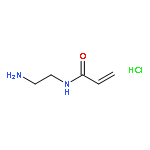
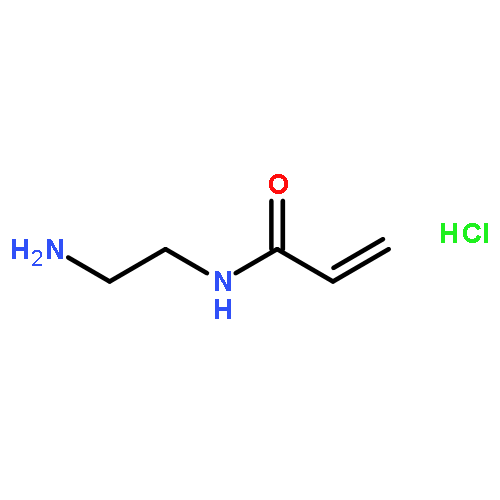
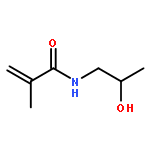
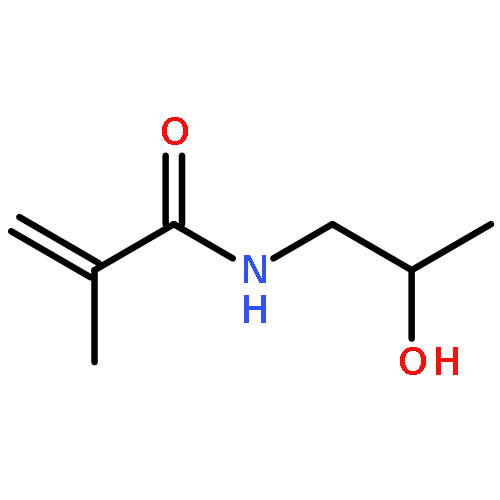
Co-reporter:Pan Gao;Hui Cao;Yi Ding;Meng Cai;Zhigang Cui;Xinhua Lu
ACS Macro Letters December 20, 2016 Volume 5(Issue 12) pp:1327-1331
Publication Date(Web):November 16, 2016
DOI:10.1021/acsmacrolett.6b00796
Analogous to cellulose, polymers whose monomer units possess both hydrogen donators and acceptors are generally insoluble in ambient water because of hydrogen bonding (HB). Herein we present stimuli-responsive long aqueous cylindrical vesicles (nanotubes) synthesized directly using HB-driven polymerization-induced self-assembly (PISA) under visible-light-mediated RAFT aqueous dispersion polymerization at 25 °C. The PISA undergoes an unprecedented film/silk-to-ribbon-to-vesicle transition and films/silks/ribbons formed at low DPs (∼25–85) of core-forming block in free-flowing aqueous solution. Pore-switchable nanotubes are synthesized by electrostatic repulsive perturbation of the HB association in anisotropic vesicular membranes via inserting minor ionized monomer units into the core-forming block. These nanotubes are synthesized at >35% solids, and tubular membranes are more sensitive than spherical counterparts in response to aqueous surroundings. This facile, robust, and general strategy paves a new avenue toward scale-up production of advanced intelligent nanomaterials.
Co-reporter:Zhigang Cui;Hui Cao;Yi Ding;Pan Gao;Xinhua Lu
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 24) pp:3755-3763
Publication Date(Web):2017/06/20
DOI:10.1039/C7PY00582B
Herein, we present supramolecular compartmentalization of an ABC triblock copolymer single-chain nanoparticle (SCNP) using coordination-driven orthogonal intramolecular self-assembly in aqueous solution. A well-defined ABC-type triblock copolymer was synthesized via visible-light-mediated RAFT aqueous sequential block copolymerization. It possesses protonated imidazolium-motifs in the A-, NH3+-motifs in C-, and hydroxyl-motifs in B-block. Coordination-driven orthogonal self-assembly locally occurring upon deprotonation (A-block within pH 3.3–4.4 and C-block within pH 5.3–6.4) has been evidenced by UV-vis spectroscopy, 1H NMR, dynamic light scattering (DLS), NMR diffusion-ordered spectroscopy (DOSY), and atomic force microscopy (AFM). A blank regime of pH 4.4–5.3 guarantees locally orthogonal intramolecular self-assembly without mutual interference. Dialysis and pH-cycling results illustrate that the inherent different subdomains are discrete and can individually respond to an external stimulus, with potential biomimetic functions in terms of sub-10 nm ultrafine internal heterogeneity.
