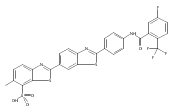
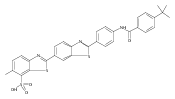
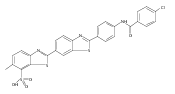
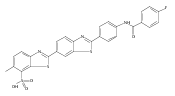
Co-reporter:Leah K. Forsberg, Weiya Liu, Jeffrey Holzbeierlein, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 18(Issue 18) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.bmcl.2017.07.030
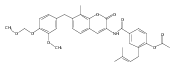
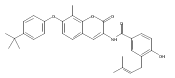
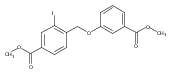
Heat Shock Protein 90 (Hsp90) is a molecular chaperone under clinical investigation for the treatment of neurodegenerative diseases and cancer. Neuroprotective Hsp90 C-terminal inhibitors (novologues) contain a biaryl ring system, and include KU-596, which was modified and investigated for potential anti-cancer activity. Incorporation of a benzamide group onto the biaryl novologues in lieu of the acetamide yielded compounds that manifest anti-cancer activity. Further exploration of the central phenyl ring led to compounds with enhanced anti-proliferative activity. The design, synthesis, and evaluation of these new analogs against breast and prostate cancer cell lines is reported herein, where it was found that 8b and 10 manifest potent anti-proliferative activity and a robust degradation of Hsp90 client-dependent proteins.Download high-res image (105KB)Download full-size image
Co-reporter:Sanket J. Mishra, Suman Ghosh, Andrew R. Stothert, Chad A. Dickey, and Brian S. J. Blagg
ACS Chemical Biology 2017 Volume 12(Issue 1) pp:
Publication Date(Web):December 13, 2016
DOI:10.1021/acschembio.6b00747
Glucose regulated protein 94 kDa, Grp94, is the endoplasmic reticulum (ER) localized isoform of heat shock protein 90 (Hsp90) that is responsible for the trafficking and maturation of toll-like receptors, immunoglobulins, and integrins. As a result, Grp94 has emerged as a therapeutic target to disrupt cellular communication, adhesion, and tumor proliferation, potentially with fewer side effects compared to pan-inhibitors of all Hsp90 isoforms. Although, the N-terminal ATP binding site is highly conserved among all four Hsp90 isoforms, recent cocrystal structures of Grp94 have revealed subtle differences between Grp94 and other Hsp90 isoforms that has been exploited for the development of Grp94-selective inhibitors. In the current study, a structure-based approach has been applied to a Grp94 nonselective compound, SNX 2112, which led to the development of 8j (ACO1), a Grp94-selective inhibitor that manifests ∼440 nM affinity and >200-fold selectivity against cytosolic Hsp90 isoforms.
Co-reporter:Rachel E. Davis;Zheng Zhang
MedChemComm (2010-Present) 2017 vol. 8(Issue 3) pp:593-598
Publication Date(Web):2017/03/23
DOI:10.1039/C6MD00377J
Inhibition of the Hsp90 C-terminus is an attractive therapeutic paradigm for the treatment of cancer, however the developmental space of C-terminal inhibitors is limited. It was hypothesized that the combination of two previously identified scaffolds into a single structure could provide a platform for which to probe the three-dimensional space within the Hsp90 C-terminal binding pocket. The resulting chimeric compounds displayed anti-proliferative activity at low micromolar concentrations and manifested inhibitory activity in an Hsp90-dependent rematuration assay. Initial structure–activity relationships suggest that this new scaffold binds Hsp90 in a conformation different from that of the parent compounds, and consequently, provides a new opportunity to develop more efficacious inhibitors of the Hsp90 C-terminal binding pocket.
Co-reporter:Jessica A. Hall; Sahithi Seedarala; Huiping Zhao; Gaurav Garg; Suman Ghosh
Journal of Medicinal Chemistry 2016 Volume 59(Issue 3) pp:925-933
Publication Date(Web):January 8, 2016
DOI:10.1021/acs.jmedchem.5b01354
Heat shock protein 90 (Hsp90) inhibition by modulation of its N- or C-terminal binding site has become an attractive strategy for the development of anticancer chemotherapeutics. The first Hsp90 C-terminus inhibitor, novobiocin, manifested a relatively high IC50 value of ∼700 μM. Therefore, investigation of the novobiocin scaffold has led to analogues with improved antiproliferative activity (nanomolar concentrations) against several cancer cell lines. During these studies, novobiocin analogues that do not inhibit Hsp90 were identified; however, these analogues demonstrated potent antiproliferative activity. Compound 2, a novobiocin analogue, was identified as a MAPK pathway signaling disruptor that lacked Hsp90 inhibitory activity. In addition, structural modifications of compound 2 were identified that segregated Hsp90 inhibition from MAPK signaling disruption. These studies indicate that compound 2 represents a novel scaffold for disruption of MAPK pathway signaling and may serve as a useful structure for the generation of new anticancer agents.
Co-reporter:Vincent M. Crowley; Anuj Khandelwal; Sanket Mishra; Andrew R. Stothert; Dustin J. E. Huard; Jinbo Zhao; Aaron Muth; Adam S. Duerfeldt; James L. Kizziah; Raquel L. Lieberman; Chad A. Dickey
Journal of Medicinal Chemistry 2016 Volume 59(Issue 7) pp:3471-3488
Publication Date(Web):March 22, 2016
DOI:10.1021/acs.jmedchem.6b00085
Glucose regulated protein 94 (Grp94) is the endoplasmic reticulum resident of the heat shock protein 90 kDa (Hsp90) family of molecular chaperones. Grp94 associates with many proteins involved in cell adhesion and signaling, including integrins, Toll-like receptors, immunoglobulins, and mutant myocilin. Grp94 has been implicated as a target for several therapeutic areas including glaucoma, cancer metastasis, and multiple myeloma. While 85% identical to other Hsp90 isoforms, the N-terminal ATP-binding site of Grp94 possesses a unique hydrophobic pocket that was used to design isoform-selective inhibitors. Incorporation of a cis-amide bioisostere into the radamide scaffold led to development of the original Grp94-selective inhibitor, BnIm. Structure–activity relationship studies have now been performed on the aryl side chain of BnIm, which resulted in improved analogues that exhibit better potency and selectivity for Grp94. These analogues also manifest superior antimigratory activity in a metastasis model as well as enhanced mutant myocilin degradation in a glaucoma model compared to BnIm.
Co-reporter:Mercy Anyika, Mason McMullen, Leah K. Forsberg, Rick T. Dobrowsky, and Brian S. J. Blagg
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 1) pp:67
Publication Date(Web):December 9, 2015
DOI:10.1021/acsmedchemlett.5b00331
KU-32 and KU-596 are novobiocin-derived, C-terminal heat shock protein 90 (Hsp90) modulators that induce Hsp70 levels and manifest neuroprotective activity. However, the synthetically complex noviose sugar requires 10 steps to prepare, which makes translational development difficult. In this study, we developed a series of “noviomimetic” analogues of KU-596, which contain noviose surrogates that can be easily prepared, while maintaining the ability to induce Hsp70 levels. Both sugar and sugar analogues were designed, synthesized, and evaluated in a luciferase reporter assay, which identified compound 37, a benzyl containing noviomimetic, as the most potent inducer of Hsp70.Keywords: C-terminal inhibition; Heat Shock Protein 70; Heat Shock Protein 90; neuroprotection; noviomimetics
Co-reporter:Suman Ghosh, Yang Liu, Gaurav Garg, Mercy Anyika, Nolan T. McPherson, Jiacheng Ma, Rick T. Dobrowsky, and Brian S. J. Blagg
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 8) pp:813
Publication Date(Web):July 5, 2016
DOI:10.1021/acsmedchemlett.6b00224
Novobiocin is a natural product that binds the Hsp90 C-terminus and manifests Hsp90 inhibitory activity. Structural investigations on novobiocin led to the development of both anti-cancer and neuroprotective agents. The varied pharmacological activity manifested by these novobiocin analogs prompted the investigation of structure–function studies to identify these contradictory effects, which revealed that modifications to the amide side chain produce either anti-cancer or neuroprotective activity. Compounds that exhibit neuroprotective activity contain a short alkyl or cycloalkyl amide side chain. In contrast, anti-cancer agents contain five or more carbons, disrupt interactions between Hsp90α and Aha1, and induce the degradation of Hsp90-dependent client proteins.Keywords: Aha1; anti-cancer; Hsp90; Hsp90α; neuroprotection; novobiocin; structure−activity relationship;
Co-reporter:Katherine M. Byrd;Chitra Subramanian;Jacqueline Sanchez;Hashim F. Motiwala;Weiya Liu; Mark S. Cohen; Jeffrey Holzbeierlein; Brian S. J. Blagg
Chemistry - A European Journal 2016 Volume 22( Issue 20) pp:6921-6931
Publication Date(Web):
DOI:10.1002/chem.201504955
Abstract
Development of heat shock protein 90 (Hsp90) C-terminal inhibitors has emerged as an exciting strategy for the treatment of cancer. Previous efforts have focused on modifications to the natural products novobiocin and coumermycin. Moreover, variations in both the sugar and amide moieties have been extensively studied, whereas replacements for the coumarin core have received less attention. Herein, 24 cores were synthesized with varying distances and angles between the sugar and amide moieties. Compounds that exhibited good anti-proliferative activity against multiple cancer cell lines and Hsp90 inhibitory activity, were those that placed the sugar and amide moieties between 7.7 and 12.1 Å apart along with angles of 180°.
Co-reporter:Suman Ghosh, Heather E. Shinogle, Gaurav Garg, George A. Vielhauer, Jeffrey M. Holzbeierlein, Rick T. Dobrowsky, and Brian S. J. Blagg
ACS Chemical Biology 2015 Volume 10(Issue 2) pp:577
Publication Date(Web):November 17, 2014
DOI:10.1021/cb5008713
Human Hsp90 isoforms are molecular chaperones that are often up-regulated in malignances and represent a primary target for Hsp90 inhibitors undergoing clinical evaluation. Hsp90α is a stress-inducible isoform of Hsp90 that plays a significant role in apoptosis and metastasis. Though Hsp90α is secreted into the extracellular space under metastatic conditions, its role in cancer biology is poorly understood. We report that Hsp90α associates with the Aha1 co-chaperone and found this complex to localize in secretory vesicles and at the leading edge of migrating cells. Knockdown of Hsp90α resulted in a defect in cell migration. The functional role of Hsp90α/Aha1 was studied by treating the cells with various novobiocin-based Hsp90 C-terminal inhibitors. These inhibitors disrupted the Hsp90α/Aha1 complex, caused a cytoplasmic redistribution of Hsp90α and Aha1, and decreased cell migration. Structure–function studies determined that disruption of Hsp90α/Aha1 association and inhibition of cell migration correlated with the presence of a benzamide side chain, since an acetamide substituted analog was less effective. Our results show that disruption of Hsp90α/Aha1 interactions with novobiocin-based Hsp90 C-terminal inhibitors may limit the metastatic potential of tumors.
