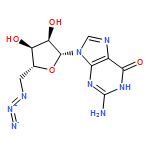
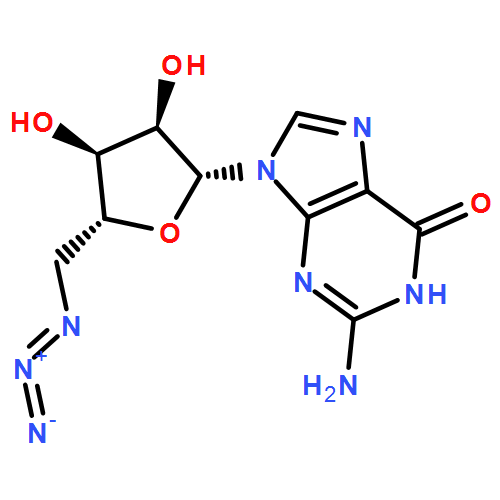
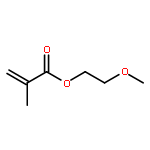
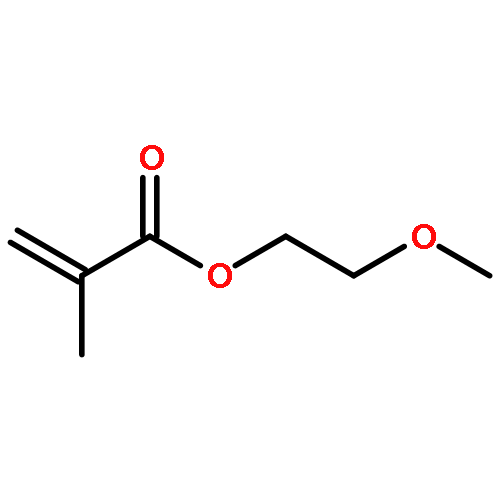
Co-reporter:Dr. Xiangcheng Pan;Sushil Lathwal;Stephanie Mack;Jiajun Yan; Subha R. Das; Krzysztof Matyjaszewski
Angewandte Chemie 2017 Volume 129(Issue 10) pp:2784-2787
Publication Date(Web):2017/03/01
DOI:10.1002/ange.201611567
AbstractA DNA synthesizer was successfully employed for preparation of well-defined polymers by atom transfer radical polymerization (ATRP), in a technique termed AutoATRP. This method provides well-defined homopolymers, diblock copolymers, and biohybrids under automated photomediated ATRP conditions. PhotoATRP was selected over other ATRP methods because of mild reaction conditions, ambient temperature, tolerance to oxygen, and no need to introduce reducing agents or radical initiators. Both acrylate and methacrylate monomers were successfully polymerized with excellent control in the DNA synthesizer. Diblock copolymers were synthesized with different targeted degrees of polymerization and with high retention of chain-end functionality. Both hydrophobic and hydrophilic monomers were grafted from DNA. The DNA-polymer hybrids were characterized by SEC and DLS. The AutoATRP method provides an efficient route to prepare a range of different polymeric materials, especially polymer-biohybrids.
Co-reporter:Saadyah Averick, Ryan A. Mehl, Subha R. Das, Krzysztof Matyjaszewski
Journal of Controlled Release 2015 Volume 205() pp:45-57
Publication Date(Web):10 May 2015
DOI:10.1016/j.jconrel.2014.11.030
The use of reversible deactivation radical polymerization (RDRP) methods has significantly expanded the field of bioconjugate synthesis. RDRP procedures have allowed the preparation of a broad range of functional materials that could not be realized using prior art poly(ethylene glycol) functionalization. The review of procedures for synthesis of biomaterials is presented with a special focus on the use of RDRP to prepare biohybrids with proteins, DNA and RNA.
Co-reporter:Munira F. Fouz, Kosuke Mukumoto, Saadyah Averick, Olivia Molinar, Brooke M. McCartney, Krzysztof Matyjaszewski, Bruce A. Armitage, and Subha R. Das
ACS Central Science 2015 Volume 1(Issue 8) pp:431
Publication Date(Web):November 4, 2015
DOI:10.1021/acscentsci.5b00259
Bright signal outputs are needed for fluorescence detection of biomolecules at their native expression levels. Increasing the number of labels on a probe often results in crowding-induced self-quenching of chromophores, and maintaining the function of the targeting moiety (e.g., an antibody) is a concern. Here we demonstrate a simple method to accommodate thousands of fluorescent dye molecules on a single antibody probe while avoiding the negative effects of self-quenching. We use a bottlebrush polymer from which extend hundreds of duplex DNA strands that can accommodate hundreds of covalently attached and/or thousands of noncovalently intercalated fluorescent dyes. This polymer–DNA assembly sequesters the intercalated fluorophores against dissociation and can be tethered through DNA hybridization to an IgG antibody. The resulting fluorescent nanotag can detect protein targets in flow cytometry, confocal fluorescence microscopy, and dot blots with an exceptionally bright signal that compares favorably to commercially available antibodies labeled with organic dyes or quantum dots.
