10.1021/ol401354a
An efficient stereocontrolled construction of the fully substituted isoindolone subunit of muironolide A is described. The approach is centered on the intramolecular Diels–Alder reaction between the enol form of the β-keto amide and an α,β,γ,δ-unsaturated ester, followed by the installation of the cyclohexene double bond.
Co-reporter:Aaron T. Herrmann ; Lindsay L. Smith
Journal of the American Chemical Society 2012 Volume 134(Issue 16) pp:6976-6979
Publication Date(Web):April 9, 2012
DOI:10.1021/ja302552e
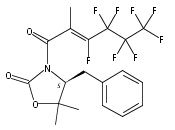
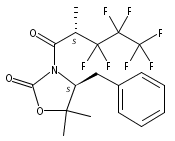
A Ru-catalyzed direct thermal trifluoromethylation and perfluoroalkylation of N-acyloxazolidinones has been developed. The reaction is experimentally simple and requires inexpensive reagents while providing good yields of products with good levels of stereocontrol. Preliminary studies have shown notable compatibility with functional groups, aromatics, and certain heteroaromatic substituents. The described method provides a useful alternative for the synthesis of fluorinated materials in an experimentally convenient manner.
Co-reporter:Craig E. Stivala, Zhenhua Gu, Lindsay L. Smith, and Armen Zakarian
Organic Letters 2012 Volume 14(Issue 3) pp:804-807
Publication Date(Web):January 19, 2012
DOI:10.1021/ol203342e
The synthesis of two complex subunits en route to spirolide C is described. A key alkyllithium addition to an aldehyde joins the fragments, which are advanced in order to investigate a ring-closing metathesis to form the 23-membered all-carbon macrocyclic framework.
Co-reporter:Jeffrey J. Jackson, Craig E. Stivala, Bogdan I. Iorga, Jordi Molgó, and Armen Zakarian
The Journal of Organic Chemistry 2012 Volume 77(Issue 22) pp:10435-10440
Publication Date(Web):November 1, 2012
DOI:10.1021/jo301632d
Pinnatoxins belong to the cyclic imine (CI) group of marine toxins with a unique toxicological profile. The need for structural integrity of the aliphatic 7-membered cyclic imine for the potent bioactivity of pinnatoxins has been experimentally demonstrated. In this study, we probe interconversion of the natural cyclic imine and its open form, pinnatoxin A amino ketone (PnTX AK), under physiologically relevant aqueous conditions. Our studies demonstrate the high stability of PnTX A. The unusual stability of the imine ring in PnTX A has implications for its oral toxicity and detoxification. These studies, as well the access to PnTX amino ketone, were enabled by the total synthesis of (+)-pinnatoxin A completed previously in our laboratory.
Co-reporter:Z. Gu;T. Saito;A. Zakarian
Chemistry of Heterocyclic Compounds 2012 Volume 48( Issue 1) pp:11-16
Publication Date(Web):2012 April
DOI:10.1007/s10593-012-0961-y
This review describes selected approaches to the synthesis of 4H-5,6-dihydro-1,2-oxazines and application of these methods to the synthesis of natural products containing this heterocyclic ring system. The focus is on applications and on interesting aspects of chemistry related to this subclass of natural products, as well as newer methods for the synthesis of oxazines.
Co-reporter:Romulo Araoz ; Denis Servent ; Jordi Molgó ; Bogdan I. Iorga ; Carole Fruchart-Gaillard ; Evelyne Benoit ; Zhenhua Gu ; Craig Stivala
Journal of the American Chemical Society 2011 Volume 133(Issue 27) pp:10499-10511
Publication Date(Web):June 6, 2011
DOI:10.1021/ja201254c
Pinnatoxins belong to an emerging class of potent marine toxins of the cyclic imine group. Detailed studies of their biological effects have been impeded by unavailability of the complex natural product from natural sources. This work describes the development of a robust, scalable synthetic sequence relying on a convergent strategy that delivered a sufficient amount of the toxin for detailed biological studies and its commercialization for use by other research groups and regulatory agencies. A central transformation in the synthesis is the highly diastereoselective Ireland–Claisen rearrangement of a complex α,α-disubstituted allylic ester based on a unique mode for stereoselective enolization through a chirality match between the substrate and the lithium amide base. With synthetic pinnatoxin A, a detailed study has been performed that provides conclusive evidence for its mode of action as a potent inhibitor of nicotinic acetylcholine receptors selective for the human neuronal α7 subtype. The comprehensive electrophysiological, biochemical, and computational studies support the view that the spiroimine subunit of pinnatoxins is critical for blocking nicotinic acetylcholine receptor subtypes, as evidenced by analyzing the effect of a synthetic analogue of pinnatoxin A containing an open form of the imine ring. Our studies have paved the way for the production of certified standards to be used for mass-spectrometric determination of these toxins in marine matrices and for the development of tests to detect these toxins in contaminated shellfish.