Co-reporter:Tao Huang, Zhigang Cui, Yi Ding, Xinhua Lu and Yuanli Cai
Polymer Chemistry 2016 vol. 7(Issue 1) pp:176-183
Publication Date(Web):16 Oct 2015
DOI:10.1039/C5PY01524C
Aqueous synthesis of a well-defined polyion or polyelectrolyte via reversible degenerative radical polymerisation (RDRP) in dilute solution at or below room temperature is increasingly imperative for emerging biological applications, such as in situ grafting of polyelectrolytes onto heat-sensitive proteins and DNA/RNA. However, the polymerisation generally suffers from a time-consuming and less effective process because of electrostatic repulsion. This problem can be circumvented by media-adjustable electrostatic association. Herein we report the use of electrostatic association for rapid and quantitative aqueous RAFT of histamine-based ionic monomers in dilute solutions under visible light irradiation at and below 25 °C. We demonstrate that electrostatic interactions dictate polymerisation rates, including the initialization period and chain growth. The use of bulky counter-anions and the addition of methanol and sodium chloride can enhance electrostatic association, and thus promote the chain-growth rate. More importantly, abnormal chain-growth acceleration can be achieved in a cold solution at 8 °C, which proceeds faster than at 25 °C, because of ion-pairing association. These external regulations over aqueous RDRP show general implications for rapid and quantitative aqueous synthesis of well-defined polyelectrolytes in dilute solutions at and below room temperature.
Co-reporter:Jie Han;Kaiyi Zhou;Xuechao Zhu;Qiuping Yu;Yi Ding;Xinhua Lu
Macromolecular Rapid Communications 2016 Volume 37( Issue 15) pp:1275-1281
Publication Date(Web):
DOI:10.1002/marc.201600214
Co-reporter:Kaiyi Zhou, Hui Cao, Pan Gao, Zhigang Cui, Yi Ding, and Yuanli Cai
Macromolecules 2016 Volume 49(Issue 6) pp:2189-2196
Publication Date(Web):March 8, 2016
DOI:10.1021/acs.macromol.6b00152
Autocatalytic self-sorting in the biomimetic poly(cystamine methacrylamide hydrochloride) (PCysMA) is presented, whose units comprise lysine-mimetic alkylammonium ions and cystine-mimetic alkyl disulfide spacers. The block copolymer with poly(2-hydroxypropylmethacrylamide) was synthesized directly by RAFT in acidic water under visible light irradiation at 25 °C. Disulfide exchange can be initiated by the terminal thiolates as generated by alkalization-induced aminolysis. 65–67% CysMA units sort into hydrophobic polymer disulfides and water-soluble cystamine at pH 10.5. Moreover, intermediate reactions occur in the presence of copper ions, i.e., Cu(II)–NH2 coordination, aminolysis, NH2-to-SH substitution, and cupric-to-cuprous reduction in metal centers, thus autocatalytic self-sorting with essentially 100% conversion at pH 8.8. UV–vis spectroscopy, 1H NMR, atomic absorption spectroscopy, and elemental analysis confirmed this ideal self-sorting. Dynamic light scattering and atomic force microscopy identified supramolecular-to-supracolloidal self-assembly with concomitant release of cystamine molecules and intermediate cuprous complexes. Such a self-sorting underlines an amazing prospect for the use of a single polymer to achieve artificial reaction complexity, hierarchy, and metabolic process, with minimal synthetic efforts.