Co-reporter:Huiping Zhao, Gaurav Garg, Jinbo Zhao, Elisabetta Moroni, Antwan Girgis, Lucas S. Franco, Swapnil Singh, Giorgio Colombo, Brian S.J. Blagg
European Journal of Medicinal Chemistry 2015 Volume 89() pp:442-466
Publication Date(Web):7 January 2015
DOI:10.1016/j.ejmech.2014.10.034
•A small library of biphenylamide derivatives was designed and analogs synthesized.•Compounds were evaluated for anti-proliferative activity against cancer cell lines.•Hsp90 inhibition was confirmed by Western blot analysis.•Several analogs showed low nanomolar inhibitory activity.Modulation of Hsp90 C-terminal function represents a promising therapeutic approach for the treatment of cancer and neurodegenerative diseases. Current drug discovery efforts toward Hsp90 C-terminal inhibition focus on novobiocin, an antibiotic that was transformed into an Hsp90 inhibitor. Based on structural information obtained during the development of novobiocin derivatives and molecular docking studies, scaffolds containing a biphenyl moiety in lieu of the coumarin ring present in novobiocin were identified as new Hsp90 C-terminal inhibitors. Structure–activity relationship studies produced new derivatives that inhibit the proliferation of breast cancer cell lines at nanomolar concentrations, which corresponded directly with Hsp90 inhibition.
Co-reporter:Jessica A. Hall; Sahithi Seedarala; Nichole Rice; Lucas Kopel; Fathi Halaweish
Journal of Natural Products 2015 Volume 78(Issue 4) pp:873-879
Publication Date(Web):March 10, 2015
DOI:10.1021/acs.jnatprod.5b00054
Heat shock protein 90 (Hsp90) facilitates the maturation of many newly synthesized and unfolded proteins (clients) via the Hsp90 chaperone cycle, in which Hsp90 forms a heteroprotein complex and relies upon cochaperones, immunophilins, etc., for assistance in client folding. Hsp90 inhibition has emerged as a strategy for anticancer therapies due to the involvement of clients in many oncogenic pathways. Inhibition of chaperone function results in client ubiquitinylation and degradation via the proteasome, ultimately leading to tumor digression. Small molecule inhibitors perturb ATPase activity at the N-terminus and include derivatives of the natural product geldanamycin. However, N-terminal inhibition also leads to induction of the pro-survival heat shock response (HSR), in which displacement of the Hsp90-bound transcription factor, heat shock factor-1, translocates to the nucleus and induces transcription of heat shock proteins, including Hsp90. An alternative strategy for Hsp90 inhibition is disruption of the Hsp90 heteroprotein complex. Disruption of the Hsp90 heteroprotein complex is an effective strategy to prevent client maturation without induction of the HSR. Cucurbitacin D, isolated from Cucurbita texana, and 3-epi-isocucurbitacin D prevented client maturation without induction of the HSR. Cucurbitacin D also disrupted interactions between Hsp90 and two cochaperones, Cdc37 and p23.
Co-reporter:Gaurav Garg, Huiping Zhao, and Brian S. J. Blagg
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 2) pp:204
Publication Date(Web):December 12, 2014
DOI:10.1021/ml5004475
Hsp90 C-terminal inhibitors represent a novel and alternative chemotherapeutic approach for the treatment of cancer. Novobiocin was the first natural product identified as an Hsp90 C-terminal inhibitor; however, it manifests poor antiproliferative activity. In contrast to N-terminal inhibitors, novobiocin does not induce the pro-survival heat shock response. Structural investigations on novobiocin have elucidated some structure–activity relationships and several promising compounds. On the basis of structure–activity relationships and computational studies, a library of ring-constrained novobiocin analogues was designed, synthesized, and evaluated in antiproliferative assays. Results obtained from these studies provide insights into the Hsp90 C-terminal binding site, and new analogues that were developed manifest low micromolar to mid-nanomolar antiproliferative activity resulting from Hsp90 inhibition.Keywords: breast cancer; Heat shock protein 90; Hsp90 C-terminal inhibitors; ring-constrained novobiocin analogues; structure−activity relationship
Co-reporter:Jessica A. Hall, Bhaskar Reddy Kusuma, Gary E. L. Brandt, and Brian S. J. Blagg
ACS Chemical Biology 2014 Volume 9(Issue 4) pp:976
Publication Date(Web):January 22, 2014
DOI:10.1021/cb400906e
The molecular chaperone Hsp90 requires the assistance of immunophilins, co-chaperones, and partner proteins for the conformational maturation of client proteins. Hsp90 inhibition represents a promising anticancer strategy due to the dependence of numerous oncogenic signaling pathways upon Hsp90 function. Historically, small molecules have been designed to inhibit ATPase activity at the Hsp90 N-terminus; however, these molecules also induce the pro-survival heat shock response (HSR). Therefore, inhibitors that exhibit alternative mechanisms of action that do not elicit the HSR are actively sought. Small molecules that disrupt Hsp90-co-chaperone interactions can destabilize the Hsp90 complex without induction of the HSR, which leads to inhibition of cell proliferation. In this article, selective inhibition of F1F0 ATP synthase by cruentaren A was shown to disrupt the Hsp90-F1F0 ATP synthase interaction and result in client protein degradation without induction of the HSR.
Co-reporter:Huiping Zhao, Elisabetta Moroni, Giorgio Colombo, and Brian S. J. Blagg
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 1) pp:84-88
Publication Date(Web):November 26, 2013
DOI:10.1021/ml400404s
Inhibition of Hsp90 C-terminal function is an advantageous therapeutic paradigm for the treatment of cancer. Currently, the majority of Hsp90 C-terminal inhibitors are derived from novobiocin, a natural product traditionally used as an antibiotic. Assisted by molecular docking studies, a scaffold containing a biphenyl moiety in lieu of the coumarin ring system found in novobiocin was identified for development of new Hsp90 C-terminal inhibitors. Initial structure–activity studies led to derivatives that manifest good antiproliferative activity against two breast cancer cell lines through Hsp90 inhibition. This platform serves as a scaffold upon which new Hsp90 C-terminal inhibitors can be readily assembled for further investigation.Keywords: biphenyl; breast cancer; Heat shock protein 90; Hsp90 C-terminal inhibitors; novobiocin;
Co-reporter:Bhaskar Reddy Kusuma, Anuj Khandelwal, Wen Gu, Douglas Brown, Weiya Liu, George Vielhauer, Jeffrey Holzbeierlein, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 4) pp:1441-1449
Publication Date(Web):15 February 2014
DOI:10.1016/j.bmc.2013.12.056
Since Hsp90 modulates all six hallmarks of cancer simultaneously, it has become an attractive target for the development of cancer chemotherapeutics. In an effort to develop more efficacious compounds for Hsp90 inhibition, novobiocin analogues were prepared by replacing the central coumarin core with naphthalene, quinolinone, and quinoline surrogates. These modifications allowed for modification of the 2-position, which was previously unexplored. Biological evaluation of these compounds suggests a hydrophobic pocket about the 2-position of novobiocin. Anti-proliferative activities of these analogues against multiple cancer cell lines identified 2-alkoxyquinoline derivatives to exhibit improved activity.
Co-reporter:Huiping Zhao, Mercy Anyika, Antwan Girgis, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 15) pp:3633-3637
Publication Date(Web):1 August 2014
DOI:10.1016/j.bmcl.2014.05.020
Co-reporter:Jinbo Zhao, Huiping Zhao, Jessica A. Hall, Douglas Brown, Eileen Brandes, Joseph Bazzill, Patrick T. Grogan, Chitra Subramanian, George Vielhauer, Mark S. Cohen and Brian S. J. Blagg
MedChemComm 2014 vol. 5(Issue 9) pp:1317-1323
Publication Date(Web):27 Jun 2014
DOI:10.1039/C4MD00102H
Hsp90 C-terminal inhibitors are advantageous for the development of new cancer chemotherapeutics due to their ability to segregate client protein degradation from induction of the prosurvival heat shock response, which is a major detriment associated with Hsp90 N-terminal inhibitors under clinical investigation. Based upon prior SAR trends, a 1,2,3-triazole side chain was placed in lieu of the aryl side chain and attached to both the coumarin and biphenyl scaffold. Antiproliferative studies against SKBr3 and MCF-7 breast cancer cell lines demonstrated these triazole-containing compounds to exhibit improved activity. These compounds were shown to manifest Hsp90 inhibitory activity through Western blot analysis and represent a new scaffold upon which more potent inhibitors can be pursued.
Co-reporter:Aaron Muth, Vincent Crowley, Anuj Khandelwal, Sanket Mishra, Jinbo Zhao, Jessica Hall, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry 2014 22(15) pp: 4083-4098
Publication Date(Web):
DOI:10.1016/j.bmc.2014.05.075
Co-reporter:Huiping Zhao, Elisabetta Moroni, Bin Yan, Giorgio Colombo, and Brian S. J. Blagg
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 1) pp:57-62
Publication Date(Web):November 29, 2012
DOI:10.1021/ml300275g
Hsp90 is an attractive therapeutic target for the treatment of cancer. Extensive structural modifications to novobiocin, the first Hsp90 C-terminal inhibitor discovered, have produced a library of novobiocin analogues and revealed some structure–activity relationships. On the basis of the most potent novobiocin analogues generated from prior studies, a three-dimensional quantitative structure–activity (3D QSAR) model was built. In addition, a new set of novobiocin analogues containing various structural features supported by the 3D QSAR model were synthesized and evaluated against two breast cancer cell lines. Several new inhibitors produced antiproliferative activity at midnanomolar concentrations, which results through Hsp90 inhibition.Keywords: 3D QSAR; breast cancer; heat shock protein 90; Hsp90 inhibitors; novobiocin;
Co-reporter:Huiping Zhao, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 2) pp:552-557
Publication Date(Web):15 January 2013