Co-reporter:Craig E. Stivala
Journal of the American Chemical Society 2011 Volume 133(Issue 31) pp:11936-11939
Publication Date(Web):July 11, 2011
DOI:10.1021/ja205107x
A direct, highly enantioselective alkylation of arylacetic acids via enediolates using a readily available chiral lithium amide as a stereodirecting reagent has been developed. This approach circumvents the traditional attachment and removal of chiral auxiliaries used currently for this type of transformation. The protocol is operationally simple, and the chiral reagent is readily recoverable.
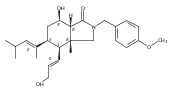
Co-reporter:Zhenhua Gu and Armen Zakarian
Organic Letters 2011 Volume 13(Issue 5) pp:1080-1082
Publication Date(Web):January 27, 2011
DOI:10.1021/ol1031238
An approach toward the synthesis of maoecrystal V is described. The synthetic strategy for this approach was designed to address unique challenges posed by the strained tetrahydrofuran ring at the center of the target structure.
Co-reporter:Aaron T. Herrmann, Steven R. Martinez, and Armen Zakarian
Organic Letters 2011 Volume 13(Issue 14) pp:3636-3639
Publication Date(Web):June 16, 2011
DOI:10.1021/ol201283n
A new protecting-group-free synthesis of the marine monocyclic ether (+)-brevisamide is reported. The enantioselective synthesis utilizes a key asymmetric Henry reaction and an Achmatowicz rearrangement for the formation of the tetrahydropyran ring. A penultimate Stille cross-coupling allows for an efficient installation of the conjugated (E,E)-diene side chain ultimately delivering (+)-brevisamide.
Co-reporter:Elizabeth A. Ilardi ;Dr. Armen Zakarian
Chemistry – An Asian Journal 2011 Volume 6( Issue 9) pp:2260-2263
Publication Date(Web):
DOI:10.1002/asia.201100338
Co-reporter:Dr. Zhenhua Gu;Aaron T. Herrmann ;Dr. Armen Zakarian
Angewandte Chemie 2011 Volume 123( Issue 31) pp:7274-7277
Publication Date(Web):
DOI:10.1002/ange.201101364
Co-reporter:Dr. Zhenhua Gu;Aaron T. Herrmann ;Dr. Armen Zakarian
Angewandte Chemie International Edition 2011 Volume 50( Issue 31) pp:7136-7139
Publication Date(Web):
DOI:10.1002/anie.201101364
Co-reporter:Aaron T. Herrmann ; Tatsuo Saito ; Craig E. Stivala ; Janine Tom
Journal of the American Chemical Society 2010 Volume 132(Issue 17) pp:5962-5963
Publication Date(Web):April 13, 2010
DOI:10.1021/ja101673v
A hydroxyl group-directed, highly regio- and stereoselective transposition of allylic alcohols based on rhenium catalysis has been developed. The method is suitable for a direct isomerization of acetals into the thermodynamically preferred isomer as long as one of the hydroxyl groups is allylic. This method will expand the scope of rhenium-catalyzed alcohol transpositions for complex molecule synthesis.
Co-reporter:Stéphane Beaumont ; Elizabeth A. Ilardi ; Lucas R. Monroe
Journal of the American Chemical Society 2010 Volume 132(Issue 5) pp:1482-1483
Publication Date(Web):January 15, 2010
DOI:10.1021/ja910154f
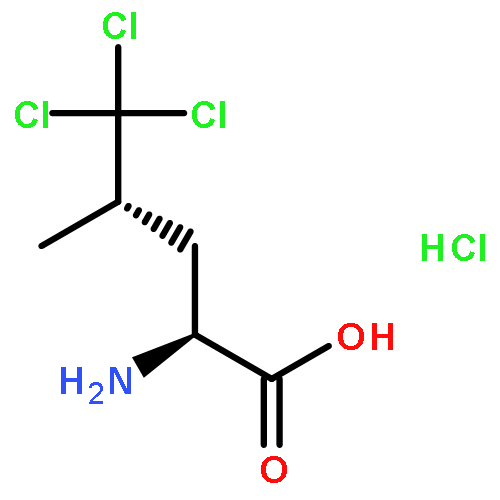
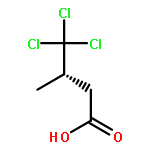
A direct ruthenium-catalyzed radical chloroalkylation of N-acyl oxazolidinones capitalizing on valence tautomerism of titanium enolates has been developed. The chloroalkylation method served as the centerpiece in the enantioselective total synthesis of trichloroleucine-derived marine natural product neodysidenin.
Co-reporter:Zhenhua Gu and Armen Zakarian
Organic Letters 2010 Volume 12(Issue 19) pp:4224-4227
Publication Date(Web):August 26, 2010
DOI:10.1021/ol101523z
A total synthesis of rhazinilam based on a transannular cyclization strategy is described. Using a Heck reaction, the axial chirality of a halogenated 13-membered lactam can be exploited to create the quaternary chiral stereogenic center in the target molecule with high enantiospecificity.