Co-reporter:Yanyan Jiang, Na Xu, Jie Han, Qiuping Yu, Lei Guo, Pan Gao, Xinhua Lu and Yuanli Cai
Polymer Chemistry 2015 vol. 6(Issue 27) pp:4955-4965
Publication Date(Web):29 May 2015
DOI:10.1039/C5PY00656B
Self-assembly of amphiphilic block copolymers in water suffers from the undesired encapsulation of hydrophobic reactive motifs in a core-forming block, which deteriorates their performance as aqueous catalysts. This problem can be circumvented by polymerisation-induced self-assembly (PISA). Herein, we report a new strategy for one-pot synthesis of reactive block copolymer nanoparticles whose hydrophobic reactive motifs decorate the surrounding core–shell interfaces. We demonstrate fast RAFT aqueous dispersion polymerisation of a commercially available specialty monomer, diacetone acrylamide (DAAM), under visible light irradiation at 25 °C. PISA is induced by polymerisation via sequential dehydration, phase separation and reaction acceleration, thus achieving complete conversion in 30 min. The replacement of minimal DAAM by an NH3+-monomer induces slight hydration of the core-forming block, and thus a low polydispersity of the resulting statistic-block copolymer. Moreover, simultaneous in situ self-assembly and chain growth favour the adjustment of newly-added NH3+-units outward to core–shell interfaces while the major DAAM units collapse into hydrophobic PISA-cores. Both lead to timely and selective self-assembly into the new reactive nanoparticles whose NH3+-motifs decorate the surrounding core–shell interfaces. These nanoparticles are well-suited for fabrication of advanced nanoreactors whose hydrophobic dative metal centres decorate the surrounding interfaces via simultaneous imine conversion and Zn(II)-coordination. Such PISA-nanostructures endow hydrophobic metal centres with a huge and accessible specific surface area and are stabilized by water-soluble shells. Therefore, this strategy holds fascinating potential for the fabrication of metalloenzyme-inspired aqueous catalysts.
Co-reporter:Hui Shi, Kaiyi Zhou, Qiuping Yu, Zhigang Cui, Yanyan Jiang, Xinhua Lu and Yuanli Cai
Polymer Chemistry 2015 vol. 6(Issue 42) pp:7455-7463
Publication Date(Web):26 Aug 2015
DOI:10.1039/C5PY01092F
Direct aqueous synthesis and programmable self-assembly and reconstructions of a well-defined reactive cystamine-block copolymer in response to conditions of the aqueous environment, such as air and solution pH, are presented in this article. A variety of well-defined poly(cystamine methacrylamide hydrochloride) (PCysMA) polymers and copolymers with 2-hydroxypropylmethacrylamide (HPMA) and 2-aminoethylmethacrylamide hydrochloride (AEMA) can be achieved via fast and well-controlled aqueous RAFT under visible light irradiation at 25 °C. PHPMA-b-PCysMA assembles into spherical PCysMA-core micelles upon NH3+-to-NH2 conversion. Moreover, progressive reactions in the PCysMA block, including reduction, ionization and oxidation, can be induced stepwise by reduction in argon gas saturated acidic water, exposure to air and stepwise alkalization. These reactions lead to the stepwise conversion of the intermolecular interactions from electrostatic repulsion (ionized PCysMA block) into hydrogen-bonding associations (reduction-generated thiol block), electrostatic repulsion (as-ionized thiolate block), and hydrophobic cross-linking (oxidation-generated disulfides), leading to the programmable self-assembly and reconstruction of a water-soluble block copolymer into compound-encapsulated nanowires, compound-released nanowires, shortened nanorods/spheres, swollen nanowires, nanorods, and branched nanowires/networks. All the phase transformations stem from the environment-mediated reaction complexity of the CysMA unit, and hold potential for biological and other emerging fields.