DOI:10.1016/j.bmcl.2012.11.022
Hsp90 is a promising therapeutic target for the treatment of cancer. Novobiocin is the first Hsp90 C-terminal inhibitor ever identified and recent structure–activity relationship studies on the noviose sugar identified several commercially available amines as suitable surrogates. In an effort to further understand this region of the molecule, analogues containing various N′-amino substituents were prepared and evaluated against two breast cancer cell lines for determination of their efficacy. Compound 37j manifested the most potent anti-proliferative activity from these studies and induced Hsp90-dependent client protein degradation at mid nano-molar concentrations.
Co-reporter:Anuj Khandelwal, Jessica A. Hall, and Brian S. J. Blagg
The Journal of Organic Chemistry 2013 Volume 78(Issue 16) pp:7859-7884
Publication Date(Web):July 8, 2013
DOI:10.1021/jo401027r
Epigallocatechin-3-gallate (EGCG), the principal polyphenol isolated from green tea, was recently shown to inhibit Hsp90; however, structure–activity relationships for this natural product have not yet been produced. Herein, we report the synthesis and biological evaluation of EGCG analogues to establish structure–activity relationships between EGCG and Hsp90. All four rings as well as the linker connecting the C- and the D-rings were systematically investigated, which led to the discovery of compounds that inhibit Hs90 and display improvement in efficacy over EGCG. Antiproliferative activity of all the analogues was determined against MCF-7 and SKBr3 cell lines and Hsp90 inhibitory activity of the four most potent analogues was further evaluated by Western blot analyses and degradation of Hsp90-dependent client proteins. The prenyl-substituted aryl ester of 3,5-dihydroxychroman-3-ol ring system was identified as a novel scaffold that exhibits Hsp90 inhibitory activity.
Co-reporter:Adam S. Duerfeldt ; Laura B. Peterson ; Jason C. Maynard ; Chun Leung Ng ; Davide Eletto ; Olga Ostrovsky ; Heather E. Shinogle ; David S. Moore ; Yair Argon ; Christopher V. Nicchitta
Journal of the American Chemical Society 2012 Volume 134(Issue 23) pp:9796-9804
Publication Date(Web):May 29, 2012
DOI:10.1021/ja303477g
Heat shock protein 90 (Hsp90) represents a promising therapeutic target for the treatment of cancer and other diseases. Unfortunately, results from clinical trials have been disappointing as off-target effects and toxicities have been observed. These detriments may be a consequence of pan-Hsp90 inhibition, as all clinically evaluated Hsp90 inhibitors simultaneously disrupt all four human Hsp90 isoforms. Using a structure-based approach, we designed an inhibitor of Grp94, the ER-resident Hsp90. The effect manifested by compound 2 on several Grp94 and Hsp90α/β (cytosolic isoforms) clients were investigated. Compound 2 prevented intracellular trafficking of the Toll receptor, inhibited the secretion of IGF-II, affected the conformation of Grp94, and suppressed Drosophila larval growth, all Grp94-dependent processes. In contrast, compound 2 had no effect on cell viability or cytosolic Hsp90α/β client proteins at similar concentrations. The design, synthesis, and evaluation of 2 are described herein.
Co-reporter:Bhaskar Reddy Kusuma, Gary E. L. Brandt, and Brian S. J. Blagg
Organic Letters 2012 Volume 14(Issue 24) pp:6242-6245
Publication Date(Web):December 3, 2012
DOI:10.1021/ol302999v
Cruentaren A, an antifungal benzolactone produced by the myxobacterium Byssovorax cruenta, is highly cytotoxic against various human cancer cell lines and a highly selective inhibitor of mitochondrial F-ATPase. A convergent and efficient synthesis of cruentaren A is reported, based upon a diastereoselective alkylation, a series of stereoselective aldol reactions utilizing Myers’ pseudoephedrine propionamide, an acyl bromide mediated esterification, and a ring-closing metathesis (RCM) as the key steps. The RCM reaction was applied for the first time toward the total synthesis of cruentaren A, which led to a convergent and efficient synthesis of the natural product.
Co-reporter:Bhaskar Reddy Kusuma ; Liang Zhang ; Teather Sundstrom ; Laura B. Peterson ; Rick T. Dobrowsky
Journal of Medicinal Chemistry 2012 Volume 55(Issue 12) pp:5797-5812
Publication Date(Web):June 15, 2012
DOI:10.1021/jm300544c
Compound 2 (KU-32) is a first-generation novologue (a novobiocin-based, C-terminal, heat shock protein 90 (Hsp90) inhibitor) that decreases glucose-induced death of primary sensory neurons and reverses numerous clinical indices of diabetic peripheral neuropathy in mice. The current study sought to exploit the C-terminal binding site of Hsp90 to determine whether the optimization of hydrogen bonding and hydrophobic interactions of second-generation novologues could enhance neuroprotective activity. Using a series of substituted phenylboronic acids to replace the coumarin lactone of 2, we identified that electronegative atoms placed at the meta-position of the B-ring exhibit improved cytoprotective activity, which is believed to result from favorable interactions with Lys539 in the Hsp90 C-terminal binding pocket. Consistent with these results, a meta-3-fluorophenyl substituted novologue (13b) exhibited a 14-fold lower ED50 for protection against glucose-induced toxicity of primary sensory neurons compared to 2.
Co-reporter:Laura B. Peterson, Jeffrey D. Eskew, George A. Vielhauer, and Brian S. J. Blagg
Molecular Pharmaceutics 2012 Volume 9(Issue 6) pp:1841-1846
Publication Date(Web):May 3, 2012
DOI:10.1021/mp300138n
Heat shock protein 90 (Hsp90) has emerged as a promising therapeutic target for the treatment of cancer. Several Hsp90 inhibitors have entered clinical trials. However, some toxicological detriments have arisen, such as cardiotoxicity resulting from hERG inhibition following the administration of Hsp90 inhibitors. We sought to investigate this toxicity as hERG has been previously reported as a client protein that depends upon Hsp90 for its maturation and functional trafficking. In this study we show that hERG depends upon a single Hsp90 isoform. hERG preferentially co-immunoprecipitated with Hsp90α, and genetic knockdown of Hsp90α, but not Hsp90β, resulted in a trafficking-defective hERG channel. This study demonstrates the importance of delineating the isoform dependence of Hsp90 client proteins and provides rationale for the design of isoform-selective Hsp90 inhibitors that avoid detrimental effects.Keywords: hERG; Hsp90; isoform;
Co-reporter:Huiping Zhao, Bin Yan, Laura B. Peterson, and Brian S. J. Blagg
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 4) pp:327
Publication Date(Web):February 26, 2012
DOI:10.1021/ml300018e
The potential therapeutic benefits associated with Hsp90 modulation for the treatment of cancer and neurodegenerative diseases highlight the importance of identifying novel Hsp90 scaffolds. KU-398, a novobiocin analogue, and silybin were recently identified as new Hsp90 inhibitors. Consequently, a library of 3-arylcoumarin derivatives that incorporated the structural features of KU-398 and silybin was designed, synthesized, and evaluated against two breast cancer cell lines. Western blot analysis confirmed that the resulting 3-arylcoumarin hybrids target the Hsp90 protein folding machinery.Keywords: 3-arylcoumarin; breast cancer; heat shock protein 90; Hsp90 inhibitors; novobiocin; silybin
Co-reporter:Bhaskar Reddy Kusuma ; Laura B. Peterson ; Huiping Zhao ; George Vielhauer ; Jeffrey Holzbeierlein
Journal of Medicinal Chemistry 2011 Volume 54(Issue 18) pp:6234-6253
Publication Date(Web):August 23, 2011
DOI:10.1021/jm200553w
The design, synthesis, and biological evaluation of conformationally constrained coumermycin A1 analogues are reported. Compounds were evaluated against both breast cancer (SKBr3 and MCF7) and prostate cancer (PC3 mm2, A549, and HT29) cell lines. Non-noviosylated coumermycin A1 analogues that manifest potent antiproliferative activity resulting from Hsp90 inhibition are provided, wherein replacement of the stereochemically complex noviose sugar with readily available piperidine rings resulted in ∼100 fold increase in antiproliferative activities as compared to coumermycin A1, producing small molecule Hsp90 inhibitors that exhibit nanomolar activities.
Co-reporter:Huiping Zhao ; Alison C. Donnelly ; Bhaskar R. Kusuma ; Gary E. L. Brandt ; Douglas Brown ; Roger A. Rajewski ̂; George Vielhauer ; Jeffrey Holzbeierlein ; Mark S. Cohen
Journal of Medicinal Chemistry 2011 Volume 54(Issue 11) pp:3839-3853
Publication Date(Web):May 9, 2011
DOI:10.1021/jm200148p
Development of the DNA gyrase inhibitor, novobiocin, into a selective Hsp90 inhibitor was accomplished through structural modifications to the amide side chain, coumarin ring, and sugar moiety. These species exhibit ∼700-fold improved anti-proliferative activity versus the natural product as evaluated by cellular efficacies against breast, colon, prostate, lung, and other cancer cell lines. Utilization of structure–activity relationships established for three novobiocin synthons produced optimized scaffolds, which manifest midnanomolar activity against a panel of cancer cell lines and serve as lead compounds that manifest their activities through Hsp90 inhibition.
Co-reporter:Robert L. Matts, Anshuman Dixit, Laura B. Peterson, Liang Sun, Sudhakar Voruganti, Palgunan Kalyanaraman, Steve D. Hartson, Gennady M. Verkhivker, and Brian S. J. Blagg
ACS Chemical Biology 2011 Volume 6(Issue 8) pp:800
Publication Date(Web):May 6, 2011
DOI:10.1021/cb200052x
The Hsp90 chaperone machine is required for the folding, activation, and/or stabilization of more than 50 proteins directly related to malignant progression. Hsp90 contains small molecule binding sites at both its N- and C-terminal domains; however, limited structural and biochemical data regarding the C-terminal binding site is available. In this report, the small molecule binding site in the Hsp90 C-terminal domain was revealed by protease fingerprinting and photoaffinity labeling utilizing LC–MS/MS. The identified site was characterized by generation of a homology model for hHsp90α using the SAXS open structure of HtpG and docking the bioactive conformation of NB into the generated model. The resulting model for the bioactive conformation of NB bound to Hsp90α is presented herein.
Co-reporter:Gary E. L. Brandt and Brian S. J. Blagg
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 10) pp:735
Publication Date(Web):July 21, 2011
DOI:10.1021/ml200108y