Co-reporter:Stéphane Beaumont;Elizabeth A. Ilardi;Nicholas D. C. Tappin
European Journal of Organic Chemistry 2010 Volume 2010( Issue 30) pp:5743-5765
Publication Date(Web):
DOI:10.1002/ejoc.201000842
Abstract
This microreview provides a compilation of synthetic approaches and total syntheses of pinnatoxin A in a survey of the literature up to early 2010. Pinnatoxin A is the first discovered and representative member of a fascinating group of potent marine toxins that share a spiroimine subunit as a unifying structural element.
Co-reporter:Stéphane Beaumont;Elizabeth A. Ilardi;Nicholas D. C. Tappin
European Journal of Organic Chemistry 2010 Volume 2010( Issue 30) pp:
Publication Date(Web):
DOI:10.1002/ejoc.201090082
Abstract
The cover picture shows the building blocks for the total synthesis of pinnatoxin A as the focal point, culminating with the marine natural product at the top of the stairs. The background reflects a highlight of the Santa Barbara Mission, located in Santa Barbara, California. Founded by Franciscan Friar Fermin de Lasuen on the Feast of St. Barbara in 1786, the mission is the principal cultural and historic landmark of the city. The spiral symbolizes the universal pattern of growth and evolution, as well as the ongoing process of discovery. Details of the total synthesis of pinnatoxin A are presented in the Microreview by A. Zakarian et al. on p. 3743 ff.
Co-reporter:Dr. Zhenhua Gu ;Dr. Armen Zakarian
Angewandte Chemie International Edition 2010 Volume 49( Issue 50) pp:9702-9705
Publication Date(Web):
DOI:10.1002/anie.201005354
Co-reporter:Dr. Zhenhua Gu ;Dr. Armen Zakarian
Angewandte Chemie 2010 Volume 122( Issue 50) pp:9896-9899
Publication Date(Web):
DOI:10.1002/ange.201005354
Co-reporter:Elizabeth A. Ilardi, Craig E. Stivala and Armen Zakarian
Chemical Society Reviews 2009 vol. 38(Issue 11) pp:3133-3148
Publication Date(Web):02 Sep 2009
DOI:10.1039/B901177N
Among the fundamental chemical transformations in organic synthesis, the [3,3]-sigmatropic rearrangement occupies a unique position as a powerful, reliable, and well-defined method for the stereoselective construction of carbon–carbon or carbon–heteroatom bonds. While many other reactions can unite two subunits and create a new bond, the strengths of sigmatropic rearrangements derive from their ability to enable structural reorganization with unmatched build-up of complexity. Recent applications that illustrate [3,3]-sigmatropic processes as a key concept in the synthesis of complex natural products are described in this tutorial review, covering literature from about 2001 through early 2009.
Co-reporter:Elizabeth A. Ilardi, Michael J. Isaacman, Ying-chuan Qin, Sommer A. Shelly, Armen Zakarian
Tetrahedron 2009 65(16) pp: 3261-3269
Publication Date(Web):
DOI:10.1016/j.tet.2008.10.048
Co-reporter:Chong-Dao Lu Dr. Dr.
Angewandte Chemie 2008 Volume 120( Issue 36) pp:6935-6937
Publication Date(Web):
DOI:10.1002/ange.200801652
Co-reporter:Chong-Dao Lu Dr. Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 36) pp:6829-6831
Publication Date(Web):
DOI:10.1002/anie.200801652
Co-reporter:Elizabeth A. Ilardi, Craig E. Stivala and Armen Zakarian
Chemical Society Reviews 2009 - vol. 38(Issue 11) pp:NaN3148-3148
Publication Date(Web):2009/09/02
DOI:10.1039/B901177N
Among the fundamental chemical transformations in organic synthesis, the [3,3]-sigmatropic rearrangement occupies a unique position as a powerful, reliable, and well-defined method for the stereoselective construction of carbon–carbon or carbon–heteroatom bonds. While many other reactions can unite two subunits and create a new bond, the strengths of sigmatropic rearrangements derive from their ability to enable structural reorganization with unmatched build-up of complexity. Recent applications that illustrate [3,3]-sigmatropic processes as a key concept in the synthesis of complex natural products are described in this tutorial review, covering literature from about 2001 through early 2009.
.jpg)
![Rhodium, tetrakis[m-[(aR)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909393-65-3.png)
![Rhodium, tetrakis[m-[(aR)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909393-65-3_b.png)
![Rhodium, tetrakis[m-[(aS)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909389-99-7.png)
![Rhodium, tetrakis[m-[(aS)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909389-99-7_b.png)

