Co-reporter:Qiuping Yu, Yi Ding, Hui Cao, Xinhua Lu, and Yuanli Cai
ACS Macro Letters 2015 Volume 4(Issue 11) pp:1293
Publication Date(Web):November 5, 2015
DOI:10.1021/acsmacrolett.5b00699
Polyion complexation (PIC) as the driving force of polymerization-induced self-assembly (PISA), that is, PIC–PISA, is explored. Reversible addition–fragmentation chain transfer (RAFT) dispersion polymerization of NH3+-monomer 2-aminoethylacrylamide hydrochloride (AEAM) can be achieved in water under visible light irradiation at 25 °C, using nonionic poly2-hydroxypropylmethacrylamide (PHPMA) macromolecular chain transfer agent in the presence of anionic poly(sodium 2-acrylamido-2-methylpropanesulfonate) (PAMPS) PIC-template. Sphere-to-network transition occurs, owing to the PIC of PAMPS with growing chains upon reaction close to isoelectric point (IEP); thereafter, the increase of electrostatic repulsion promotes the split of networks and the rupture of spheres into fragments. Therefore, the free-flowing solution becomes viscous liquid and free-standing physical gel, and then back into viscous and free-flowing liquid. Such a PIC–PISA is appealing for gene delivery because the size and surface charge are variable on demand and at high solids.
Co-reporter:Zhengguang Zhu;Na Xu;Qiuping Yu;Lei Guo;Hui Cao;Xinhua Lu
Macromolecular Rapid Communications 2015 Volume 36( Issue 16) pp:1521-1527
Publication Date(Web):
DOI:10.1002/marc.201500281
Co-reporter:Lei Guo;Yi Ding;Jie Han;Na Xu;Xinhua Lu
Macromolecular Rapid Communications 2015 Volume 36( Issue 16) pp:1505-1510
Publication Date(Web):
DOI:10.1002/marc.201500219
Co-reporter:Na Xu, Jie Han, Zhengguang Zhu, Bo Song, Xinhua Lu and Yuanli Cai
Soft Matter 2015 vol. 11(Issue 27) pp:5546-5553
Publication Date(Web):26 May 2015
DOI:10.1039/C5SM00546A
An emerging strategy towards the sophistication of supramolecular nanomaterials is the use of supracolloidal self-assembly, in which micelles or colloids are used as building blocks. Binding directionality can produce nanostructures with attractive properties. Herein, we present a new directional supracolloidal self-assembly by virtue of dynamic covalent bonds and metal coordination in water. Conjugation of a ligand precursor to a water-soluble block copolymer through dynamic covalent bonds leads to the dehydration and micellization of the functionalized polymer. Reversible reaction facilitates the permeation of metal ions into core–shell interfaces. Conversely, metal-coordination promotes reaction over the interfaces. Cu(II)-coordination occurs overwhelmingly inside each isolated micelle. However, Zn(II)-coordination induced a directional self-assembly whose nanostructures evolve stepwise from nanorods, nanowires, necklaces, and finally to supracolloidal networks scaling-up to several tens of micrometres. Post-reactions of simultaneous dynamic covalent bond conversion and Zn(II)-coordination over the core–shell interfaces endow these supracolloidal networks with a huge specific surface area for hydrophobic dative metal centres accessible to substrates in water. Water-soluble shells play important roles in directional supracolloidal assembly and in the stabilization of nanostructures. Thus the directional self-assembly provides a versatile platform to produce metallo-hybridized nanomaterials that are promising as enzyme-inspired aqueous catalysts.
