Co-reporter:Junseok Lee;Dan C. Sorescu;Kenneth D. Jordan
The Journal of Physical Chemistry C November 13, 2008 Volume 112(Issue 45) pp:17672-17677
Publication Date(Web):2017-2-22
DOI:10.1021/jp807467x
The formation of hydroxyl chains from water dissociation on the Cu(110) surface has been studied by using a combination of scanning tunneling microscopy (STM), electron stimulated desorption ion angular distribution (ESDIAD), temperature programmed desorption (TPD), and density functional theory (DFT) calculations. Annealing the D2O-covered surface to a temperature of ∼200 K leads to desorption of D2O molecules and produces a zigzag structure due to adsorbed OD groups with a periodicity of 5 Å along the <11̅0> direction in the STM image. Coadsorption of O2 promotes the water dissociation reaction and produces hydroxyl chains with much higher coverage. ESDIAD measurements show a two-beam pattern consistent with OD(a) species inclined ∼40° with respect to the surface normal and orientated along the <001> azimuth. The calculations reveal the existence of stable chain structures comprised solely of hydroxyl groups as well as of interacting water and hydroxyl groups that are consistent with the observed STM image.
Co-reporter:Monica McEntee, Wenjie Tang, Matthew Neurock, and John T. Yates Jr.
ACS Catalysis 2015 Volume 5(Issue 2) pp:744
Publication Date(Web):December 12, 2014
DOI:10.1021/cs5014255
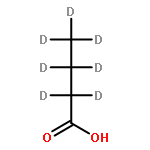
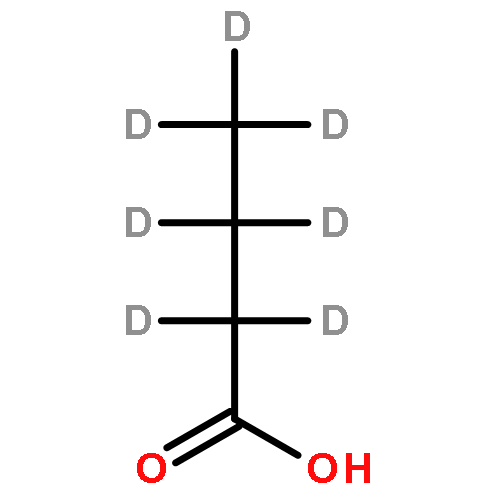
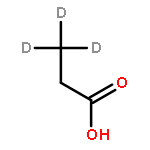
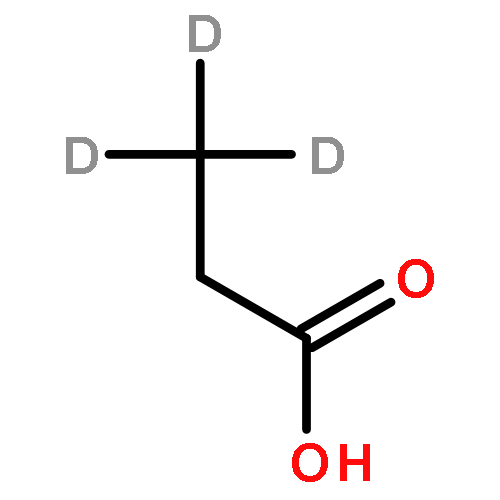
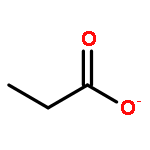
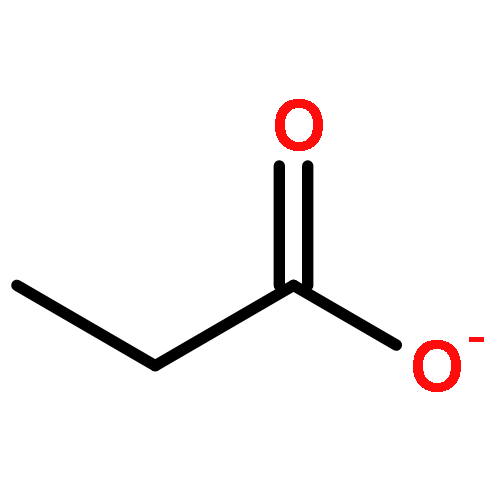
The partial oxidation of model C2–C4 (acetic, propionic, and butyric) carboxylic acids on Au/TiO2 catalysts consisting of Au particles ∼3 nm in size was investigated using transmission infrared spectroscopy and density functional theory. All three acids readily undergo oxidative dehydrogenation on Au/TiO2. Propionic and butyric acid dehydrogenate at the C2–C3 positions, whereas acetic acid dehydrogenates at the C1–C2 position. The resulting acrylate and crotonate intermediates are subsequently oxidized to form β-keto acids that decarboxylate. All three acids form a gold ketenylidene intermediate, Au2C═C═O, along the way to their full oxidation to form CO2. Infrared measurements of Au2C═C═O formation as a function of time provides a surface spectroscopic probe of the kinetics for the activation and oxidative dehydrogenation of the alkyl groups in the carboxylate intermediates that form. The reaction proceeds via the dissociative adsorption of the acid onto TiO2, the adsorption and activation of O2 at the dual perimeter sites on the Au particles (Au–O–O-Ti), and the subsequent activation of the C2–H and C3–H bonds of the bound propionate and butyrate intermediates by the weakly bound and basic oxygen species on Au to form acrylate and crotonate intermediates, respectively. The C═C bond of the unsaturated acrylate and crotonate intermediates is readily oxidized to form an acid at the beta (C3) position, which subsequently decarboxylates. This occurs with an overall activation energy of 1.5–1.7 ± 0.2 eV, ultimately producing the Au2C═C═O species for all three carboxylates. The results suggest that the decrease in the rate in moving from acetic to propionic to butyric acid is due to an increase