Co-reporter:Torben Steenbock, David A. Shultz, Martin L. Kirk, and Carmen Herrmann
The Journal of Physical Chemistry A 2017 Volume 121(Issue 1) pp:
Publication Date(Web):December 6, 2016
DOI:10.1021/acs.jpca.6b07270
Increasing interactions between spin centers in molecules and molecular materials is a desirable goal for applications such as single-molecule magnets for information storage or magnetic metal–organic frameworks for adsorptive separation and targeted drug delivery and release. To maximize these interactions, introducing unpaired spins on bridging ligands is a concept used in several areas where such interactions are otherwise quite weak, in particular, lanthanide-based molecular magnets and magnetic metal–organic frameworks. Here, we use Kohn–Sham density functional theory to study how much the ground spin state is stabilized relative to other low-lying spin states by creating an additional spin center on the bridge for a series of simple model compounds. The di- and triradical structures consist of nitronyl nitroxide (NNO) and semiquinone (SQ) radicals attached to a meta-phenylene(R) bridge (where R = −NH•/–NH2, −O•/OH, −CH2•/CH2). These model compounds are based on a fully characterized SQ–meta-phenylene–NNO diradical with moderately strong antiferromagnetic coupling. Replacing closed-shell substituents CH3 and NH2 with their radical counterparts CH2• and NH• leads to an increase in stabilization of the ground state with respect to other low-lying spin states by a factor of 3–6, depending on the exchange–correlation functional. For OH compared with O• substituents, no conclusions can be drawn as the spin state energetics depend strongly on the functional. This could provide a basis for constructing sensitive test systems for benchmarking theoretical methods for spin state energy splittings. Reassuringly, the stabilization found for a potentially synthesizable complex (up to a factor of 3.5) is in line with the simple model systems (where a stabilization of up to a factor of 6.2 was found). Absolute spin state energy splittings are considerably smaller for the potentially stable system than those for the model complexes, which points to a dependence on the spin delocalization from the radical substituent on the bridge.
Co-reporter:Torben Steenbock, Jos Tasche, Alexander I. Lichtenstein, and Carmen Herrmann
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 12) pp:5651-5664
Publication Date(Web):October 27, 2015
DOI:10.1021/acs.jctc.5b00349
Exchange spin coupling is usually evaluated in quantum chemistry from the energy difference between a high-spin determinant and a Broken-Symmetry (BS) determinant in combination with Kohn–Sham density functional theory (KS-DFT), based on the work of Noodleman. As an alternative, an efficient approximate approach relying on Green’s functions has been developed by one of the authors. This approach stems from solid-state physics and has never been systematically tested for molecular systems. We rederive a version of the Green’s-function approach originally suggested by Han, Ozaki, and Yu. This new derivation employs local projection operators as common in quantum chemistry for defining local properties such as partial charges, rather than using a dual basis as in the Han–Ozaki–Yu approach. The result is a simple postprocessing procedure for KS-DFT calculations, which in contrast to the BS energy-difference approach requires the electronic structure of only one spin state. We show for several representative small molecules, diradicals, and dinuclear transition metal complexes that this method gives qualitatively consistent results with the BS energy-difference approach as long as it is applied to high-spin determinants and as long as structural relaxation effects in different spin states do not play an important role.
Co-reporter:Suranjan Shil
Inorganic Chemistry 2015 Volume 54(Issue 24) pp:11733-11740
Publication Date(Web):December 3, 2015
DOI:10.1021/acs.inorgchem.5b01707
Ferrocene is an interesting coupler for designing magnetic molecules because of its rich chemistry and controllable oxidation state. In this work we have calculated the exchange spin coupling of a ferrocene-coupled nitronyl nitroxide diradical in its neutral and oxidized state (in which an additional spin center is introduced on the metallocene subunit). We do so by carrying out spin-unrestricted Kohn–Sham density functional theory (KS-DFT) calculations with different approximate exchange-correlation functionals and basis sets. We find that the neutral complex is weakly ferromagnetically coupled (in contrast to experimental results on single crystals), whereas the spin centers in the cationic complex are strongly antiferromagnetically coupled, resulting in an overall ferrimagnetic arrangement of the spins. Our calculations suggest that the magnetic exchange occurs through a spin alternation mechanism and that the lowest unoccupied molecular orbital (LUMO) plays an important role. The ferromagnetic behavior of the neutral complex is very sensitive to rotating one Cp ring versus the other. In the case of the cationic complex, the magnetic coupling is nearly independent of such structural changes. Thus, oxidation allows for switching between a weakly coupled and a strongly coupled, robust overall ferrimagnetic spin arrangement.