![Pyridine, 2,6-bis[(4S,5R)-4,5-dihydro-4,5-diphenyl-2-oxazolyl]-](/data/chemimg/1180200/292625-77-5.png)
![Pyridine, 2,6-bis[(4S,5R)-4,5-dihydro-4,5-diphenyl-2-oxazolyl]-](/data/chemimg/1180200/292625-77-5_b.png)
![1-[TERT-BUTYL(DIMETHYL)SILYL]OXYHEX-5-EN-3-OL](http://img.cochemist.com/ccimg/261700/261633-45-8.png)
![1-[TERT-BUTYL(DIMETHYL)SILYL]OXYHEX-5-EN-3-OL](http://img.cochemist.com/ccimg/261700/261633-45-8_b.png)



![BENZENEETHANOL, 3-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-](http://img.cochemist.com/ccimg/160800/160701-58-6.png)
![BENZENEETHANOL, 3-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-](http://img.cochemist.com/ccimg/160800/160701-58-6_b.png)
![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4.png)
![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4_b.png)












![2,6-Bis[(4S)-4-tert-butyl-2-oxazolin-2yl]pyridine](/data/chemimg/119100/118949-63-6.png)
![2,6-Bis[(4S)-4-tert-butyl-2-oxazolin-2yl]pyridine](/data/chemimg/119100/118949-63-6_b.png)





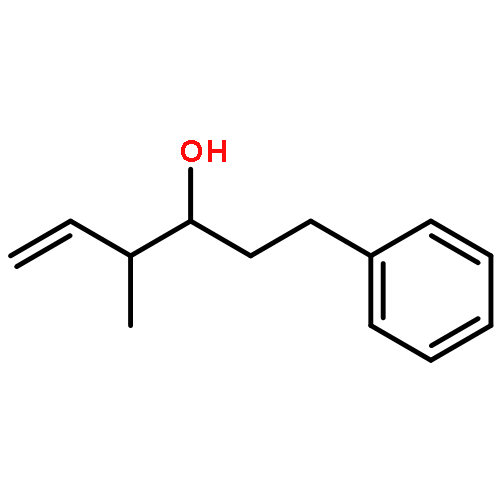






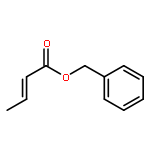









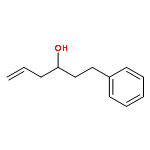
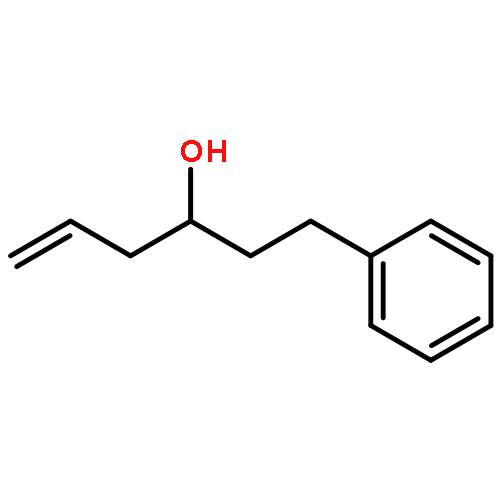





























