•Electronic formation mechanism of oxygen species is well interpreted.•CO is directly oxidized by superoxide.•A tridentate carbonate forms when CO reacts with peroxide.•Catalytic effects of doped Zr ions in ceria-based catalysts are elucidated.Exploring the catalytic effects of Zr doping ion on ceria-based catalysts and its corresponding oxidation mechanism are of great significance. Using density functional theory method, the possible oxygen species and their catalytic roles on Ce1 − xZrxO2(111) surfaces are systematically investigated. For O2 adsorbed on the stoichiometric Ce1 − xZrxO2(111), it remains the electronic character of free molecule. When O2 adsorbs at the top site of Ce on the partially reduced Ce1 − xZrxO2(111), a paramagnetic superoxide forms; while a diamagnetic peroxide appears if O2 fills into the surface oxygen vacancy. Using CO as a probe molecule, the oxidation mechanism on Ce1 − xZrxO2(111) is explored. Our results indicate that CO is directly oxidized to CO2 by superoxide; while a tridentate carbonate forms when CO reacts with peroxide. Compared with ceria, one of the most important catalytic effects of doped Zr ion is to lower the oxygen vacancy formation energy and improve its oxidative activity.Electronic formation mechanism of O2− and O22− on the partially reduced Ce1 − xZrxO2(111).
Co-reporter:Bo-Tao Teng, Jia-Jian Lang, Xiao-Dong Wen, Ce Zhang, Maohong Fan, and H. Gordon Harris
The Journal of Physical Chemistry C 2013 Volume 117(Issue 37) pp:18986-18993
Publication Date(Web):August 5, 2013
DOI:10.1021/jp4056279
Understanding the O2 adsorption and oxidative activity on gold-based catalysts is of great significance for gold catalysis. According to the adsorption behaviors of O2 on Au(111)+, 3Au/Au(111)+, Au19+, and Au9/CeO2(111), the electronic nature of why O2 weakly interacts with free positively charged Au substrate while it strongly interacts with Au cluster supported on ceria is well-explained herein. The ceria support serves as an electronic repository, where it gains and stores electrons from the supported metal cluster and releases them when the metal cluster interacts with molecular O2. The possible oxygen species on gold-based catalysts have been systematically confirmed for the first time. On the Au9/CeO2 modeling catalyst, a peroxide species forms when O2 locates at the hollow site of Au9, while a superoxide forms for O2 at the top site of a Ce atom. It is very interesting to find that Ce3+ ion distributions in Aun/CeO2 catalysts have diverse possibilities. The superoxide close to Au9 has the highest oxidative activity. The interface between the Au cluster and ceria surface is the active site for Aun/CeO2 catalysts. The present work sheds light on understanding the oxidative mechanism of metal/support catalysts, as well as the development of new catalysts with high performance at relatively low temperature.
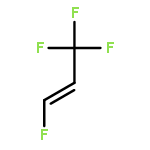
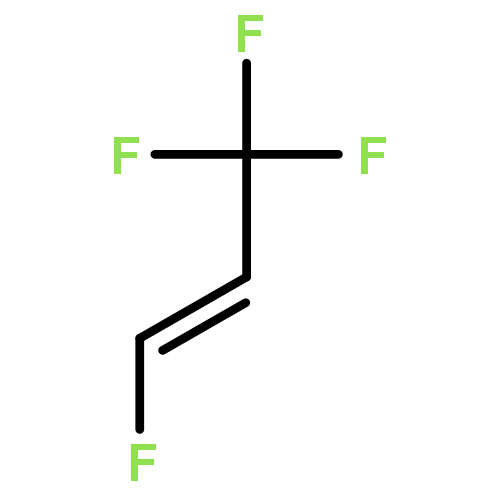
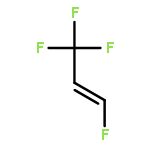
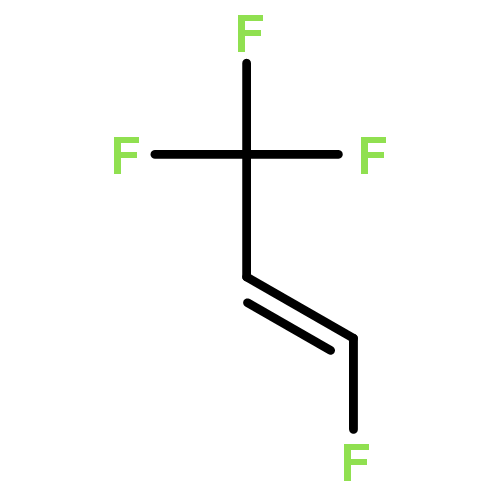
The adsorption and reaction behaviors of CF3CH2I on Ag(111) were systematically studied by density functional theory (DFT) calculations. Physical adsorption of CF3CH2I on Ag(111) occurs due to the weak interactions between surface Ag atoms and iodine atom of CF3CH2I; while strong chemisorption occurs for CF3CH2 fragment on Ag(111). Electronic analysis indicates that the singly occupied molecular orbital (SOMO) of CF3CH2 strongly interacts with the surface Ag atoms. It is very interesting to find that the most stable structures of CF3CH2 on Ag(111) locate at the top site, instead of the hollow sites. This might be attributed to the facts that CF3CH2 adsorbed at the top site will maximize the sp3-type hybridization, and the possible weak interaction between the fluorine lone pair electrons of p orbitals for CF3CH2 and surface Ag(111) occurs, which is supported by the charge density difference (CDD) analysis with a low isosurface value. We propose that the charge density difference (CDD) analysis with a high or low isosurface value can be widely applied to analyze the strong or weak electronic interactions upon adsorption. Transition state calculations suggested that the energy barrier of CF bond rupture for CF3CH2I on Ag(111) (1.44 eV) is much higher than that of CI bond breakage for CF3CH2I (0.43 eV); and the activation energy of the CF bond dissociation for CF3CH2(a) is 0.67 eV.Highlights► The adsorption behaviors of CF3CH2I and CF3CH2 on Ag(111) were studied. ► Stable structures of CF3CH2 on Ag(111) are related with the orientation of F atoms. ► The density of states and charge density difference were analyzed. ► Transition states were calculated for CI and CF bond rupture of CF3CH2I on Ag(111).