in the free energy of activation for the formation of the Au2C═C═O species on Au/TiO2 with an increasing size of the alkyl substituent. The formation of Au2C═C═O proceeds for carboxylic acids that are longer than C2 without a deuterium kinetic isotope effect, demonstrating that C–H bond scission is not involved in the rate-determining step; the rate instead appears to be controlled by C–O bond scission. The adsorbed Au2C═C═O intermediate species can be hydrogenated to produce ketene, H2C═C═O(g), with an activation energy of 0.21 ± 0.05 eV. These studies show that selective oxidative dehydrogenation of the alkyl side chains of fatty acids can be catalyzed by nanoparticle Au/TiO2 at temperatures near 400 K.Keywords: Au/TiO2; carboxylic acid oxidation; catalysis; decarboxylation; ketenylidene; oxidative-dehydrogenation
Co-reporter:Monica McEntee ; Wenjie Tang ; Matthew Neurock ; Jr
Journal of the American Chemical Society 2014 Volume 136(Issue 13) pp:5116-5120
Publication Date(Web):March 5, 2014
DOI:10.1021/ja500928h
The oxidative-dehydrogenation of carboxylic acids to selectively produce unsaturated acids at the second and third carbons regardless of alkyl chain length was found to occur on a Au/TiO2 catalyst. Using transmission infrared spectroscopy (IR) and density functional theory (DFT), unsaturated acrylate (H2C═CHCOO) and crotonate (CH3CH═CHCOO) were observed to form from propionic acid (H3CCH2COOH) and butyric acid (H3CCH2CH2COOH), respectively, on a catalyst with ∼3 nm diameter Au particles on TiO2 at 400 K. Desorption experiments also show gas phase acrylic acid is produced. Isotopically labeled 13C and 12C propionic acid experiments along with DFT calculated frequency shifts confirm the formation of acrylate and crotonate. Experiments on pure TiO2 confirmed that the unsaturated acids were not produced on the TiO2 support alone, providing evidence that the sites for catalytic activity are at the dual Au–Ti4+ sites at the nanometer Au particles’ perimeter. The DFT calculated energy barriers between 0.3 and 0.5 eV for the reaction pathway are consistent with the reaction occurring at 400 K on Au/TiO2.
Co-reporter:Ana Stevanovic ; Shiliang Ma ; Jr.
The Journal of Physical Chemistry C 2014 Volume 118(Issue 36) pp:21275-21280
Publication Date(Web):August 16, 2014
DOI:10.1021/jp507156p
Photoluminescence spectroscopy was employed to study the photoinduced charge transfer between TiO2 nanoparticles and Au nanoparticles under vacuum. We found that small coverages of 3 nm Au nanoparticles deposited on TiO2 significantly diminish the 540 nm (2.3 eV) photoluminescence emission from TiO2 because of the redistribution of the photoexcited charge to Au nanoparticles that are capable of accepting negative charges behind the Schottky barrier. The lack of development of the photoluminescence emission of Au/TiO2 during continuous UV irradiation occurs because of a short circuit established through Au nanoparticles in which transferred electrons in Au recombine nonradiatively with holes in TiO2. The photoexcited electron transfer from TiO2 to the Au nanoparticles occurs beyond the Au particle perimeter over a distance of at least 4 nm. The quenching of photoluminescence by resonance energy transfer from TiO2 to Au nanoparticles is unimportant as Au plasmonic absorption is not observed.
Co-reporter:Ana Stevanovic ; Shiliang Ma ; Jr.