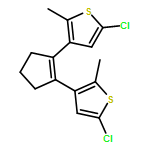
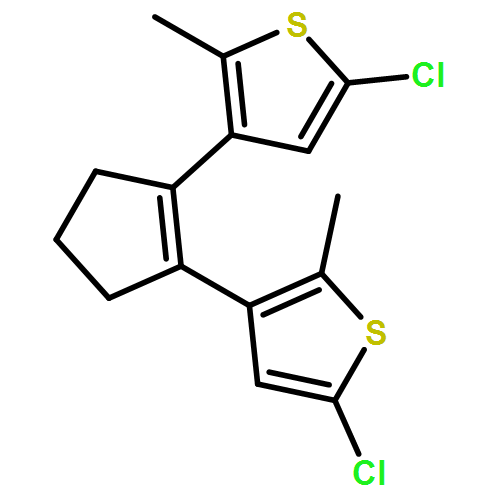
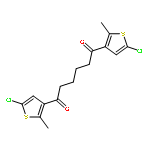
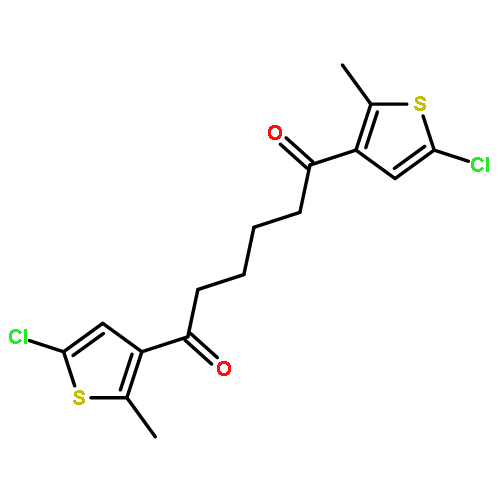
Co-reporter:Torben Steenbock;Alejra Escribano; Jürgen Heck; Carmen Herrmann
ChemPhysChem 2015 Volume 16( Issue 7) pp:1491-1501
Publication Date(Web):
DOI:10.1002/cphc.201402842
Abstract
Photoswitching is an intriguing way of incorporating functionality into molecules or their subunits. Dithienylethene switches are particularly promising, but have so far mostly been studied with five-membered ring (cyclopentenyl) backbones. We aim at comparing the switching properties of backbones with five and six carbon atoms in the ring. A major advantage is that cyclohexenyl rings offer new options for chiral functionalization. A slight change in the reaction conditions of a McMurry ring closure reaction leads to the formation of dithienyl derivatives with a cyclohexene backbone in reasonable yield. Density functional theory calculations were carried out, demonstrating the similarity of both compounds. Experimental results confirm the theoretical outcomes.
Co-reporter:Dr. Daniela Stummer; Carmen Herrmann; Andrea Rentmeister
ChemistryOpen 2015 Volume 4( Issue 3) pp:295-301
Publication Date(Web):
DOI:10.1002/open.201402104
Abstract
Bioorthogonal click reactions are powerful tools to specifically label biomolecules in living cells. Considerable progress has been made in site-specific labeling of proteins and glycans in complex biological systems, but equivalent methods for mRNAs are rare. We present a chemo-enzymatic approach to label the 5’ cap of eukaryotic mRNAs using a bioorthogonal photoclick reaction. Herein, the N7-methylated guanosine of the 5’ cap is enzymatically equipped with an allyl group using a variant of the trimethylguanosine synthase 2 from Giardia lamblia (GlaTgs2). To elucidate whether the resulting N2-modified 5’ cap is a suitable dipolarophile for photoclick reactions, we used Kohn–Sham density functional theory (KS-DFT) and calculated the HOMO and LUMO energies of this molecule and nitrile imines. Our in silico studies suggested that combining enzymatic allylation of the cap with subsequent labeling in a photoclick reaction was feasible. This could be experimentally validated. Our approach generates a turn-on fluorophore site-specifically at the 5’ cap and therefore presents an important step towards labeling of eukaryotic mRNAs in a bioorthogonal manner.