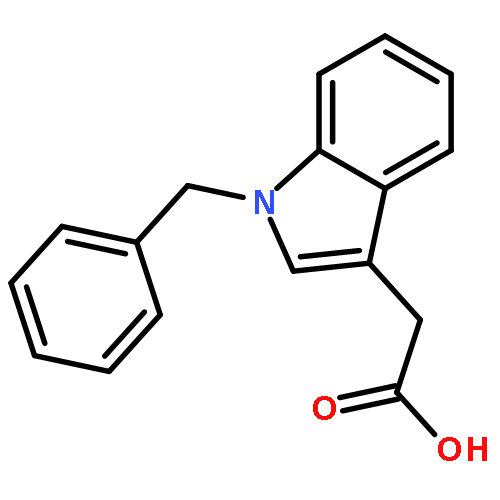
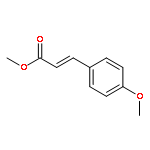
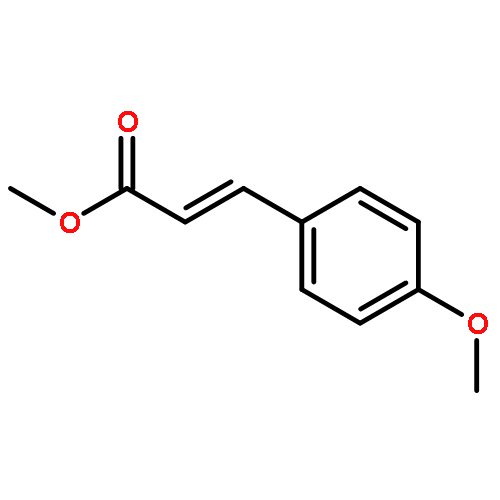




![2,6-Bis((3aS,8aR)-8,8a-dihydro-3aH-indeno[1,2-d]oxazol-2-yl)pyridine](/data/chemimg/119100/185346-09-2.png)
![2,6-Bis((3aS,8aR)-8,8a-dihydro-3aH-indeno[1,2-d]oxazol-2-yl)pyridine](/data/chemimg/119100/185346-09-2_b.png)
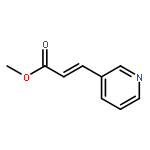

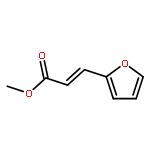
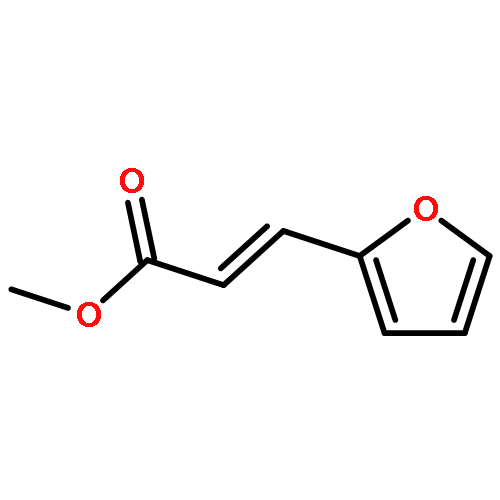


