The Journal of Physical Chemistry C 2014 Volume 118(Issue 41) pp:23614-23620
Publication Date(Web):September 24, 2014
DOI:10.1021/jp508061w
Photoluminescence (PL) spectroscopy was employed to study the effect of embedded single-walled carbon nanotubes (SWNTs) on charge transport in powdered TiO2. It was found that 1–5 wt % SWNTs mixed with TiO2 accept electrons from photoexcited TiO2 and quench the PL intensity from TiO2. The PL quenching efficiency by SWNTs is proportional to the fractional occupancy of TiO2 particles which experience electrical contact with SWNTs. Ultraviolet light was used to cause surface charging of the SWNT/TiO2 sample, and the charging/discharging rate was measured using PL. The PL charging/discharging rate of all SWNT/TiO2 mixtures is identical to that of pure TiO2, indicating that SWNTs only accept photoexcited electron from TiO2 but do not transport electrons under conditions of the experiment. This is due to the effect of positively charged TiO2 particles which immobilize the photoexcited electrons transferred to SWNTs at the interface between first-layer TiO2 and SWNTs, inhibiting charge transport through SWNT channels. The inhibition of charge transport through SWNTs may be removed by applying an external voltage to overcome the built-in electric field which prevents current flow in the SWNTs.
Co-reporter:Lynn Mandeltort ; De-Li Chen ; Wissam A. Saidi ; J. Karl Johnson ; Milton W. Cole ; Jr.
Journal of the American Chemical Society 2013 Volume 135(Issue 20) pp:7768-7776
Publication Date(Web):April 29, 2013
DOI:10.1021/ja402928s
Single-walled carbon nanotubes (SWNTs) exhibit high surface areas and precisely defined pores, making them potentially useful materials for gas adsorption and purification. A thorough understanding of the interactions between adsorbates and SWNTs is therefore critical to predicting adsorption isotherms and selectivities. Metallic (M-) and semiconducting (S-) SWNTs have extremely different polarizabilities that might be expected to significantly affect the adsorption energies of molecules. We experimentally and theoretically show that this expectation is contradicted, for both a long chain molecule (n-heptane) and atoms (Ar, Kr, and Xe). Temperature-programmed desorption experiments are combined with van der Waals corrected density functional theory, examining adsorption on interior and exterior sites of the SWNTs. Our calculations show a clear dependence of the adsorption energy on nanotube diameter but not on whether the tubes are conducting or insulating. We find no significant experimental or theoretical difference in adsorption energies for molecules adsorbed on M- and S-SWNTs having the same diameter. Hence, we conclude that the differences in polarizabilities between M- and S-SWNTs have a negligible influence on gas adsorption for spherical molecules as well as for highly anisotropic molecules such as n-heptane. We expect this conclusion to apply to all types of adsorbed molecules where van der Waals interactions govern the molecular interaction with the SWNT.
Co-reporter:Ana Stevanovic and John T. Yates, Jr.
The Journal of Physical Chemistry C 2013 Volume 117(Issue 46) pp:24189-24195
Publication Date(Web):October 17, 2013
DOI:10.1021/jp407765r
Photoluminescence spectroscopy was employed to observe electron transport between TiO2 particles. Ultraviolet (UV) irradiation (3.88 eV) was shown to positively enhance the photovoltage of TiO2 particles at the powder surface, causing an enhancement of their photoluminescence (PL) at 530 nm. The charging of the TiO2 particles on the powder surface by UV irradiation partially discharges in the dark, where the displaced bulk negative charge diffuses back toward the TiO2 surface. This charge flow partially restores upward band bending, causing the PL intensity to decrease. The rate of the discharging process was used to estimate the electron migration mobility (∼10–10 m2 V–1 s–1 at 300 K) between TiO2 particles in the TiO2 matrix. Electron migration between TiO2 particles is temperature-dependent with an activation energy of 0.015 ± 0.008 eV. In addition, it was found that the adsorption of an immobile electron-donor molecule (NH3), attracts negative charge on the TiO2 surface which does not exhibit mobility behavior, in contrast to mobile electrons produced by UV. These measurements were carried out in high vacuum in the absence of oxygen and surface impurities detectable by IR spectroscopy.
Co-reporter:Zhen Zhang, Ke Cao, and John T. Yates Jr.
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 4) pp:674-679
Publication Date(Web):February 5, 2013
DOI:10.1021/jz400101f
The dissociative adsorption of water at oxygen-vacancy defect sites on the TiO2(110) surface spatially redistributes the defect electron density originally present at subsurface sites near the defect sites. This redistribution of defect-electrons makes them more accessible to Ti4+ ions surrounding the defects. The redistribution of electron density decreases the O+ desorption yield from surface lattice O2- ions in TiO2, as excited by electron-stimulated desorption (ESD). A model in which OH formation on defect sites redistributes defect electrons to neighboring Ti4+ sites is proposed. This switches off the Knotek–Feibelman mechanism for ESD of O+ ions from lattice sites. Conversely, enhanced O+ reneutralization could also be induced by redistribution of defect electrons. The redistribution of surface electrons by adsorption is further verified by the use of donor and acceptor molecules that add or remove electron density.Keywords: electron distribution; electron stimulated desorption; H2O; oxygen vacancy; TiO2;
Co-reporter:Zhen Zhang and John T. Yates Jr.