Co-reporter:Saadyah E. Averick ; Eduardo Paredes ; Sourav K. Dey ; Kristin M. Snyder ; Nikos Tapinos ; Krzysztof Matyjaszewski
Journal of the American Chemical Society 2013 Volume 135(Issue 34) pp:12508-12511
Publication Date(Web):August 12, 2013
DOI:10.1021/ja404520j
Short interfering ribonucleic acids (siRNAs) are important agents for RNA interference (RNAi) that have proven useful in gene function studies and therapeutic applications. However, the efficacy of exogenous siRNAs for gene knockdown remains hampered by their susceptibility to cellular nucleases and impermeability to cell membranes. We report here new covalent polymer-escort siRNA constructs that address both of these constraints simultaneously. By simple postsynthetic click conjugation of polymers to the passenger strand of an siRNA duplex followed by annealing with the complementary guide strand, we obtained siRNA in which one strand includes terminal polymer escorts. The polymer escorts both confer protection against nucleases and facilitate cellular internalization of the siRNA. These autotransfecting polymer-escort siRNAs are viable in RNAi and effective in knocking down reporter and endogenous genes.
Co-reporter:Eduardo Paredes, Xiaojuan Zhang, Harshad Ghodke, Vamsi K. Yadavalli, and Subha R. Das
ACS Nano 2013 Volume 7(Issue 5) pp:3953
Publication Date(Web):April 19, 2013
DOI:10.1021/nn305787m
Nanotechnology based on the highly specific pairing of nucleobases in DNA has been used to generate a wide variety of well-defined two- and three-dimensional assemblies, both static and dynamic. However, control over the junction angles to achieve them has been limited. To achieve higher order assemblies, the strands of the DNA duplex are typically made to deviate at junctions with configurations based on crossovers or non-DNA moieties. Such strand crossovers tend to be intrinsically unstructured with the overall structural rigidity determined by the architecture of the nanoassembly, rather than the junction itself. Specific approaches to define nanoassembly junction angles are based either on the cooperative twist- and strain-promoted tuning of DNA persistence length leading to bent DNA rods for fairly large nano-objects, or de novo synthesis of individual junction inserts that are typically non-DNA and based on small organic molecules or metal-coordinating ligand moieties. Here, we describe a general strategy for direct control of junction angles in DNA nanostructures that are completely tunable about the DNA helix. This approach is used to define angular vertices through readily accessible backbone-branched DNAs (bbDNAs). We demonstrate how such bbDNAs can be used as a new building block in DNA nanoconstruction to obtain well-defined nanostructures. Angular control through readily accessible bbDNA building block provides a general and versatile approach for incorporating well-defined junctions in nanoconstructs and expands the toolkit toward achieving strain free, highly size- and shape-tunable DNA based architectures.Keywords: backbone-branched DNA; click chemistry; DNA nanostructures; nanoassembly · junction angles
Co-reporter:Eduardo Paredes, Subha R. Das
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 16) pp:5313-5316
Publication Date(Web):15 August 2012
DOI:10.1016/j.bmcl.2012.06.027
The copper(I) catalyzed azide–alkyne cycloaddition ‘click’ reaction yields a specific product under mild conditions and in some of the most chemically complex environments. This reaction has been used extensively to tag DNA, proteins, glycans and only recently RNA. Click reactions in aqueous buffer typically include a ligand for Cu(I), however we find that acetonitrile as a minor co-solvent can serve this role. Here we investigate the click labeling of RNA and DNA in aqueous buffer to determine the relationship between the stoichoimetry of Cu(I) and the acetonitrile co-solvent that affects nucleic acid stability. We find that very low concentrations of acetonitrile perform equally well and obviate the need for any additional Cu(I) stabilizing ligand. These pseudo-ligandless reaction conditions are optimal for nucleic acids click conjugations.