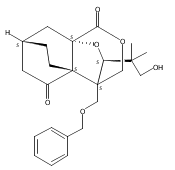



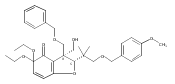



























































































































































































































































































































































































































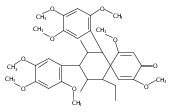










































































































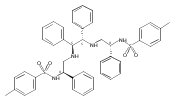







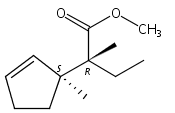



























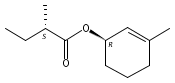



























































































































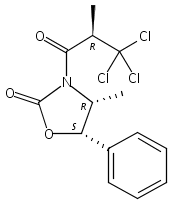















































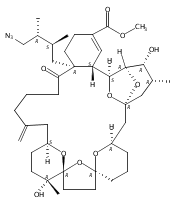



























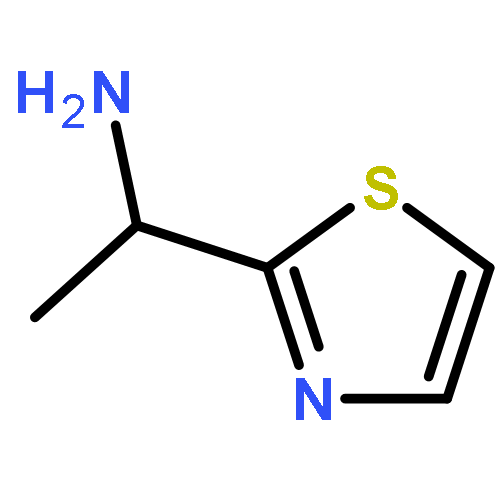
































![Benzene, 1-methoxy-4-[(4-pentynyloxy)methyl]-](http://img.cochemist.com/ccimg/197300/197219-01-5.png)
![Benzene, 1-methoxy-4-[(4-pentynyloxy)methyl]-](http://img.cochemist.com/ccimg/197300/197219-01-5_b.png)














![2,2-dimethyl-n-[(1r)-1-phenyl-2-piperidin-1-ylethyl]propan-1-amine](http://img.cochemist.com/ccimg/153900/153837-28-6.png)
![2,2-dimethyl-n-[(1r)-1-phenyl-2-piperidin-1-ylethyl]propan-1-amine](http://img.cochemist.com/ccimg/153900/153837-28-6_b.png)










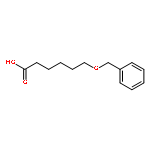










![2-Butene,1-[(1,1-dimethylethyl)thio]-, (E)- (9CI)](http://img.cochemist.com/ccimg/107200/107183-90-4.png)
![2-Butene,1-[(1,1-dimethylethyl)thio]-, (E)- (9CI)](http://img.cochemist.com/ccimg/107200/107183-90-4_b.png)




![Phosphonic acid, P-[(2,2-dimethyl-4-oxo-4H-1,3-dioxin-6-yl)methyl]-, diethyl ester](/data/chemimg/3762300/81956-28-7.png)
![Phosphonic acid, P-[(2,2-dimethyl-4-oxo-4H-1,3-dioxin-6-yl)methyl]-, diethyl ester](/data/chemimg/3762300/81956-28-7_b.png)












![(5S)-4-methoxy-5-(1-methylethyl)-1-[(2E,5S)-6,6,6-trichloro-3-methoxy-5-methylhex-2-enoyl]-1,5-dihydro-2H-pyrrol-2-one](http://img.cochemist.com/ccimg/63100/63079-71-0.png)
![(5S)-4-methoxy-5-(1-methylethyl)-1-[(2E,5S)-6,6,6-trichloro-3-methoxy-5-methylhex-2-enoyl]-1,5-dihydro-2H-pyrrol-2-one](http://img.cochemist.com/ccimg/63100/63079-71-0_b.png)








![(S)-2-(2-Fluoro-[1,1'-biphenyl]-4-yl)propanoic acid](http://img.cochemist.com/ccimg/51600/51543-39-6.png)
![(S)-2-(2-Fluoro-[1,1'-biphenyl]-4-yl)propanoic acid](http://img.cochemist.com/ccimg/51600/51543-39-6_b.png)


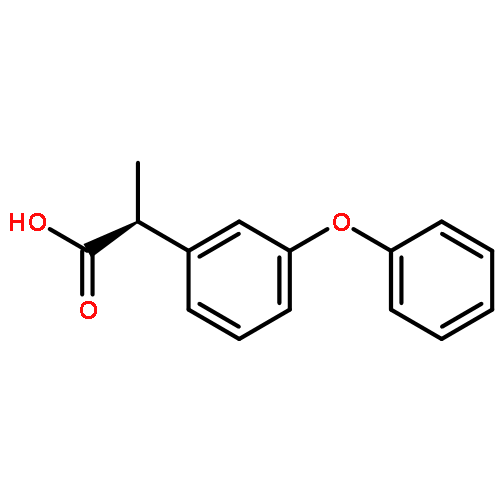
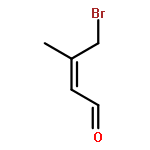