Chemical Reviews 2012 Volume 112(Issue 10) pp:5520
Publication Date(Web):July 11, 2012
DOI:10.1021/cr3000626
Co-reporter:Isabel Xiaoye Green ; Wenjie Tang ; Matthew Neurock ; Jr.
Journal of the American Chemical Society 2012 Volume 134(Issue 33) pp:13569-13572
Publication Date(Web):August 7, 2012
DOI:10.1021/ja305911e
Chemisorbed acetate species derived from the adsorption of acetic acid have been oxidized on a nano-Au/TiO2 (∼3 nm diameter Au) catalyst at 400 K in the presence of O2(g). It was found that partial oxidation occurs to produce gold ketenylidene species, Au2═C═C═O. The reactive acetate intermediates are bound at the TiO2 perimeter sites of the supported Au/TiO2 catalyst. The ketenylidene species is identified by its measured characteristic stretching frequency ν(CO) = 2040 cm–1 and by 13C and 18O isotopic substitution comparing to calculated frequencies found from density functional theory. The involvement of dual catalytic Ti4+ and Au perimeter sites is postulated on the basis of the absence of reaction on a similar nano-Au/SiO2 catalyst. This observation excludes low coordination number Au sites as being active alone in the reaction. Upon raising the temperature to 473 K, the production of CO2 and H2O is observed as both acetate and ketenylidene species are further oxidized by O2(g). The results show that partial oxidation of adsorbed acetate to adsorbed ketenylidyne can be cleanly carried out over Au/TiO2 catalysts by control of temperature.
Co-reporter:Isabel Xiaoye Green ; Wenjie Tang ; Monica McEntee ; Matthew Neurock ; Jr.
Journal of the American Chemical Society 2012 Volume 134(Issue 30) pp:12717-12723
Publication Date(Web):June 27, 2012
DOI:10.1021/ja304426b
TiO2-supported gold nanoparticles exhibit surprising catalytic activity for oxidation reactions compared to noble bulk gold which is inactive. The catalytic activity is localized at the perimeter of the Au nanoparticles where Au atoms are atomically adjacent to the TiO2 support. At these dual-catalytic sites an oxygen molecule is efficiently activated through chemical bonding to both Au and Ti4+ sites. A significant inhibition by a factor of 22 in the CO oxidation reaction rate is observed at 120 K when the Au is preoxidized, caused by the oxygen-induced positive charge produced on the perimeter Au atoms. Theoretical calculations indicate that induced positive charge occurs in the Au atoms which are adjacent to chemisorbed oxygen atoms, almost doubling the activation energy for CO oxidation at the dual-catalytic sites in agreement with experiments. This is an example of self-inhibition in catalysis by a reactant species.
Co-reporter:Lynn Mandeltort and John T. Yates, Jr.
The Journal of Physical Chemistry C 2012 Volume 116(Issue 47) pp:24962-24967
Publication Date(Web):October 31, 2012
DOI:10.1021/jp308101c
The diffusion of dilute metallic lithium across the surface and into the bulk of atomically clean highly oriented pyrolytic graphite in ultrahigh vacuum is reported. Auger electron spectroscopy and a surface-dependent chemical reaction were utilized to monitor the coverage, oxidation state, and diffusion of Li. A very small diffusion activation energy (0.16 ± 0.02 eV; 15.4 ± 1.8 kJ/mol) is found for Li surface diffusion across the graphite basal plane. A model involving diffusion-rate-limited Li atom transport across the basal plane of graphite through step edge sites into the interior is proposed. These measurements indicate that the diffusion coefficient, D, for Li is very large (D(300 K) ≈ 5 × 10–6 cm2 s–1).
Co-reporter:John T. Yates Jr.
Catalysis Letters 2012 Volume 142( Issue 11) pp:1412
Publication Date(Web):2012 November
DOI:10.1007/s10562-012-0888-z
Co-reporter:Lynn Mandeltort, Pabitra Choudhury, J. Karl Johnson, and John T. Yates Jr.