Co-reporter:Saadyah E. Averick, Eduardo Paredes, Debasish Grahacharya, Bradley F. Woodman, Shigeki J. Miyake-Stoner, Ryan A. Mehl, Krzysztof Matyjaszewski, and Subha R. Das
Langmuir 2012 Volume 28(Issue 4) pp:1954-1958
Publication Date(Web):January 6, 2012
DOI:10.1021/la204077v
Protein–polymer hybrids (PPHs) represent an important and rapidly expanding class of biomaterials. Typically in these hybrids the linkage between the protein and the polymer is covalent. Here we describe a straightforward approach to a noncovalent PPH that is mediated by DNA. Although noncovalent, the DNA-mediated approach affords the highly specific pairing and assembly properties of DNA. To obtain the protein–DNA conjugate for assembly of the PPH, we report here the first direct copper catalyzed azide–alkyne cycloaddition-based protein–DNA conjugation. This significantly simplifies access to protein–DNA conjugates. The protein–DNA conjugate and partner polymer–DNA conjugate are readily assembled through annealing of the cDNA strands to obtain the PPH, the assembly of which was confirmed via dynamic light scattering and fluorescence spectroscopy.
Co-reporter:Saadyah Averick, Eduardo Paredes, Wenwen Li, Krzysztof Matyjaszewski, and Subha R. Das
Bioconjugate Chemistry 2011 Volume 22(Issue 10) pp:2030
Publication Date(Web):August 26, 2011
DOI:10.1021/bc200240q
Polymer biomolecule hybrids represent a powerful class of highly customizable nanomaterials. Here, we report star-polymer conjugates with DNA using a “ligandless” Cu(I) promoted azide–alkyne cycloaddition click reaction. The multivalency of the star-polymer architecture allows for the concomitant conjugation of other molecules along with the DNA, and the conjugation method provides control over the DNA orientation. The star-polymer DNA nanoparticles are shown to assemble into higher-order nanoassemblies through hybridization. Further, we show that the DNA strands can be utilized in controlled disassembly of the nanostructures.
Co-reporter:Eduardo Paredes ; Subha R. Das
ChemBioChem 2011 Volume 12( Issue 1) pp:125-131
Publication Date(Web):
DOI:10.1002/cbic.201000466
Abstract
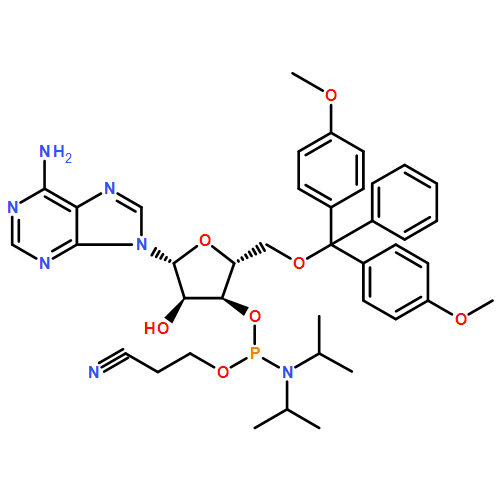
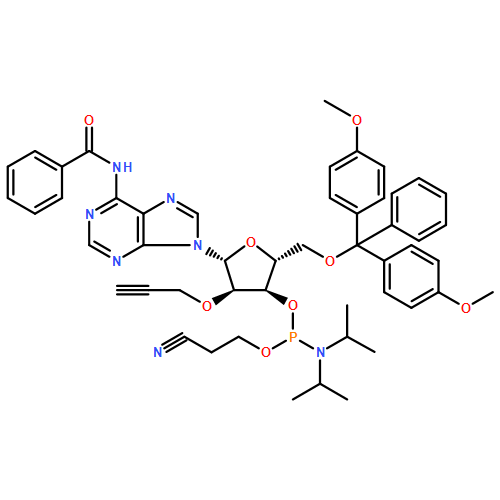
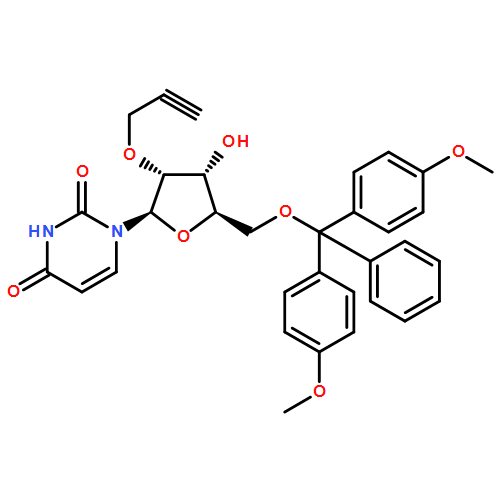
The copper(I)-promoted azide–alkyne cycloaddition reaction (click chemistry) is shown to be compatible with RNA (with free 2′-hydroxyl groups) in spite of the intrinsic lability of RNA. RNA degradation is minimized through stabilization of the CuI in aqueous buffer with acetonitrile as cosolvent and no other ligand; this suggests the general possibility of “ligandless” click chemistry. With the viability of click chemistry validated on synthetic RNA bearing “click”-reactive alkynes, the scope of the reaction is extended to in-vitro-transcribed or, indeed, any RNA, as a click-reactive azide is incorporated enzymatically. Once clickable groups are installed on RNA, they can be rapidly click labeled or conjugated together in click ligations, which may be either templated or nontemplated. In click ligations the resultant unnatural triazole-linked RNA backbone is not detrimental to RNA function, thus suggesting a broad applicability of click chemistry in RNA biological studies.