Macrocyclic natural products are a powerful class of leadlike chemical entities. Despite commonly violating Lipinski's “rule of 5”, these compounds often demonstrate superior druglike physicochemical and pharmacokinetic attributes when compared to their acyclic counterparts. However, the elaborate structural architectures of such molecules require rigorous synthetic investigation that complicates analogue development and their application to drug discovery programs. To circumvent these limitations, a conformation-based approach using limited structure–activity relationships and molecular modeling was implemented to design simplified analogues of trienomycin A, in which the corresponding analogues could be prepared in a succinct manner to rapidly identify essential structural components necessary for biological activity. Trienomycin A is a member of the ansamycin family of natural products that possesses potent anticancer activity. These studies revealed a novel trienomycin A analogue, monoenomycin, which manifests potent anticancer activity.Keywords: ansamycin synthesis; anticancer; conformation; structure−activity relationship; Trienomycin A
Co-reporter:Robert L. Matts, Gary E.L. Brandt, Yuanming Lu, Anshuman Dixit, Mehdi Mollapour, Suiquan Wang, Alison C. Donnelly, Leonard Neckers, Gennady Verkhivker, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 1) pp:684-692
Publication Date(Web):1 January 2011
DOI:10.1016/j.bmc.2010.10.029
Several Hsp90 modulators have been identified including the N-terminal ligand geldanamycin (GDA), the C-terminal ligand novobiocin (NB), and the co-chaperone disruptor celastrol. Other Hsp90 modulators elicit a mechanism of action that remains unknown. For example, the natural product gedunin and the synthetic anti-spermatogenic agent H2-gamendazole, recently identified Hsp90 modulators, manifest biological activity through undefined mechanisms. Herein, we report a series of biochemical techniques used to classify such modulators into identifiable categories. Such studies provided evidence that gedunin and H2-gamendazole both modulate Hsp90 via a mechanism similar to celastrol, and unlike NB or GDA.
Co-reporter:Bhaskar Reddy Kusuma, Adam S. Duerfeldt, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 23) pp:7170-7174
Publication Date(Web):1 December 2011
DOI:10.1016/j.bmcl.2011.09.073
Novobiocin analogs lacking labile glycosidic ether have been designed, synthesized and evaluated for Hsp90 inhibitory activity. Replacement of the synthetically complex noviose sugar with simple aromatic side chains produced analogs that maintain moderate cytotoxic activity against MCF7 and SkBR3 breast cancer cell-lines. Rationale for the preparation of des-noviose novobiocin analogs in addition to their synthesis and biological evaluation are presented herein.
Co-reporter:Huiping Zhao, Gary E. Brandt, Lakshmi Galam, Robert L. Matts, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 9) pp:2659-2664
Publication Date(Web):1 May 2011
DOI:10.1016/j.bmcl.2010.12.088
Through Hsp90-dependent firefly luciferase refolding and Hsp90-dependent heme-regulated eIF2α kinase (HRI) activation assays, silybin was identified as a novel Hsp90 inhibitor. Subsequently, a library of silybin analogues was designed, synthesized and evaluated. Initial SAR studies identified the essential, non-essential and detrimental functionalities on silybin that contribute to Hsp90 inhibition.Figure options
Co-reporter:Jared R. Mays, Stephanie A. Hill, Justin T. Moyers, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 1) pp:249-266
Publication Date(Web):1 January 2010
DOI:10.1016/j.bmc.2009.10.061
The natural products novobiocin and derrubone have both demonstrated Hsp90 inhibition and structure–activity relationships have been established for each scaffold. Given these compounds share several key structural features, we hypothesized that incorporation of elements from each could provide insight to structural features important for Hsp90 inhibition. Thus, chimeric analogues of novobiocin and derrubone were constructed and evaluated. These studies confirmed that the functionality present at the 3-position of the isoflavone plays a critical role in determining Hsp90 inhibition and suggests that the bicyclic ring system present in both novobiocin and derrubone do not share similar modes of binding.
Co-reporter:Huiping Zhao, Bhaskar Reddy Kusuma, and Brian S. J. Blagg
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 7) pp:311
Publication Date(Web):July 13, 2010
DOI:10.1021/ml100070r
Structural modifications to the coumarin core and benzamide side chain of novobiocin have successfully transformed the natural product from a selective DNA gyrase inhibitor into a potent inhibitor of the Hsp90 C-terminus. However, no structure−activity relationship studies have been conducted on the noviose appendage, which represents the rate-limiting synthon in the preparation of analogues. Therefore, a series of sugar mimics and nonsugar derivatives were synthesized and evaluated to identify simplified compounds that exhibit Hsp90 inhibition. Evaluation against two breast cancer cell lines demonstrated that replacement of the stereochemical complex noviose with simplified alkyl amines increased antiproliferative activity, resulting in novobiocin analogues that manifest IC50 values in the midnanomolar range.Keywords (keywords): breast cancer; Heat shock protein 90; Hsp90 inhibitors; novobiocin; stucture−activity relationships
Co-reporter:Adam S. Duerfeldt, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 17) pp:4983-4987
Publication Date(Web):1 September 2010
DOI:10.1016/j.bmcl.2010.06.108
The 90 kDa heat shock proteins (Hsp90) represent a class of molecular chaperones responsible for the maturation and stabilization of many oncogenic proteins. Disrupting the ability of ATP to bind and facilitate the operation of Hsp90 has emerged as a promising approach toward cancer chemotherapeutic development. While numerous Hsp90 inhibitory scaffolds have been identified, progress through the clinic has revealed many obstacles that should be addressed in future analogue development. Recent reports of the complications, pitfalls, and downstream effects associated with Hsp90 inhibition are discussed herein, in hopes of providing a reference that can be used to guide the future design of Hsp90 inhibitory scaffolds.
Co-reporter:Laura B. Peterson, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 13) pp:3957-3960
Publication Date(Web):1 July 2010
DOI:10.1016/j.bmcl.2010.04.140
A series of triazole-containing novobiocin analogues has been designed, synthesized and their inhibitory activity determined. These compounds contain a triazole ring in lieu of the amide moiety present in the natural product. The anti-proliferative effects of these compounds were evaluated against two breast cancer cell lines (SKBr-3 and MCF-7), and manifested activities similar to their amide-containing counterparts. In addition, Hsp90-dependent client protein degradation was observed via Western blot analyses, supporting a common mode of Hsp90 inhibition for both structural classes.
Co-reporter:Alison C. Donnelly, Huiping Zhao, Bhaskar Reddy Kusuma and Brian S. J. Blagg
MedChemComm 2010 vol. 1(Issue 2) pp:165-170
Publication Date(Web):30 Jul 2010
DOI:10.1039/C0MD00063A
Studies on the natural product, novobiocin, have elucidated specific modifications that increase Hsp90 inhibition. Through diversification of the sugar appendage, coumarin core and benzamide side chain of novobiocin, structurally unique scaffolds have been synthesized. These structural adaptations have produced potent cytotoxic agents, such as KU135, which are prepared more simply than those that contain the noviose sugar. These analogues have been evaluated against two cancer cell lines and demonstrated low micromolar anti-proliferative activity.
Co-reporter:Adam S. Duerfeldt, Gary E. L. Brandt and Brian S. J. Blagg
Organic Letters 2009 Volume 11(Issue 11) pp:2353-2356
Publication Date(Web):May 12, 2009
DOI:10.1021/ol900783m
Conformationally constrained cis-amide chimeric inhibitors of Hsp90 have been synthesized and evaluated for their Hsp90 inhibitory activity. These new compounds exhibited Hsp90 ATPase inhibition and induced Hsp90-dependent client protein degradation in a dose-dependent manner. Biological data reported herein suggests that amide bond isomerization of geldanamycin derivatives plays an important role in affinity for the heteroprotein complex present in cancer cells.
Co-reporter:Adam S. Duerfeldt and Brian S. J. Blagg
ACS Chemical Biology 2009 Volume 4(Issue 4) pp:245
Publication Date(Web):April 17, 2009
DOI:10.1021/cb9000712
Resistance to Hsp90 inhibition has become an important concern as several clinical trials are currently in progress for the treatment of cancer. A summary of known mechanisms of resistance to Hsp90 inhibitors is provided, including the recent solution of the Humicola fuscoatra Hsp90 structure, the organism responsible for the biosynthesis of radicicol. Through careful analyses of Hsp90 structures, a plausible mechanism for resistance to Hsp90 inhibitors has been obtained by single mutations about the N-terminal ATP-binding site.
Co-reporter:Yuanming Lu, Sabah Ansar, Mary L. Michaelis, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 4) pp:1709-1715
Publication Date(Web):15 February 2009
DOI:10.1016/j.bmc.2008.12.047
Alzheimer’s disease (AD) neuropathology is characterized by loss of synapses and neurons, neuritic plaques consisting of β-amyloid (Aβ) peptides, and neurofibrillary tangles consisting of intracellular aggregates of hyperphosphorylated tau protein in susceptible brain regions. Aβ oligomers trigger a cascade of pathogenic events including tau hyperphosphorylation and aggregation, inflammatory reactions, and excitotoxicity that contribute to the progression of AD. The molecular chaperone Hsp90 facilitates the folding of newly synthesized and denatured proteins and is believed to play a role in neurodegenerative disorders in which the defining pathology results in misfolded proteins and the accumulation of protein aggregates. Some agents that inhibit Hsp90 protect neurons against Aβ toxicity and tau aggregation, and assays for rapidly screening potential Hsp90 inhibitors are of interest. We used the release of the soluble cytosolic enzyme lactate dehydrogenase (LDH) as an indicator of the loss of cell membrane integrity and cytotoxicity resulting from exposure to Aβ peptides to evaluate the neuroprotective properties of novel novobiocin analogues and established Hsp90 inhibitors. Compounds were assessed for potency in protecting proliferating and differentiated SH-SY5Y neuronal cells against Aβ-induced cell death; the potential toxicity of each agent alone was also determined. The data indicated that several of the compounds decreased Aβ toxicity even at low nanomolar concentrations and, unexpectedly, were more potent in protecting the undifferentiated cells against Aβ. The novobiocin analogues alone were not toxic even up to 10 μM concentrations whereas GDA and the parent compound, novobiocin, were toxic at 1 and 10 μM, respectively. The results suggest that novobiocin analogues may provide novel leads for the development of neuroprotective drugs.A series of novobiocin analogues, including A4, A4-dimer and KU32 from our laboratory, along with several other previously identified Hsp90 natural product inhibitors, were evaluated their ability to protect neuronal cells against Aβ-induced toxicity utilizing an LDH activity assay developed for high-throughput screening.