Co-reporter:Ann Christin Jahnke, Jonny Proppe, Mariana Spulber, Cornelia G. Palivan, Carmen Herrmann, and Oliver S. Wenger
The Journal of Physical Chemistry A 2014 Volume 118(Issue 47) pp:11293-11303
Publication Date(Web):November 13, 2014
DOI:10.1021/jp5082164
A series of selenophenes with redox-active amine end-capping groups was synthesized and investigated. A combination of cyclic voltammetry, optical absorption, EPR spectroscopy, and quantum-chemical calculations based on Kohn–Sham density functional theory was used to explore charge delocalization in the monocationic mixed-valence forms of these selenophenes, and the results were compared to those obtained from analogous studies of structurally identical thiophenes. The striking finding is that the comproportionation constant (Kc) for the experimentally investigated biselenophene is more than 2 orders of magnitude lower than for its bithiophene counterpart (in CH3CN with 0.1 M TBAPF6), and the electronic coupling between the two amine end-capping groups in the mixed-valent biselenophene monocation is only roughly half as strong as in the corresponding bithiophene monocation. These are surprisingly large differences given the structural similarity between the respective biselenophene and bithiophene molecules. However, the computationally determined comproportionation constants for biselenophene and bithiophene are almost identical, and the electronic coupling in the monocationic biselenophene is only slightly smaller than that in the monocationic bithiophene. We assume that the external electric field may be responsible for the differences in monocation stabilities between experiment and computation. Our findings indicate that charge delocalization across individual selenophenes tends to be less pronounced than across individual thiophenes, and this may have important implications for long-range charge transfer across selenophene oligomers or polymers.
Co-reporter:Hendrik Schlicke; Dr. Carmen Herrmann
ChemPhysChem 2014 Volume 15( Issue 18) pp:4011-4018
Publication Date(Web):
DOI:10.1002/cphc.201402561
Abstract
Conductance switching through chemical modification of a molecular bridge is a major goal in molecular electronics, with the potential to lead to molecule-based functional devices. In terms of switching speed, mechanisms that rely on only minor rearrangements of molecular structures are particularly promising. We demonstrate, based on density functional theory calculations combined with a coherent tunneling approach, how protonation and deprotonation of amine-substituted or amine-bridged model molecular wires can switch off and on π-sites and thus: a) remove or introduce interference features in the electron transmission, and b) decrease or increase coupling along a chain. This mechanism may also be relevant for interactions between molecular bridges and metal cations, for example, in sensor applications.
Co-reporter:Carmen Herrmann and Jan Elmisz
Chemical Communications 2013 vol. 49(Issue 89) pp:10456-10458
Publication Date(Web):13 Aug 2013
DOI:10.1039/C3CC45125A
A concept that plays an important role in chemistry in general and in molecular spintronics in particular is electronic communication through molecular bridges. An improved understanding of this concept may help to transfer knowledge between different areas of chemistry and nanoscience. We aim at finding the limits of electronic communication as a property of the bridge by comparing and rationalizing trends in exchange spin coupling in diradicals and in conductance in dithiolate–gold junctions which share common sets of molecular bridges.
Co-reporter:Carmen Herrmann and Jan Elmisz
Chemical Communications 2013 - vol. 49(Issue 89) pp:NaN10458-10458
Publication Date(Web):2013/08/13
DOI:10.1039/C3CC45125A
A concept that plays an important role in chemistry in general and in molecular spintronics in particular is electronic communication through molecular bridges. An improved understanding of this concept may help to transfer knowledge between different areas of chemistry and nanoscience. We aim at finding the limits of electronic communication as a property of the bridge by comparing and rationalizing trends in exchange spin coupling in diradicals and in conductance in dithiolate–gold junctions which share common sets of molecular bridges.





![Benzene, 1,3,5-tris[2-(trimethylsilyl)ethynyl]-](/data/chemimg/3030600/18772-58-2.png)
![Benzene, 1,3,5-tris[2-(trimethylsilyl)ethynyl]-](/data/chemimg/3030600/18772-58-2_b.png)