Co-reporter:Eduardo Paredes, Molly Evans, Subha R. Das
Methods (June 2011) Volume 54(Issue 2) pp:251-259
Publication Date(Web):1 June 2011
DOI:10.1016/j.ymeth.2011.02.008
Advances in RNA nanotechnology will depend on the ability to manipulate, probe the structure and engineer the function of RNA with high precision. This article reviews current abilities to incorporate site-specific labels or to conjugate other useful molecules to RNA either directly or indirectly through post-synthetic labeling methodologies that have enabled a broader understanding of RNA structure and function. Readily applicable modifications to RNA can range from isotopic labels and fluorescent or other molecular probes to protein, lipid, glycoside or nucleic acid conjugates that can be introduced using combinations of synthetic chemistry, enzymatic incorporation and various conjugation chemistries. These labels, conjugations and ligations to RNA are quintessential for further investigation and applications of RNA as they enable the visualization, structural elucidation, localization, and biodistribution of modified RNA.
.jpg)
























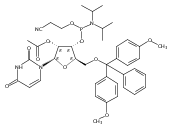
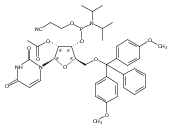




![3H-Indolium, 3-[5-[(aminohydroxyphosphinyl)oxy]pentyl]-2-[3-(1,3-dihydro-1,3,3-trimethyl-2H-indol-2-ylidene)-1-propen-1-yl]-1,3-dimethyl-](/data/chemimg/178400/1354898-55-7.png)
![3H-Indolium, 3-[5-[(aminohydroxyphosphinyl)oxy]pentyl]-2-[3-(1,3-dihydro-1,3,3-trimethyl-2H-indol-2-ylidene)-1-propen-1-yl]-1,3-dimethyl-](/data/chemimg/178400/1354898-55-7_b.png)






















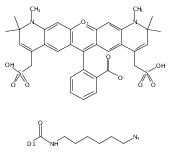
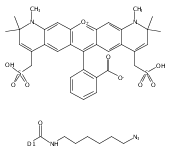






















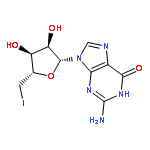
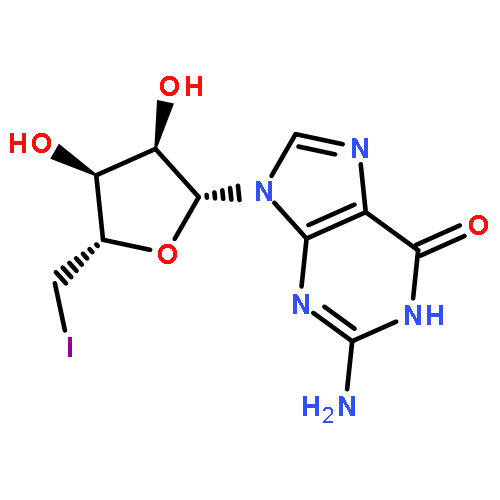
![Ethanaminium, N-?ethyl-?N,?N-?dimethyl-?2-?[(2-?methyl-?1-?oxo-?2-?propen-?1-?yl)?oxy]?-?, bromide (1:1)](/data/chemimg/3779200/52243-12-6.png)
![Ethanaminium, N-?ethyl-?N,?N-?dimethyl-?2-?[(2-?methyl-?1-?oxo-?2-?propen-?1-?yl)?oxy]?-?, bromide (1:1)](/data/chemimg/3779200/52243-12-6_b.png)