Co-reporter:M. Kyle Hadden, Stephanie A. Hill, Jason Davenport, Robert L. Matts, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 2) pp:634-640
Publication Date(Web):15 January 2009
DOI:10.1016/j.bmc.2008.11.064
High-throughput screening of a library of diverse molecules has identified the 1,4-naphthoquinone scaffold as a new class of Hsp90 inhibitors. The synthesis and evaluation of a rationally-designed series of analogues containing the naphthoquinone core scaffold has provided key structure–activity relationships for these compounds. The most active inhibitors exhibited potent in vitro activity with low micromolar IC50 values in anti-proliferation and Her2 degradation assays. In addition, 3g, 12, and 13a induced the degradation of oncogenic Hsp90 client proteins, a hallmark of Hsp90 inhibition. The identification of these naphthoquinones as Hsp90 inhibitors provides a new scaffold upon which improved Hsp90 inhibitors can be developed.
Co-reporter:Michael W. Amolins, Laura B. Peterson, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 1) pp:360-367
Publication Date(Web):1 January 2009
DOI:10.1016/j.bmc.2008.10.057
The natural product curcumin has long been recognized for its medicinal properties and is utilized for the treatment of many diseases. However, it remains unknown whether this activity is based on its presumably promiscuous scaffold, or if it results from the Michael acceptor properties of the α,β-unsaturated 1,3-diketone moiety central to its structure. To probe this issue, electron-rich pyrazole and isoxazole analogues were prepared and evaluated against two breast cancer cell lines, which resulted in the identification of several compounds that exhibit low micromolar to mid nanomolar anti-proliferative activity. A conjugate addition study was also performed to compare the relative electrophilicity of the diketone, pyrazole and isoxazole analogues.
Co-reporter:Vinod D. Jadhav, Adam S. Duerfeldt, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 24) pp:6845-6850
Publication Date(Web):15 December 2009
DOI:10.1016/j.bmcl.2009.10.091
Bicyclic radester analogues have been synthesized and evaluated for Hsp90 inhibitory activity. These analogues induce concentration-dependent degradation of Hsp90-dependent client proteins with the six-membered bicyclic analogues manifesting increased activity versus the five-membered counterparts.
Co-reporter:M. Kyle Hadden and Brian S. J. Blagg
The Journal of Organic Chemistry 2009 Volume 74(Issue 13) pp:4697-4704
Publication Date(Web):June 3, 2009
DOI:10.1021/jo900278g
Previously, we reported the Hsp90 inhibitory activity of radamide, an open chain amide chimera of geldanamycin and radicicol. Attempts to further expand upon structure−activity relationships for this class of Hsp90 inhibitors led to the preparation of a series of radamide analogues focused on differing tether lengths and quinone mimics. In addition, the cup-shaped conformation adopted by the two natural products when bound to the Hsp90 N-terminal ATP binding pocket suggests that conformationally biased compounds may demonstrate improved binding and inhibition. The preparation and evaluation of radamide analogues with cis/trans α,β-unsaturated amides yielded compounds that exhibit improved antiproliferative activity. In addition, several analogues demonstrated the ability to induce degradation of Hsp90-dependent oncogenic signaling proteins in vitro, a hallmark of Hsp90 N-terminal inhibition.
Co-reporter:Gary E. L. Brandt ; Matthew D. Schmidt ; Thomas E. Prisinzano
Journal of Medicinal Chemistry 2008 Volume 51(Issue 20) pp:6495-6502
Publication Date(Web):September 25, 2008
DOI:10.1021/jm8007486
Gedunin (1), a tetranortriterpenoid isolated from the Indian neem tree (Azadirachta indica), was recently shown to manifest anticancer activity via inhibition of the 90 kDa heat shock protein (Hsp90) folding machinery and to induce the degradation of Hsp90-dependent client proteins similar to other Hsp90 inhibitors. The mechanism of action by which gedunin induces client protein degradation remains undetermined, however, prior studies have demonstrated that it does not bind competitively versus ATP. In an effort to further probe the mechanism of action, 19 semisynthetic derivatives of gedunin were prepared and their antiproliferative activity against MCF-7 and SkBr3 breast cancer cells determined. Although no compound was found to exhibit antiproliferative activity more effective than the natural product, functionalities critical for antiproliferative activity have been identified.
Co-reporter:M. Kyle Hadden, Lakshmi Galam, Jason E. Gestwicki, Robert L. Matts and Brian S. J. Blagg
Journal of Natural Products 2007 Volume 70(Issue 12) pp:2014-2018
Publication Date(Web):November 17, 2007
DOI:10.1021/np070190s
High-throughput screening of a library of diverse molecules has identified derrubone (1), an isoflavone natural product from Derris robusta, as a potent Hsp90 inhibitor. Subsequent testing in several cellular-based assays established 1 as a low micromolar inhibitor in vitro. In addition, derrubone induced the degradation of numerous Hsp90 client proteins, a hallmark effect resulting from Hsp90 inhibition. The identification of 1 as an Hsp90 inhibitor provides a new natural product scaffold upon which the development of novel Hsp90 inhibitors can be pursued.
Co-reporter:Christopher Avila, Boris A. Kornilayev, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 4) pp:1134-1142
Publication Date(Web):15 February 2006
DOI:10.1016/j.bmc.2005.09.027
The Hsp90 molecular chaperone is responsible for the conformational maturation of nascent polypeptides and the rematuration of denatured proteins. Inhibition of Hsp90 represents a promising approach towards the treatment of cancer because numerous signaling cascades can be simultaneously targeted by disruption of the Hsp90-mediated process. Hsp90’s ATPase activity is essential to the Hsp90-mediated protein folding process, consequently, a coupled assay was developed and optimized for determination of Hsp90’s inherent ATPase activity. Using maltose phosphorylase, glucose oxidase, and horseradish peroxidase as components of this assay, a highly reproducible assay with a Z-factor of 0.87 has been produced.
Co-reporter:Subhabrata Chaudhury;Timothy R. Welch;Brian S. J. Blagg
ChemMedChem 2006 Volume 1(Issue 12) pp:
Publication Date(Web):25 OCT 2006
DOI:10.1002/cmdc.200600112
Blocking the chaperones: Heat shock protein 90 (Hsp90) has emerged as a promising target for the treatment of cancer, Alzheimer's and Parkinson's diseases, motor impairments, multiple sclerosis, and other disorders. Progress toward the development of both N- and C-terminal inhibitors is described.
Co-reporter:Michael J. Urban, Rick T. Dobrowsky, Brian S.J. Blagg
Trends in Pharmacological Sciences (March 2012) Volume 33(Issue 3) pp:129-137
Publication Date(Web):1 March 2012
DOI:10.1016/j.tips.2011.11.001
Dysfunctional insulin and insulin-like growth factor-I (IGF-I) signaling contributes to the pathological progression of diabetes, diabetic peripheral neuropathy (DPN), Alzheimer's (AD), Parkinson's (PD) and Huntington's diseases (HD). Despite their prevalence, there are limited therapeutic options available for the treatment of these neurodegenerative disorders. Therefore, establishing a link between insulin/IGF-I and the pathoetiology of these diseases may provide alternative approaches toward their management. Many of the heat shock proteins (Hsps) are well-known molecular chaperones that solubilize and clear damaged proteins and protein aggregates. Recent studies suggest that modulating Hsps may represent a promising therapeutic avenue for improving insulin and IGF-I signaling. Pharmacological induction of the heat shock response (HSR) may intersect with insulin/IGF-I signaling to improve aspects of neurodegenerative phenotypes. Herein, we review the intersection between Hsps and the insulin/IGF systems under normal and pathological conditions. The discussion will emphasize the potential of non-toxic HSR inducers as viable therapeutic agents.
Co-reporter:Gaurav Garg, Huiping Zhao, Brian S.J. Blagg
Bioorganic & Medicinal Chemistry (15 January 2017) Volume 25(Issue 2) pp:
Publication Date(Web):15 January 2017
DOI:10.1016/j.bmc.2016.11.030
Hsp90 is a promising therapeutic target for the development of anti-cancer agents due to its integral role in the stability and function of proteins associated with all ten hallmarks of cancer. Novobiocin, a coumarin antibiotic, was the first natural product identified that targeted the Hsp90 C-terminal domain and manifested anti-proliferative activity (SKBr3 IC50 ∼ 700 μM). Subsequent structural investigations on novobiocin led to analogues with significantly improved anti-proliferative activity against multiple cancer cell lines. In an effort to develop more efficacious and diverse analogues, it was recently found that the coumarin ring of novobiocin could be replaced with the biphenyl core without compromising activity. Based on these prior studies, a series of alkylamino biphenylamides was designed, synthesized and evaluated for anti-proliferative activity against two breast cancer cell lines. SAR studies demonstrated that the incorporation of an alkylamino side chain onto the biphenyl core improved anti-proliferative activity and resulted in compounds that exhibit sub-micromolar to mid-nanomolar activity through Hsp90 inhibition. Importantly, these studies indicate the presence of a hydrophilic region about the central core that can be exploited for the design of new inhibitors.
.jpg)






