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 12) pp:1680-1683
Publication Date(Web):June 5, 2012
DOI:10.1021/jz300578x
The reaction of methyl radicals with the basal plane of graphite has been observed to occur with an activation energy of less than 0.3 eV. This reaction is initiated by Li-induced CH3Cl dissociation to produce CH3 radicals on the graphite surface. It is found that ∼3/4 of the methyl radicals remain on the graphite surface up to 700 K at puckered sp3 carbon sites, while 1/4 of the CH3 radicals participate in CH4 formation and small amounts of C2 and C3 hydrocarbon formation. CH3 radicals become mobile over an activation energy barrier of ∼0.7 eV.Keywords: DFT; graphite; Li-doped graphite; methyl; sp2−sp3; surface reactions; TPD;
Co-reporter:Lynn Mandeltort, Pabitra Choudhury, J. Karl Johnson, and John T. Yates, Jr.
The Journal of Physical Chemistry C 2012 Volume 116(Issue 34) pp:18347-18357
Publication Date(Web):August 9, 2012
DOI:10.1021/jp3063367
The reaction of submonolayer Li atoms with CH3Cl at 100 K on a highly oriented pyrolytic graphite (HOPG) surface has been studied under ultrahigh vacuum. We exploit the low defect density of the high quality HOPG used here (∼109 defects cm–2) to eliminate the effects of step edges and defects on the graphite surface chemistry. Li causes C–Cl bond scission in CH3Cl, liberating CH3 radicals below 130 K. Ordinarily, two CH3 species would couple to form products such as C2H6, but in the presence of graphite, CH3 preferentially adsorbs on the flat basal plane of Li-treated graphite. A C–CH3 bond of 1.2 eV is formed, which is enhanced relative to CH3 binding to clean graphite (0.52 eV) due to donation of electrons from Li into the graphite and back-donation from graphite to CH3. A low yield of C1, C2, and C3 hydrocarbon products above 330 K is found along with a low yield of H2. The low yield of these products indicates that the majority of the CH3 groups are irreversibly bound to the basal plane of graphite, and only a small fraction participate in the production of C1–C3 volatile products or in extensive dehydrogenation. Spin-polarized density functional theory calculations indicate that CH3 binds to the Li-treated surface with an activation energy of 0.3 eV to form a C–CH3 adsorbed surface species with sp3 hybridization of the graphite, and the methyl carbon atoms is involved in bond formation. Bound CH3 radicals become mobile with 0.7 eV activation energy and can participate in combination reactions for the production of small yields of C1–C3 hydrocarbon products. We show that alkyl radical attachment to the graphite surface is kinetically preferred over hydrocarbon product desorption.
Co-reporter:Ana Stevanovic ; Michael Büttner ; Zhen Zhang ; Jr.
Journal of the American Chemical Society 2011 Volume 134(Issue 1) pp:324-332
Publication Date(Web):November 21, 2011
DOI:10.1021/ja2072737
The photoluminescence (PL) of TiO2 at 529.5 nm (2.34 eV) has been found to be a sensitive indicator of UV-induced band structure modification. As UV irradiation occurs, the positive surface potential changes and shifts the depth of the depletion layer. In addition, reversible band bending due to the adsorption of the electron-donor NH3 and CO molecules has been observed in measurements combining PL with FTIR surface spectroscopy. It has been found that the O2 molecule acts in two ways: as a reversibly adsorbed electron-acceptor molecule and as an irreversibly adsorbed molecule that heals natural oxygen vacancy defects in the near-surface region.
Co-reporter:Isabel Xiaoye Green;Wenjie Tang;Matthew Neurock Jr.
Science 2011 Volume 333(Issue 6043) pp:736-739
Publication Date(Web):05 Aug 2011
DOI:10.1126/science.1207272
The low-temperature oxidation of carbon monoxide proceeds initially with oxygen molecules that bridge titanium and gold sites.
Co-reporter:Junseok Lee ; Zhen Zhang ; Xingyi Deng ; Dan C. Sorescu ; Christopher Matranga ; Jr.
The Journal of Physical Chemistry C 2011 Volume 115(Issue 10) pp:4163-4167
Publication Date(Web):February 23, 2011
DOI:10.1021/jp1112697
The interaction between CO and O adsorbed on TiO2(110) has been investigated using scanning tunneling microscopy (STM), electron stimulated desorption (ESD), temperature programmed desorption (TPD), and density functional theory (DFT). Coadsorption of CO and O produces CO−O and CO−O−CO surface complexes via weak attractive interaction as revealed by STM and DFT. The effect of the adsorbates interaction can also be observed in two ensemble-averaged techniques, ESD and TPD, strongly supporting the STM and DFT results. The CO molecules adsorbed near chemisorbed O cause a ∼70% decrease in the O+ ion yield in ESD. The interaction between CO and O is considered to be electrostatic in nature due to the charge rearrangement upon chemisorption.
Co-reporter:Zhen Zhang ; Wenjie Tang ; Matthew Neurock ; Jr.