Co-reporter:Xianghua Tang, Jie Han, Zhengguang Zhu, Xinhua Lu, Hong Chen and Yuanli Cai
Polymer Chemistry 2014 vol. 5(Issue 13) pp:4115-4123
Publication Date(Web):04 Mar 2014
DOI:10.1039/C4PY00146J
We report facile synthesis, sequence-tuned thermoresponsive behaviours and reaction-induced reorganization of ketone-based hydrophilic reactive polymers. The well-controlled and living RAFT polymerisation of commercially-available diacetone acrylamide (DAAM), in methanol media on irradiation with visible light at 25 °C, leads to well-defined keto-polymers over wide degrees of polymerisation from 50–1000 and a block copolymer with poly(N,N-dimethyl acrylamide) (PDMA). Statistical copolymerisation with DMA gives a gradient copolymer, P(DAAM-grad-DMA), whose aqueous solubility can be finely tuned. Binary copolymers that have the tailored structures of PDMA-b-P(DAAM-grad-DMA) can be achieved via a one-pot iterative process. Aqueous solutions of the P(DAAM-grad-DMA) copolymers exhibit typical thermoresponsive behaviours on heating, with cloud points that can be widely tuned from 16 °C to permanently water-soluble. Adjusting the sequence from gradient, “short-block”-gradient, “middle-block”-gradient, “long-block”-gradient to block structures can induce diversity in the dehydration, heating-triggered phase transition, assembly and reaction-induced dynamic reorganization. These water-soluble reactive keto-polymers are thus promising for emerging biological applications on demand in physiological aqueous solution.
Co-reporter:Guangxiang Li;Na Xu;Qiuping Yu;Xinhua Lu;Hong Chen
Macromolecular Rapid Communications 2014 Volume 35( Issue 16) pp:1430-1435
Publication Date(Web):
DOI:10.1002/marc.201400153
Co-reporter:Guhuan Liu, Hui Shi, Yuru Cui, Jianyu Tong, Ying Zhao, Dujin Wang and Yuanli Cai
Polymer Chemistry 2013 vol. 4(Issue 4) pp:1176-1182
Publication Date(Web):02 Nov 2012
DOI:10.1039/C2PY20810E
We report the rapid and well-controlled aqueous RAFT polymerization of primary amine functional monomer under visible light irradiation at 25 °C. N-(2-aminoethyl)methacrylamide hydrochloride (AEMA) and N-(6-aminohexyl)methacrylamide hydrochloride (AHMA) were synthesized as prototypes of primary amine functional monomers. The results demonstrated that this polymerization proceeded rapidly in a well-controlled manner in acidic aqueous media upon irradiating with mild visible light at 25 °C. Moreover, improving the ion-pairing or association of the ionized monomers via adding less polar alcohols or improving the concentration of ionized monomers could shorten the initialization period and accelerate chain propagation, thus remarkably accelerating this polymerization. This polymerization was immediately switched on or off via turning on or off visible light. Dithioester residues at polymer chain ends could be retained or removed simply by adjusting pH. Accordingly, this paper provides a facile approach toward direct and rapid RAFT synthesis of well-defined primary amine functional polymers at room temperature.
Co-reporter:Jianyu Tong;Yi Shi;Guhuan Liu;Tao Huang;Na Xu;Zhengguang Zhu
Macromolecular Rapid Communications 2013 Volume 34( Issue 23-24) pp:1827-1832
Publication Date(Web):
DOI:10.1002/marc.201300697
Co-reporter:Zhenhua Hao;Guangxiang Li;Ke Yang
Macromolecular Rapid Communications 2013 Volume 34( Issue 5) pp:411-416
Publication Date(Web):
DOI:10.1002/marc.201200685
Abstract
Thermoresponsive synergistic hydrogen bonding (H-bonding) switched by several guest units in a water-soluble polymer is reported. Adjusting the distribution of guest units can effectively change the synergistic H-bonding inside polymer chains, thus widely switch the preorganization and thermoresponsive behavior of a water-soluble polymer. The synergistic H-bonding is also evidenced by converting less polar aldehyde groups into water-soluble oxime groups, which bring about the lowering-down of cloud point and an amplified hysteresis effect. This is a general approach toward the wide tunability of thermosensitivity of a water-soluble polymer simply by adjusting the distribution of several guest H-bonding units.