![2(3H)-Furanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]dihydro-, (4S)-](http://img.cochemist.com/ccimg/122700/122654-37-9.png)
![2(3H)-Furanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]dihydro-, (4S)-](http://img.cochemist.com/ccimg/122700/122654-37-9_b.png)





![[1,1'-Biphenyl]-4-ol, 3-methoxy-4'-nitro-](http://img.cochemist.com/ccimg/112200/112148-12-6.png)
![[1,1'-Biphenyl]-4-ol, 3-methoxy-4'-nitro-](http://img.cochemist.com/ccimg/112200/112148-12-6_b.png)












![1H-Pyrrole, 2-[(2-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/79500/79499-35-7.png)
![1H-Pyrrole, 2-[(2-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/79500/79499-35-7_b.png)





















































![Phenol, 4-[(4-nitrophenyl)ethynyl]-](http://img.cochemist.com/ccimg/19000/18938-22-2.png)
![Phenol, 4-[(4-nitrophenyl)ethynyl]-](http://img.cochemist.com/ccimg/19000/18938-22-2_b.png)





![Phenol,4-[(1E)-2-(4-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/14100/14064-83-6.png)
![Phenol,4-[(1E)-2-(4-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/14100/14064-83-6_b.png)
![[1,1'-BIPHENYL]-2-CARBONYL CHLORIDE-](http://img.cochemist.com/ccimg/14100/14002-52-9.png)
![[1,1'-BIPHENYL]-2-CARBONYL CHLORIDE-](http://img.cochemist.com/ccimg/14100/14002-52-9_b.png)



















![Oxireno[c]phenanthro[1,2-d]pyran-3,8(3aH,4bH)-dione,5-(acetyloxy)-1-(3-furanyl)-1,5,6,6a,7,10a,10b,11,12,12a-decahydro-4b,7,7,10a,12a-pentamethyl-,(1S,3aS,4aR,4bS,5R,6aR,10aR,10bR,12aS)-](http://img.cochemist.com/ccimg/2800/2753-30-2.png)
![Oxireno[c]phenanthro[1,2-d]pyran-3,8(3aH,4bH)-dione,5-(acetyloxy)-1-(3-furanyl)-1,5,6,6a,7,10a,10b,11,12,12a-decahydro-4b,7,7,10a,12a-pentamethyl-,(1S,3aS,4aR,4bS,5R,6aR,10aR,10bR,12aS)-](http://img.cochemist.com/ccimg/2800/2753-30-2_b.png)




![Bicyclo[2.2.1]heptan-2-amine](http://img.cochemist.com/ccimg/900/822-98-0.png)
![Bicyclo[2.2.1]heptan-2-amine](http://img.cochemist.com/ccimg/900/822-98-0_b.png)








































































































































































































































































































































































































































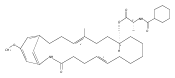










![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[3-[methyl(1-methylethyl)amino]propoxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200900/1320336-90-0.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[3-[methyl(1-methylethyl)amino]propoxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200900/1320336-90-0_b.png)
![Benzamide, N-[7-[3-(ethylmethylamino)propoxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-4-hydroxy-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200900/1320336-89-7.png)
![Benzamide, N-[7-[3-(ethylmethylamino)propoxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-4-hydroxy-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200900/1320336-89-7_b.png)


























































































































































































































































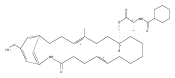
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[2-(methylamino)ethoxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-16-4.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[2-(methylamino)ethoxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-16-4_b.png)


![Benzamide, 4-(acetyloxy)-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[2-(methylamino)ethoxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-13-1.png)
![Benzamide, 4-(acetyloxy)-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[2-(methylamino)ethoxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-13-1_b.png)


![Benzamide, 4-(acetyloxy)-N-[7-[2-(dimethylamino)ethoxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200800/1239438-10-8.png)
![Benzamide, 4-(acetyloxy)-N-[7-[2-(dimethylamino)ethoxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200800/1239438-10-8_b.png)




![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-(3-piperidinyloxy)-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-06-2.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-(3-piperidinyloxy)-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-06-2_b.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[(1-methyl-3-piperidinyl)oxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-05-1.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[(1-methyl-3-piperidinyl)oxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-05-1_b.png)
![Benzamide, 4-(acetyloxy)-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-(3-piperidinyloxy)-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-04-0.png)
![Benzamide, 4-(acetyloxy)-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-(3-piperidinyloxy)-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-04-0_b.png)


![Benzamide, 4-(acetyloxy)-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[(1-methyl-3-piperidinyl)oxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-02-8.png)
![Benzamide, 4-(acetyloxy)-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[(1-methyl-3-piperidinyl)oxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239438-02-8_b.png)




![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-(4-piperidinyloxy)-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239437-99-0.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-(4-piperidinyloxy)-2H-1-benzopyran-3-yl]-](/data/chemimg/200800/1239437-99-0_b.png)
![Benzamide, 4-(acetyloxy)-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-(4-piperidinyloxy)-2H-1-benzopyran-3-yl]-](/data/chemimg/200700/1239437-97-8.png)
![Benzamide, 4-(acetyloxy)-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-(4-piperidinyloxy)-2H-1-benzopyran-3-yl]-](/data/chemimg/200700/1239437-97-8_b.png)








![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-[[(2S,4R)-tetrahydro-4-hydroxy-2H-pyran-2-yl]oxy]-2H-1-benzopyran-3-yl]-](/data/chemimg/200700/1239437-89-8.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-[[(2S,4R)-tetrahydro-4-hydroxy-2H-pyran-2-yl]oxy]-2H-1-benzopyran-3-yl]-](/data/chemimg/200700/1239437-89-8_b.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-[[(2S,3R)-tetrahydro-3-hydroxy-2H-pyran-2-yl]oxy]-2H-1-benzopyran-3-yl]-](/data/chemimg/200700/1239437-88-7.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-[[(2S,3R)-tetrahydro-3-hydroxy-2H-pyran-2-yl]oxy]-2H-1-benzopyran-3-yl]-](/data/chemimg/200700/1239437-88-7_b.png)
![Benzamide, N-[7-[(4-deoxy-β-D-erythro-pentopyranosyl)oxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-4-hydroxy-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200700/1239437-87-6.png)
![Benzamide, N-[7-[(4-deoxy-β-D-erythro-pentopyranosyl)oxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-4-hydroxy-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200700/1239437-87-6_b.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-[[(2R,4R)-tetrahydro-4-hydroxy-2H-pyran-2-yl]oxy]-2H-1-benzopyran-3-yl]-](/data/chemimg/200700/1239437-86-5.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-[[(2R,4R)-tetrahydro-4-hydroxy-2H-pyran-2-yl]oxy]-2H-1-benzopyran-3-yl]-](/data/chemimg/200700/1239437-86-5_b.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-[[(2R,3R)-tetrahydro-3-hydroxy-2H-pyran-2-yl]oxy]-2H-1-benzopyran-3-yl]-](/data/chemimg/200700/1239437-85-4.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-2-oxo-7-[[(2R,3R)-tetrahydro-3-hydroxy-2H-pyran-2-yl]oxy]-2H-1-benzopyran-3-yl]-](/data/chemimg/200700/1239437-85-4_b.png)
![Benzamide, N-[7-[(4-deoxy-α-D-erythro-pentopyranosyl)oxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-4-hydroxy-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200700/1239437-84-3.png)
![Benzamide, N-[7-[(4-deoxy-α-D-erythro-pentopyranosyl)oxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-4-hydroxy-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200700/1239437-84-3_b.png)




















![2H-1-Benzopyran-2-one, 7-[(6-deoxy-5-C-methyl-4-O-methyl-α-L-lyxo-hexopyranosyl)oxy]-8-methyl-3-[4-[(1E)-2-phenylethenyl]-1H-1,2,3-triazol-1-yl]-](/data/chemimg/200700/1238848-32-2.png)
![2H-1-Benzopyran-2-one, 7-[(6-deoxy-5-C-methyl-4-O-methyl-α-L-lyxo-hexopyranosyl)oxy]-8-methyl-3-[4-[(1E)-2-phenylethenyl]-1H-1,2,3-triazol-1-yl]-](/data/chemimg/200700/1238848-32-2_b.png)
![2H-1-Benzopyran-2-one, 7-[(6-deoxy-5-C-methyl-4-O-methyl-α-L-lyxo-hexopyranosyl)oxy]-3-[4-(3',6-dimethoxy[1,1'-biphenyl]-3-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-31-1.png)
![2H-1-Benzopyran-2-one, 7-[(6-deoxy-5-C-methyl-4-O-methyl-α-L-lyxo-hexopyranosyl)oxy]-3-[4-(3',6-dimethoxy[1,1'-biphenyl]-3-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-31-1_b.png)