The Journal of Physical Chemistry C 2011 Volume 115(Issue 48) pp:23848-23853
Publication Date(Web):November 15, 2011
DOI:10.1021/jp2067809
The influence of charge transfer from single Au atoms to TiO2(110) on the photostimulated desorption (PSD) of 18O2 has been studied using a measurement of the rate of hole transport which mediates PSD. Band bending effects observed experimentally and theoretically by density functional theory indicate that Auδ+ atoms are present. The presence of atomically dispersed Au on the TiO2(110) surface with a constant coverage of preadsorbed 18O2 depresses the photoinduced hole transport rate from the bulk to the TiO2 surface and decreases the 18O2 PSD yield. This indicates that single Au atoms donate a fraction of an electron to the surface, causing downward band bending. DFT calculations show that ∼0.2 electron transfers from single Au atoms to the O2/TiO2(110) surface and the valence and conduction bands bend ∼0.6 eV downward. With increasing Au coverage, the positive charge per Au atom decreases due to the formation of small Au clusters.
Co-reporter:John T. Yates, Jr.;Charles T. Campbell
PNAS 2011 Volume 108 (Issue 3 ) pp:911-916
Publication Date(Web):2011-01-18
DOI:10.1073/pnas.1006671107
This special issue on surface chemistry is introduced with a brief history of the field, a summary of the importance of surface
chemistry in technological applications, a brief overview of some of the most important recent developments in this field,
and a look forward to some of its most exciting future directions. This collection of invited articles is intended to provide
a snapshot of current developments in the field, exemplify the state of the art in fundamental research in surface chemistry,
and highlight some possibilities in the future. Here, we show how those articles fit together in the bigger picture of this
field.
Co-reporter:Zhen Zhang ; Jr.
Journal of the American Chemical Society 2010 Volume 132(Issue 37) pp:12804-12807
Publication Date(Web):August 31, 2010
DOI:10.1021/ja106207w
The role of electrons and holes in the electronically excited oxidation of adsorbed CO on TiO2(110) has been investigated by tuning the surface electron and hole availability by the adsorption of Cl2 or O2. The presence of an electron acceptor (Cl2 or O2) on the TiO2(110) surface causes upward band bending, increasing the excited hole availability and decreasing the excited electron availability in the near surface region. This enhances O2 desorption and depresses CO2 production during electronic excitation. This result gives clear evidence for the first time that the electronically excited CO oxidation reaction is caused by an electron-mediated process in contrast to O2 desorption which is mediated by holes.
Co-reporter:Mahesh Rajappan, Michael Büttner, Charlie Cox and John T. Yates Jr.
The Journal of Physical Chemistry A 2010 Volume 114(Issue 10) pp:3443-3448
Publication Date(Web):February 15, 2010
DOI:10.1021/jp9093436
A novel IR method for measuring the kinetics of N2O photodecomposition has been devised and used to calibrate the flux of Lyman-α (10.2 eV) radiation from a H2/Ar microwave discharge lamp. The photodecomposition of N2O occurs with a weak pressure dependence due to the operation of a wall effect consuming some photogenerated active oxygen species. This effect is removed by working at high N2O pressures. The Lyman-α flux from the lamp is 1.28 ± 0.36 × 1015 photons cm−2 s−1.
Co-reporter:Zhen Zhang, Junseok Lee, John T. Yates Jr., Ralf Bechstein, Estephania Lira, Jonas Ø. Hansen, Stefan Wendt and Flemming Besenbacher
The Journal of Physical Chemistry C 2010 Volume 114(Issue 7) pp:3059-3062
Publication Date(Web):January 25, 2010
DOI:10.1021/jp910358w
The diffusion of interstitial Ti3+ species (Tii3+) in rutile TiO2(110) from the bulk to the surface has been studied utilizing two experimental techniques. Electron-stimulated desorption of O+ ions was employed to kinetically monitor the reaction between oxygen adatoms with Tii3+ species at temperatures between 360 and 400 K. Scanning tunneling microscopy was also used to measure the Tii3+ diffusion rate. Both methods yield a rate constant kTii3+ = 5 × 10−4 s−1 at 393 K. The activation energy as measured by the rate dependence on temperature is ∼1.0 eV.
Co-reporter:John T. Yates Jr.
Topics in Catalysis 2010 Volume 53( Issue 5-6) pp:297
Publication Date(Web):2010 May
DOI:10.1007/s11244-010-9442-7
Co-reporter:Zhen Zhang and John T. Yates Jr.