Co-reporter:Junjie Deng
Macromolecular Rapid Communications 2013 Volume 34( Issue 18) pp:1459-1463
Publication Date(Web):
DOI:10.1002/marc.201300474
Co-reporter:Fen Wu, Xianghua Tang, Lei Guo, Ke Yang and Yuanli Cai
Soft Matter 2013 vol. 9(Issue 15) pp:4036-4044
Publication Date(Web):01 Mar 2013
DOI:10.1039/C3SM00020F
We report a general approach that can switch preorganization and thermoresponsive behaviour of a water-soluble polymer via light-tuned hydrogen bonding (H-bonding) “dimerization” or “polymerisation”. To this end, well-defined copolymers of 2-hydroxy-3-(4-hydroxyiminomethyl)phenoxypropyl methacrylate with oligo(ethylene glycol) methacrylate (OEGMA) were synthesized via RAFT copolymerisation of 3-(4-formylphenoxy)-2-hydroxypropyl methacrylate with OEGMA under visible light irradiation at 25 °C and fully converting the aldehyde groups into the targeted oxime groups. Photoisomerization and stability of these oxime groups, and the effect of oxime configurations on preorganization and thermoresponsive behaviour were studied using dynamic light scattering and temperature-variable 1H NMR. The results demonstrated that photoisomerization of E-oximes equilibrated at 76% Z-type formation. These oxime groups were stable against heating at 80 °C in water for 8 h. Moreover, the self-restrained H-bonding “dimerization” of E-oximes led to the small-sized micelles with negligible hysteresis, while the extensible H-bonding “polymerisation” of Z-type ones led to the enlarged micelles with remarkable hysteresis.
Co-reporter:Jian Yang, Yuhai Liu, Tao Wen, Xiaoxiao Wei, Zhiyong Li, Yuanli Cai, Yunlan Su, and Dujin Wang
Crystal Growth & Design 2012 Volume 12(Issue 1) pp:29-32
Publication Date(Web):November 28, 2011
DOI:10.1021/cg201201j
Isotactic polypropylene (iPP) fiber, the surface of which is hydrophobic, can modulate the crystallization polymorphs of calcium carbonate (CaCO3) at the air/solution interface under mild conditions. The present results provide a novel perspective on controlling the crystallization of biominerals by an insoluble matrix, and they can shed new light on understanding the biomineralization process of CaCO3 as it occurs in nature.
Co-reporter:Zhilin Liu;Yufang Tang;Nan Li;Lican Lu;Junjie Deng
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 3) pp:495-508
Publication Date(Web):
DOI:10.1002/pola.25057
Abstract
This article describes a rhodopsin-inspired photosensitive polymer whose light-tunable acid sensitivity can be widely modulated simply by adjusting the position of a single methoxy substituent in the aromatic rings of cinnamyl groups. The well-defined poly(5-ethyl-5-methacryloyloxymethyl-2-(p-methoxystyryl)-[1,3]dioxane) (PEMpMSD) and poly(5-ethyl-5-methacryloyloxymethyl-2-(o-methoxystyryl)-[1,3]dioxane) (PEMoMSD) as well as poly(5-ethyl-5-methacryloyloxymethyl-2-styryl-[1,3]dioxane) were synthesized via reversible addition-fragmentation chain transfer (RAFT) process. The results demonstrated that the para-methoxy substitution of EMpMSD monomer led to the more shortened initialization period and rapid chain propagation of RAFT process than 5-ethyl-5-methacryloyloxymethyl-2-styryl-[1,3]dioxane monomer under mild visible light radiation at 25 °C. The ortho-methoxy substitution of PEMoMSD led to high degree of photoinduced Z-isomerization over 80%. Moreover, the para-methoxy substitution of PEMpMSD led to the rapid hydrolysis of the cyclic acetal linkages in ambient acid media, while the ortho-methoxy substitution of PEMoMSD slowed down this hydrolysis. This hydrolysis slowed down on Z-isomerization particularly in PEMoMSD. These effects widely broadened the tunability of the light-modulated acid sensitivity. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2011