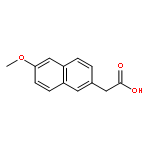

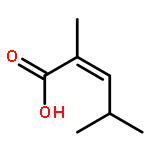
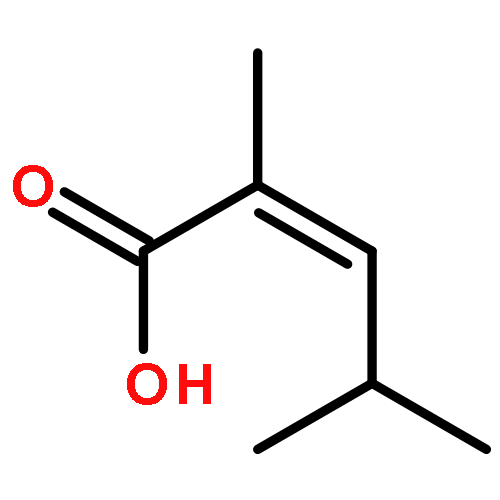




![[1,1'-Biphenyl]-4-acetic acid, a-methyl-, (S)-](http://img.cochemist.com/ccimg/10600/10532-14-6.png)
![[1,1'-Biphenyl]-4-acetic acid, a-methyl-, (S)-](http://img.cochemist.com/ccimg/10600/10532-14-6_b.png)


![2-(2-Fluoro-[1,1'-biphenyl]-4-yl)acetic acid](http://img.cochemist.com/ccimg/5100/5001-96-7.png)
![2-(2-Fluoro-[1,1'-biphenyl]-4-yl)acetic acid](http://img.cochemist.com/ccimg/5100/5001-96-7_b.png)



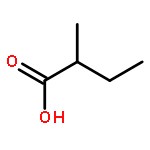

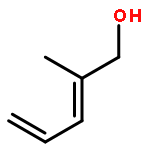
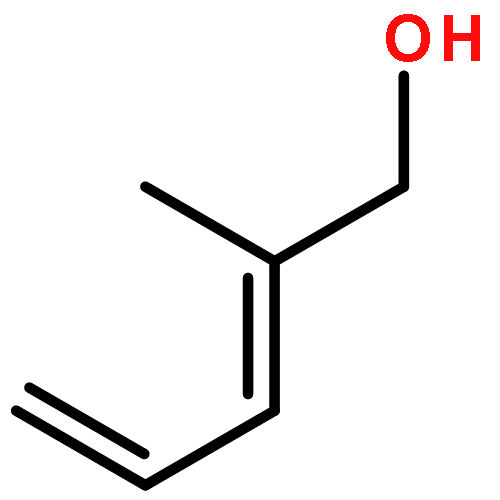

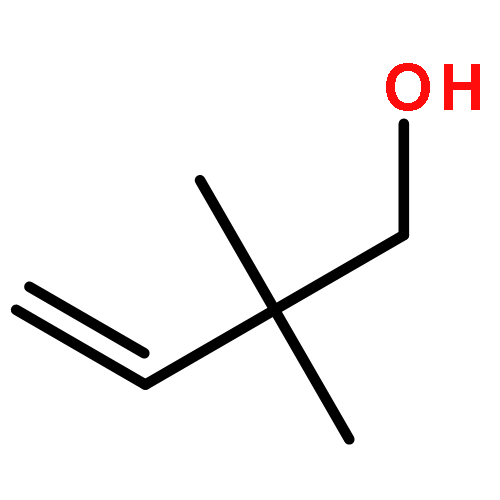
























![Ruthenium,chloro[(1,2,3,4,5-h)-1,2,3,4,5-pentamethyl-2,4-cyclopentadien-1-yl]bis(triphenylphosphine)-](http://img.cochemist.com/ccimg/92400/92361-49-4.png)
![Ruthenium,chloro[(1,2,3,4,5-h)-1,2,3,4,5-pentamethyl-2,4-cyclopentadien-1-yl]bis(triphenylphosphine)-](http://img.cochemist.com/ccimg/92400/92361-49-4_b.png)