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 14) pp:2185-2188
Publication Date(Web):July 1, 2010
DOI:10.1021/jz1007559
The role of band bending on the efficiency of charge transfer across the TiO2(110) single crystal surface has been measured in ultrahigh vacuum, in the absence of a wide range of surface site inhomogeneities, surface impurities and solvent effects, and particle size effects. The adsorption of the Cl2 (electron acceptor) molecule and the O2 (electron acceptor) molecule have been found to enhance hole transport from TiO2 to 18O2 molecules adsorbed on oxygen vacancy sites, increasing the rate of electron stimulated desorption (ESD) of 18O2. This confirms that O2-ESD is hole mediated. Conversely, adsorption of CH3OH, a donor molecule, reduces the transfer rate for holes to the adsorbed O2, reducing its rate of ESD to near zero. The maximum effect of donor and acceptor molecules occurs near 1 monolayer coverage.Keywords (keywords): acceptor; band bending; charge transfer; donor; electron stimulated desorption (ESD); oxygen; titanium dioxide (TiO2);
Co-reporter:Zhen Zhang and John T. Yates Jr.
The Journal of Physical Chemistry C 2010 Volume 114(Issue 7) pp:3098-3101
Publication Date(Web):February 2, 2010
DOI:10.1021/jp910404e
The kinetics of surface and bulk electron−hole pair recombination have been measured separately on TiO2(110) in ultrahigh vacuum under well-controlled surface conditions for the first time. Using 100 eV incident electrons, excitation of electron−hole pairs occurs within ∼6 Å of the surface, and the rate of stimulated desorption of O2 was measured as a means of determining the charge carrier recombination kinetics, which are found to be mediated by the surface and to be first-order in charge carrier concentration. Comparison with a previous O2 photodesorption experiment, where excitation by 3.4 eV photons was used for electron−hole pair generation in a deeper region below the surface (∼100 Å), shows that bulk recombination in TiO2 occurs by second-order kinetics in charge carrier concentration.
Co-reporter:John T. Yates Jr.
Surface Science 2009 Volume 603(10–12) pp:1605-1612
Publication Date(Web):1 June 2009
DOI:10.1016/j.susc.2008.11.052
Photochemistry from TiO2 surfaces is described for two cases: The UV-induced photodesorption of O2 from TiO2(1 1 0) – 1 × 1; and the hydrophilic effect caused by UV irradiation on TiO2. In both cases fundamental information about how these processes occur has been found. In the case of the O2 photodesorption kinetics, it has been found that the rate of the process is proportional to the square root of the UV flux, showing that second-order electron–hole pair recombination is dominant in governing the photodesorption rate. In addition these measurements provide an estimate of the concentration of hole traps in the TiO2 crystal. In other measurements of the UV-induced hydrophilicity, starting with the atomically-clean TiO2 surface, it has been shown that the effect occurs suddenly at a critical point during irradiation as a result of photooxidation of a monolayer of hydrocarbon (n-hexane) at equilibrium with ppm concentration of n-hexane in O2 at 1 atmosphere pressure.
Co-reporter:M. Büttner, L. Xiao, L. Mandeltort, S. Edington, J. K. Johnson and J. T. Yates Jr.
The Journal of Physical Chemistry C 2009 Volume 113(Issue 12) pp:4829-4838
Publication Date(Web):2017-2-22
DOI:10.1021/jp810139q
Doping of opened single-walled carbon nanotubes (SWNTs) with metallic Li has been carried out under ultrahigh vacuum. Li atoms enter into the interior sites of the SWNTs by surface migration with an activation energy of 0.3 ± 0.04 eV. Density functional theory calculations indicate that the Li atoms ionize inside the SWNTs to produce Li+ ions. The Li-doped SWNTs exhibit an ∼10% enhancement of the van der Waals bonding for n-heptane molecules, a model for volatile organic compounds, within the interior due to polarization of the alkane molecules in the strong electrostatic field around the Li+ ions. The investigations reveal the utility of temperature-programmed desorption from SWNTs for understanding the energetics of confined molecule adsorption in pristine and doped SWNTs. Statistical mechanical simulations using a polarizable potential for alkanes give increases in isosteric heats of adsorption at high alkane loading that are in excellent agreement with experiments. Our simulations predict that the binding energy of n-heptane in the Li-doped nanotubes can be increased by as much as a factor of 3 at very low alkane loading.
Co-reporter:John T. Yates Jr.;Kenneth D. Jordan;Dan C. Sorescu;Peter Maksymovych
Science 2008 Volume 322(Issue 5908) pp:1664-1667
Publication Date(Web):12 Dec 2008
DOI:10.1126/science.1165291
Abstract
Self-assembly of molecules on surfaces is a route toward not only creating structures, but also engineering chemical reactivity afforded by the intermolecular interactions. Dimethyldisulfide (CH3SSCH3) molecules self-assemble into linear chains on single-crystal gold surfaces. Injecting low-energy electrons into individual molecules in the self-assembled structures with the tip of a scanning tunneling microscope led to a propagating chemical reaction along the molecular chain as sulfur–sulfur bonds were broken and then reformed to produce new CH3SSCH3 molecules. Theoretical and experimental evidence supports a mechanism involving electron attachment followed by dissociation of a CH3SSCH3 molecule and initiation of a chain reaction by one or both of the resulting CH3S intermediates.