![2H-1-Benzopyran-2-one, 7-hydroxy-3-[4-(1H-indol-3-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-25-3.png)
![2H-1-Benzopyran-2-one, 7-hydroxy-3-[4-(1H-indol-3-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-25-3_b.png)
![2H-1-Benzopyran-2-one, 7-hydroxy-3-[4-(1H-indol-2-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-23-1.png)
![2H-1-Benzopyran-2-one, 7-hydroxy-3-[4-(1H-indol-2-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-23-1_b.png)


![2H-1-Benzopyran-2-one, 3-[4-(3',6-dimethoxy[1,1'-biphenyl]-3-yl)-1H-1,2,3-triazol-1-yl]-7-hydroxy-8-methyl-](/data/chemimg/200700/1238848-18-4.png)
![2H-1-Benzopyran-2-one, 3-[4-(3',6-dimethoxy[1,1'-biphenyl]-3-yl)-1H-1,2,3-triazol-1-yl]-7-hydroxy-8-methyl-](/data/chemimg/200700/1238848-18-4_b.png)




![2H-1-Benzopyran-2-one, 3-[4-(2,4-difluorophenyl)-1H-1,2,3-triazol-1-yl]-7-hydroxy-8-methyl-](/data/chemimg/200700/1238848-15-1.png)
![2H-1-Benzopyran-2-one, 3-[4-(2,4-difluorophenyl)-1H-1,2,3-triazol-1-yl]-7-hydroxy-8-methyl-](/data/chemimg/200700/1238848-15-1_b.png)


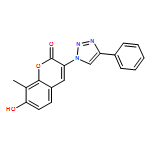

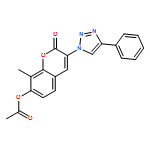
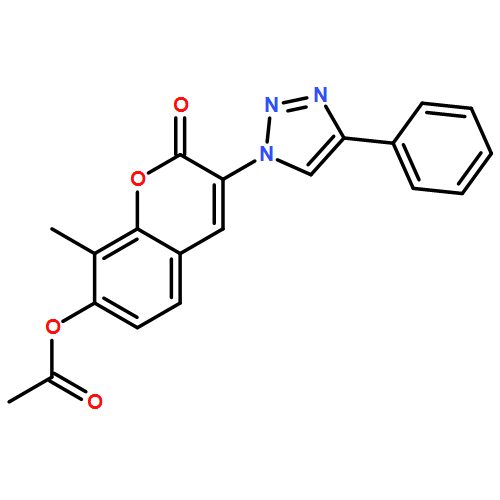
![2H-1-Benzopyran-2-one, 7-(acetyloxy)-3-[4-(1H-indol-3-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-12-8.png)
![2H-1-Benzopyran-2-one, 7-(acetyloxy)-3-[4-(1H-indol-3-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-12-8_b.png)
![2H-1-Benzopyran-2-one, 7-(acetyloxy)-3-[4-(1H-indol-2-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-11-7.png)
![2H-1-Benzopyran-2-one, 7-(acetyloxy)-3-[4-(1H-indol-2-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-11-7_b.png)


![2H-1-Benzopyran-2-one, 7-(acetyloxy)-3-[4-(3',6-dimethoxy[1,1'-biphenyl]-3-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-09-3.png)
![2H-1-Benzopyran-2-one, 7-(acetyloxy)-3-[4-(3',6-dimethoxy[1,1'-biphenyl]-3-yl)-1H-1,2,3-triazol-1-yl]-8-methyl-](/data/chemimg/200700/1238848-09-3_b.png)






![2H-1-Benzopyran-2-one, 7-(acetyloxy)-8-methyl-3-[4-(4-methylphenyl)-1H-1,2,3-triazol-1-yl]-](/data/chemimg/200700/1238848-01-5.png)
![2H-1-Benzopyran-2-one, 7-(acetyloxy)-8-methyl-3-[4-(4-methylphenyl)-1H-1,2,3-triazol-1-yl]-](/data/chemimg/200700/1238848-01-5_b.png)



















![Spiro[isobenzofuran-1(3H),9'-[9H]xanthene]-6-carboxamide, 2',7'-difluoro-3',6'-dihydroxy-N-(5-hydroxypentyl)-3-oxo-](/data/chemimg/200700/1236205-36-9.png)
![Spiro[isobenzofuran-1(3H),9'-[9H]xanthene]-6-carboxamide, 2',7'-difluoro-3',6'-dihydroxy-N-(5-hydroxypentyl)-3-oxo-](/data/chemimg/200700/1236205-36-9_b.png)
![2H-Pyrano[3,2-g]quinoline-6-methanesulfonic acid, 8,9-dihydro-9-[6-[(5-hydroxypentyl)amino]-6-oxohexyl]-8,8-dimethyl-2-oxo-4-(trifluoromethyl)-, compd. with N,N-diethylethanamine (1:1)](http://img.cochemist.com/ccimg/1236300/1236205-34-7.png)
![2H-Pyrano[3,2-g]quinoline-6-methanesulfonic acid, 8,9-dihydro-9-[6-[(5-hydroxypentyl)amino]-6-oxohexyl]-8,8-dimethyl-2-oxo-4-(trifluoromethyl)-, compd. with N,N-diethylethanamine (1:1)](http://img.cochemist.com/ccimg/1236300/1236205-34-7_b.png)
![Pyridinium, 1-[[3-[[(5-hydroxypentyl)amino]carbonyl]phenyl]methyl]-4-[5-(4-methoxyphenyl)-2-oxazolyl]-](/data/chemimg/200700/1236205-32-5.png)
![Pyridinium, 1-[[3-[[(5-hydroxypentyl)amino]carbonyl]phenyl]methyl]-4-[5-(4-methoxyphenyl)-2-oxazolyl]-](/data/chemimg/200700/1236205-32-5_b.png)


![3H,5H-Dipyrrolo[1,2-c:2',1'-f]pyrimidine-8-acetamide, 1-ethyl-N-(5-hydroxypentyl)-2,7,9-trimethyl-3,5-dioxo-](/data/chemimg/200700/1236205-30-3.png)
![3H,5H-Dipyrrolo[1,2-c:2',1'-f]pyrimidine-8-acetamide, 1-ethyl-N-(5-hydroxypentyl)-2,7,9-trimethyl-3,5-dioxo-](/data/chemimg/200700/1236205-30-3_b.png)




























































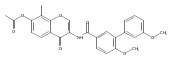















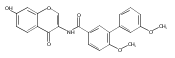

























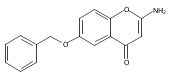


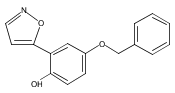
























































































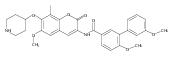















































![[1,1'-Biphenyl]-3-carboxamide, N-[7-[(6-deoxy-5-C-methyl-4-O-methyl-α-L-lyxo-hexopyranosyl)oxy]-8-methoxy-2-oxo-2H-1-benzopyran-3-yl]-3',6-dimethoxy-](/data/chemimg/200600/1083313-23-8.png)
![[1,1'-Biphenyl]-3-carboxamide, N-[7-[(6-deoxy-5-C-methyl-4-O-methyl-α-L-lyxo-hexopyranosyl)oxy]-8-methoxy-2-oxo-2H-1-benzopyran-3-yl]-3',6-dimethoxy-](/data/chemimg/200600/1083313-23-8_b.png)


![[1,1'-Biphenyl]-3-carboxamide, N-[7-[(6-deoxy-5-C-methyl-4-O-methyl-α-L-lyxo-hexopyranosyl)oxy]-6-methoxy-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-3',6-dimethoxy-](/data/chemimg/200600/1083313-07-8.png)
![[1,1'-Biphenyl]-3-carboxamide, N-[7-[(6-deoxy-5-C-methyl-4-O-methyl-α-L-lyxo-hexopyranosyl)oxy]-6-methoxy-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-3',6-dimethoxy-](/data/chemimg/200600/1083313-07-8_b.png)









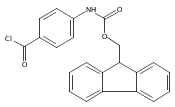






![3H,5H-Dipyrrolo[1,2-c:2',1'-f]pyrimidine-8-acetic acid, 1-ethyl-2,7,9-trimethyl-3,5-dioxo-, 2,5-dioxo-1-pyrrolidinyl ester](/data/chemimg/200600/1027512-35-1.png)
![3H,5H-Dipyrrolo[1,2-c:2',1'-f]pyrimidine-8-acetic acid, 1-ethyl-2,7,9-trimethyl-3,5-dioxo-, 2,5-dioxo-1-pyrrolidinyl ester](/data/chemimg/200600/1027512-35-1_b.png)






















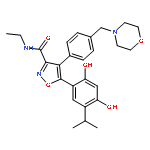

































![(2R,3R)-2-[(2R,3R)-2,3-dihydro-3-(4-hydroxy-3-methoxyphenyl)-2-(hydroxymethyl)-1,4-benzodioxin-6-yl]-2,3-dihydro-3,5,7-trihydroxy-4H-1-benzopyran-4-one](http://img.cochemist.com/ccimg/301400/301334-08-7.png)
![(2R,3R)-2-[(2R,3R)-2,3-dihydro-3-(4-hydroxy-3-methoxyphenyl)-2-(hydroxymethyl)-1,4-benzodioxin-6-yl]-2,3-dihydro-3,5,7-trihydroxy-4H-1-benzopyran-4-one](http://img.cochemist.com/ccimg/301400/301334-08-7_b.png)
![4-Pentenamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N,2-dimethyl-, (2S)-](/data/chemimg/120300/277297-37-7.png)
![4-Pentenamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N,2-dimethyl-, (2S)-](/data/chemimg/120300/277297-37-7_b.png)


















![Propanamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/192100/192060-67-6.png)
![Propanamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/192100/192060-67-6_b.png)






![Propanamide,N-[(1S,2S)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/159300/159213-03-3.png)
![Propanamide,N-[(1S,2S)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/159300/159213-03-3_b.png)














































![Benzoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](/data/chemimg/118600/107658-28-6.png)
![Benzoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](/data/chemimg/118600/107658-28-6_b.png)


![D-Alanine,N-(cyclohexylcarbonyl)-,(5R,6E,8E,10E,13S,14R,15R,16Z)-15,22-dihydroxy-5-methoxy-14,16-dimethyl-3-oxo-2-azabicyclo[18.3.1]tetracosa-1(24),6,8,10,16,20,22-heptaen-13-ylester](http://img.cochemist.com/ccimg/98900/98873-83-7.png)
![D-Alanine,N-(cyclohexylcarbonyl)-,(5R,6E,8E,10E,13S,14R,15R,16Z)-15,22-dihydroxy-5-methoxy-14,16-dimethyl-3-oxo-2-azabicyclo[18.3.1]tetracosa-1(24),6,8,10,16,20,22-heptaen-13-ylester](http://img.cochemist.com/ccimg/98900/98873-83-7_b.png)








![Ethanone, 1-[2,6-bis(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/76500/76434-54-3.png)
![Ethanone, 1-[2,6-bis(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/76500/76434-54-3_b.png)




![1H-ISOINDOLE-1,3(2H)-DIONE, 2-[2-(2,4-DIHYDROXYPHENYL)-2-OXOETHYL]-](http://img.cochemist.com/ccimg/65200/65190-77-4.png)
![1H-ISOINDOLE-1,3(2H)-DIONE, 2-[2-(2,4-DIHYDROXYPHENYL)-2-OXOETHYL]-](http://img.cochemist.com/ccimg/65200/65190-77-4_b.png)
![6-phenylimidazo[2,1-b][1,3,4]thiadiazole-2-sulfonamide](http://img.cochemist.com/ccimg/63800/63735-71-7.png)
![6-phenylimidazo[2,1-b][1,3,4]thiadiazole-2-sulfonamide](http://img.cochemist.com/ccimg/63800/63735-71-7_b.png)


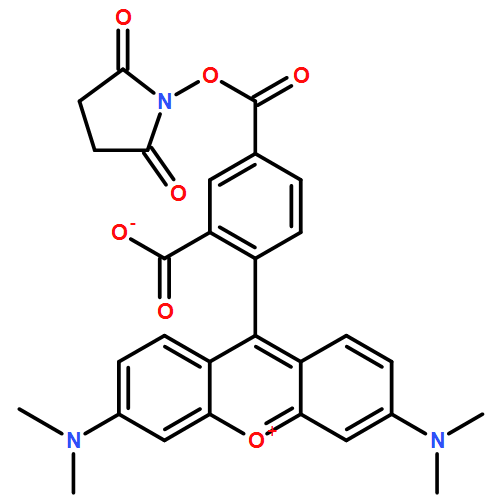
![N-[9-(2-carboxyphenyl)-6-(dimethylamino)-3H-xanthen-3-ylidene]-N-methylmethanaminium perchlorate](http://img.cochemist.com/ccimg/62700/62669-72-1.png)
![N-[9-(2-carboxyphenyl)-6-(dimethylamino)-3H-xanthen-3-ylidene]-N-methylmethanaminium perchlorate](http://img.cochemist.com/ccimg/62700/62669-72-1_b.png)









![[2,6':2',6''-Terbenzothiazole]-7-sulfonicacid, 2''-(4-aminophenyl)-6-methyl-](http://img.cochemist.com/ccimg/47900/47812-48-6.png)
![[2,6':2',6''-Terbenzothiazole]-7-sulfonicacid, 2''-(4-aminophenyl)-6-methyl-](http://img.cochemist.com/ccimg/47900/47812-48-6_b.png)
![3-(benzo[d][1,3]dioxol-5-yl)-5,7-dihydroxy-4H-chromen-4-one](http://img.cochemist.com/ccimg/40700/40624-03-1.png)
![3-(benzo[d][1,3]dioxol-5-yl)-5,7-dihydroxy-4H-chromen-4-one](http://img.cochemist.com/ccimg/40700/40624-03-1_b.png)
![6-(1,3-DIHYDROXYBUTYL)-7-METHYL-3-METHYLENE-3,3A,4,7,8,8A-HEXAHYD<WBR />RO-2H-CYCLOHEPTA[B]FURAN-2-ONE](http://img.cochemist.com/ccimg/38600/38561-68-1.png)
![6-(1,3-DIHYDROXYBUTYL)-7-METHYL-3-METHYLENE-3,3A,4,7,8,8A-HEXAHYD<WBR />RO-2H-CYCLOHEPTA[B]FURAN-2-ONE](http://img.cochemist.com/ccimg/38600/38561-68-1_b.png)
![1-[2,4,6-TRIS(METHOXYMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/36900/36804-11-2.png)
![1-[2,4,6-TRIS(METHOXYMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/36900/36804-11-2_b.png)


![2,2'-[(2,4-DIMETHYL-3-PENTANYL)IMINO]DIETHANOL](http://img.cochemist.com/ccimg/32900/32811-40-8.png)
![2,2'-[(2,4-DIMETHYL-3-PENTANYL)IMINO]DIETHANOL](http://img.cochemist.com/ccimg/32900/32811-40-8_b.png)













![Phenol, 4-[(1E)-3-hydroxy-1-propenyl]-](http://img.cochemist.com/ccimg/20700/20649-40-5.png)
![Phenol, 4-[(1E)-3-hydroxy-1-propenyl]-](http://img.cochemist.com/ccimg/20700/20649-40-5_b.png)




![6H-Oxireno[e][2]benzoxacyclotetradecin-6,12(7H)-dione,8-chloro-1a,14,15,15a-tetrahydro-9,11-dihydroxy-14-methyl-,(1aR,2Z,4E,14R,15aR)-](http://img.cochemist.com/ccimg/12800/12772-57-5.png)
![6H-Oxireno[e][2]benzoxacyclotetradecin-6,12(7H)-dione,8-chloro-1a,14,15,15a-tetrahydro-9,11-dihydroxy-14-methyl-,(1aR,2Z,4E,14R,15aR)-](http://img.cochemist.com/ccimg/12800/12772-57-5_b.png)




![ETHANONE, 1-[2,4-BIS(METHOXYMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/6600/6515-05-5.png)
![ETHANONE, 1-[2,4-BIS(METHOXYMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/6600/6515-05-5_b.png)
![[1,1'-BIPHENYL]-3,3'-DICARBONYL DICHLORIDE](http://img.cochemist.com/ccimg/6500/6423-26-3.png)
![[1,1'-BIPHENYL]-3,3'-DICARBONYL DICHLORIDE](http://img.cochemist.com/ccimg/6500/6423-26-3_b.png)






![7-Acetoxy-3-benzo[1,3]dioxol-5-yl-chromen-4-on](http://img.cochemist.com/ccimg/5600/5574-40-3.png)
![7-Acetoxy-3-benzo[1,3]dioxol-5-yl-chromen-4-on](http://img.cochemist.com/ccimg/5600/5574-40-3_b.png)


![[1,1'-Biphenyl]-3,3'-dicarboxylic acid, 6,6'-dimethoxy-, dimethyl ester](http://img.cochemist.com/ccimg/4800/4712-73-6.png)
![[1,1'-Biphenyl]-3,3'-dicarboxylic acid, 6,6'-dimethoxy-, dimethyl ester](http://img.cochemist.com/ccimg/4800/4712-73-6_b.png)





![[1,1'-Biphenyl]-3,3'-dicarboxylicacid, 3,3'-dimethyl ester](http://img.cochemist.com/ccimg/1800/1751-97-9.png)
![[1,1'-Biphenyl]-3,3'-dicarboxylicacid, 3,3'-dimethyl ester](http://img.cochemist.com/ccimg/1800/1751-97-9_b.png)


![[1,1'-Biphenyl]-3,3'-dicarboxylic acid](/data/chemimg/592200/612-87-3.png)
![[1,1'-Biphenyl]-3,3'-dicarboxylic acid](/data/chemimg/592200/612-87-3_b.png)
































![Benzamide, 4-(acetyloxy)-N-[7-[3-(dimethylamino)propoxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200500/1239438-11-9.png)
![Benzamide, 4-(acetyloxy)-N-[7-[3-(dimethylamino)propoxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200500/1239438-11-9_b.png)


![Benzamide, 4-(acetyloxy)-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[(1-methyl-4-piperidinyl)oxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200500/1239437-95-6.png)
![Benzamide, 4-(acetyloxy)-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[(1-methyl-4-piperidinyl)oxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200500/1239437-95-6_b.png)


















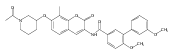


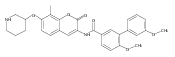











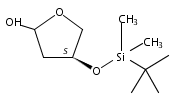








![Benzamide, N-[7-[3-(dimethylamino)propoxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-4-hydroxy-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200500/1239438-15-3.png)
![Benzamide, N-[7-[3-(dimethylamino)propoxy]-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-4-hydroxy-3-(3-methyl-2-buten-1-yl)-](/data/chemimg/200500/1239438-15-3_b.png)

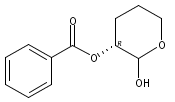




![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[(1-methyl-4-piperidinyl)oxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200500/1239437-98-9.png)
![Benzamide, 4-hydroxy-3-(3-methyl-2-buten-1-yl)-N-[8-methyl-7-[(1-methyl-4-piperidinyl)oxy]-2-oxo-2H-1-benzopyran-3-yl]-](/data/chemimg/200500/1239437-98-9_b.png)