Co-reporter:Yufang Tang;Zhilin Liu;Xuewen Wu;Guhuan Liu;Ke Yang;Yaohua Li;Lican Lu
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 14) pp:2878-2888
Publication Date(Web):
DOI:10.1002/pola.26065
Abstract
This article describes the photosensitive polymer micelles whose structural stability and acid sensitivity can be widely tuned simply via one-batch UV irradiation. To this end, the well-defined poly(5-ethyl-5-methacryloyloxy-methyl-2-styryl-[1,3]dioxane)-block-poly[poly(ethylene glycol) methacrylate] (PEMSD-b-PPEGMA) copolymers were synthesized via RAFT polymerization under mild visible light radiation at 30 °C. The results demonstrated that the irradiation of the homogeneous acetone solution with UV light only induced Z-isomerization of their cinnamyl groups, while irradiating PEMSD chains in the bulky micellar cores only induced dimerization. Moreover, the micelles of previously Z-isomerized copolymer could be effectively stabilized without changing their acid sensitivity on irradiating for shortly 3 min, while UV irradiation for 30 min could remarkably improve the acid stability of these micelles. These novel properties are of potential applications in controlled drug delivery. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Ke Yang, Xiangqian Wei, Fen Wu, Chaowei Cao, Junjie Deng and Yuanli Cai
Soft Matter 2011 vol. 7(Issue 12) pp:5861-5872
Publication Date(Web):24 May 2011
DOI:10.1039/C1SM05224A
This paper describes the sequence control over thermo-triggered micellization and the corresponding dynamic nanogels of copolymers of poly[poly(ethylene glycol) methyl ether methacrylate] (PEGMA) and 1,3-(4-formylphenoxy)-2-hydroxypropyl methacrylate (FPHPMA). To this end, the well-defined copolymers in the sequences of P(FPHPMA-ran-PEGMA), P(FPHPMA-ran-PEGMA)-b-PFPHPMA and P(FPHPMA-ran-PEGMA)-b-PPEGMA, at a molar ratio of [FPHPMA]:[PEGMA]≈1:1 and a fixed overall degree of polymerization or DP≈50, were synthesized via reversible addition-fragmentation chain transfer radical polymerization or RAFT polymerization under mild visible light radiation at 25 °C. The results demonstrated that altering the sequence from P(FPHPMA-ran-PEGMA) to P(FPHPMA-ran-PEGMA)-b-PPEGMA or P(FPHPMA-ran-PEGMA)-b-PFPHPMA led to an increase of cloud point and decrease of hydrodynamic diameter from hundreds of nm to tens of nm. Moreover, altering the copolymer sequence resulted in the thermo-triggered micellization mechanism changing from the insertion/expulsion of single chains to fusion/fission of their pre-assembled “polymerases”. The hysteresis and solvent isotopic effects varied with the sequence of copolymers. Moreover, the thermo-induced dehydration and the apparent reactivity of aldehyde functionalities in swollen micelles also varied with the sequence. These thermo-induced micelles could react with diamine to form dynamic nanogels. The pH- and/or thermo-triggered swellability of these nanogels could be adjusted by changing the sequence.

![2-Propenamide, N-[2-[(2-aminoethyl)dithio]ethyl]-2-methyl-, hydrochloride (1:1)](/data/chemimg/3781600/1453334-95-6.png)
![2-Propenamide, N-[2-[(2-aminoethyl)dithio]ethyl]-2-methyl-, hydrochloride (1:1)](/data/chemimg/3781600/1453334-95-6_b.png)
![Butanoic acid, 4-cyano-4-[[(ethylthio)thioxomethyl]thio]-](/data/chemimg/3781600/1803289-73-7.png)
![Butanoic acid, 4-cyano-4-[[(ethylthio)thioxomethyl]thio]-](/data/chemimg/3781600/1803289-73-7_b.png)
![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-2-methyl-](/data/chemimg/113500/881037-62-3.png)
![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-2-methyl-](/data/chemimg/113500/881037-62-3_b.png)
![2-Propenamide, N-[2-(1H-imidazol-5-yl)ethyl]-](/data/chemimg/1616800/10124-85-3.png)
![2-Propenamide, N-[2-(1H-imidazol-5-yl)ethyl]-](/data/chemimg/1616800/10124-85-3_b.png)