Co-reporter:Petro Kondratyuk and John T. Yates Jr.
Accounts of Chemical Research 2007 Volume 40(Issue 10) pp:995
Publication Date(Web):July 4, 2007
DOI:10.1021/ar700013c
We discuss our own studies of molecular adsorption on and inside of single-wall carbon nanotubes in the broader context of important theoretical and experimental developments in the field. We show that adsorption in the nanotube interior sites as well as in the groove and exterior sites may be resolved by various experimental methods. In addition, the changes that the adsorbate phases undergo due to confinement in the nanotube interior are discussed, particularly focusing on confined molecules of water, alkanes, and an alkene. Attention is also devoted to the use of oxidizing agents such as ozone to open the ends and walls of nanotubes for interior adsorption.
Co-reporter:Junseok Lee, Daniel B. Dougherty, John T. Yates Jr.
Surface Science 2007 Volume 601(Issue 16) pp:L91-L94
Publication Date(Web):15 August 2007
DOI:10.1016/j.susc.2007.04.248
Sequential stages of formation of a self-assembled monolayer of flat-lying 2,6-dimethylpyridine molecules on a single crystal Cu(1 1 0) surface have been observed by low-temperature scanning tunneling microscopy (LT-STM). At an adsorption temperature of 10 K, all of the molecules are randomly distributed at low coverage upon adsorption. The isolated molecules align their molecular axes parallel to the 〈0 0 1〉 azimuth of the Cu lattice. The nitrogen atom in the molecule is located at the four-fold hollow site. Upon annealing to 100 K, the molecules associate to form head-to-head dimers. The dimer units involve a pair of weak hydrogen bonds between methyl group-hydrogen atoms and N moieties on adjacent molecules, forming a core structure for further growth. In a later stage of self-assembly, single head-to-tail weak hydrogen bonds between ring C–H bonds and N moieties form in chains on the periphery of the central cores, leading to larger domains with a c(6 × 2) overlayer structure.
Co-reporter:Gregg A. Morgan Jr., Yu Kwon Kim, John T. Yates Jr.
Surface Science 2007 Volume 601(Issue 17) pp:3548-3555
Publication Date(Web):1 September 2007
DOI:10.1016/j.susc.2007.04.160
Co-reporter:Gregg A. Morgan Jr., Dan C. Sorescu, Yu Kwon Kim, John T. Yates Jr.
Surface Science 2007 Volume 601(Issue 17) pp:3533-3547
Publication Date(Web):1 September 2007
DOI:10.1016/j.susc.2007.06.019
We present a direct side-by-side comparison of the adsorption and desorption of nitrogen on the atomically-stepped Ru(1 0 9) surface and the atomically-flat Ru(0 0 1) surface. Both infrared reflection absorption spectroscopy (IRAS) and temperature programmed desorption (TPD) are employed in this study, along with density functional theory (DFT). We find that the chemisorptive terminal binding of N2 is stronger on the atomic step sites than on the terrace sites of Ru(1 0 9) as indicated by TPD and by a reduction of the singleton vibrational frequency, ν(N2), by ∼9 cm−1, comparing steps to terraces. In addition, we find that metal–metal compression effects on the terrace sites of Ru(1 0 9) cause stronger binding of N2 than found on the Ru(0 0 1) surface, as indicated by a reduction of the terrace-N2 singleton vibrational frequency by ∼11 cm−1 when compared to the singleton N2 mode on Ru(0 0 1). These spectroscopic results, comparing compressed terrace sites to Ru(0 0 1) sites and confirmed by TPD and DFT, indicate that N2 bonds primarily as a σ-donor to Ru. Using equimolar 15N2 and 14N2, it is found that dynamic dipole coupling effects present at higher N2 coverages may be partially eliminated by isotopically detuning neighbor oscillators. These experiments, considered together, indicate that the order of the bonding strength for terminal-N2 on Ru is: atomic steps > atomic terraces > Ru(0 0 1). DFT calculations also show that 4-fold coordinated N2 may be stabilized in several structures on the double-atom wide steps of Ru(1 0 9) and that this form of bonding produces substantial decreases in the N2 vibrational frequency and increases in the binding energy, compared to terminally-bound N2. These highly coordinated N2 species are not observed by IRAS.
.jpg)