Studying the structures of metal clusters on oxide supports is challenging due to their various structural possibilities. In the present work, a simple rule in which the number of Au atoms in different layers of Aux clusters is changed successively is used to systematically investigate the structures of Aux (x=1–10) clusters on stoichiometric and partially reduced CeO2(111) surface by DFT calculations. The calculations indicate that the adsorption energy of a single Au atom on the surface, the surface structure, as well as the AuAu bond strength and arrangement play the key roles in determining Aux structures on CeO2(111). The most stable Au2 and Au3 clusters on CeO2(111) are 2D vertical structures, while the most stable structures of Aux clusters (x>3) are generally 3D structures, except for Au7. The 3D structures of large Aux clusters in which the Au number in the bottom layer does not exceed that in the top layer are not stable. The differences between Aux on CeO2(111) and Mg(100) were also studied. The stabilizing effect of surface oxygen vacancies on Aux cluster structures depends on the size of Aux cluster and the relative positions of Aux cluster and oxygen vacancy. The present work will be helpful in improving the understanding of metal cluster structures on oxide supports.
Co-reporter:Yun Zhao, Bo-Tao Teng, Xiao-Dong Wen, Yue Zhao, Qiao-Ping Chen, Lei-Hong Zhao, and Meng-Fei Luo
The Journal of Physical Chemistry C 2012 Volume 116(Issue 30) pp:15986-15991
Publication Date(Web):July 14, 2012
DOI:10.1021/jp3016326
The electronic properties and oxidation roles of superoxide and peroxide species on the small and big CeO2(111) surfaces with an oxygen vacancy are systematically investigated utilizing density functional theory. When the CeO2 surface is partially reduced, a surface oxygen vacancy forms, and two Ce4+ ions of substrate are reduced to Ce3+ ones. If O2 adsorbs at the top site of Ce3+ ion, which is close to the surface oxygen vacancy, it slips into the vacancy. Then, two 4f electrons of Ce3+ ions feedback to the π2p* orbital of O2, and a diamagnetic peroxide species forms. When O2 adsorbs at the top site of Ce3+ ion apart from the surface oxygen vacancy, superoxide species with three different orientations are obtained. Meanwhile, only one Ce3+ ion is oxidized to Ce4+ since one 4f electron transfers to the π2p* orbital of O2. The migration barrier from O2–(η2-1) to O22– is 0.35 eV, indicating that superoxide might easily transform into peroxide with elevating temperature. CO is directly oxidized to CO2 by the superoxide without energy barrier, while a carbonate forms when CO reacts with peroxide. A high desorption barrier of the carbonate to form a gas CO2 molecule indicates that peroxide species might play the dominant role at relatively high temperature.
Science China Chemistry 2011 Volume 54( Issue 5) pp:
Publication Date(Web):2011 May
DOI:10.1007/s11426-011-4242-x
We report a detailed investigation of the behavior of chemisorbed hydrogen atoms (Ha) on Pt(111) by a combination of an experimental study of the Ha + Da reaction and first-principles calculations. The coverage-dependent adsorption and desorption behavior of Ha and Da on Pt(111) have been systematically established and can be well interpreted in terms of repulsive interactions between adsorbates. Ha adsorbs exclusively on the face-centered cubic (fcc) sites of Pt(111) at coverages not exceeding 1 monolayer (ML). With increasing Ha coverage, repulsive interactions between Ha increase, leading to a reduction in both the adsorption energy and the desorption activation energy. It is proposed that the lateral interactions within a Ha layer are partly induced by the local repulsive interactions due to high mobility of Ha on Pt(111). For the Ha + Da exchange reaction on Pt(111), it is found that Ha has a higher selectivity for HD formation than Da. Considering that Ha diffuses much faster than Da on Pt(111), we propose that the difference in diffusion rates between Ha and Da may determine the selectivity of Ha and Da in forming HD in the Ha + Da reaction on Pt(111).
Co-reporter:Yun Zhao ; Botao Teng ; Zongxian Yang ; Yue Zhao ; Leihong Zhao ;Mengfei Luo
The Journal of Physical Chemistry C 2011 Volume 115(Issue 33) pp:16461-16466
Publication Date(Web):July 22, 2011
DOI:10.1021/jp203640f
The adsorption behaviors and electronic properties of Sn on the CeO2(111) surface were systematically investigated using the density functional theory (DFT) method. Our results suggested that Sn on the hollow site is more stable than that on the top oxygen site at the coverage of 0.25 ML, while Sn on the top oxygen site is the most stable configuration when the coverage of Sn is equal to or higher than 0.5 ML. Charge density difference (CDD) analysis indicates that electrons transfer from the Sn adatom to the substrate, leading to the reduction of Ce4+ to Ce3+ ion, which is in agreement with the experimental spectroscopy. The reduction degree of the substrate increases with the Sn coverage, which is well supported by the CDD and spin-resolved density of states (DOS) of the most stable xSn/CeO2(111) configurations. The adsorption of Sn can partially activate the surface oxygen of ceria. The tentative study of a probe molecule CO adsorption on the Sn/CeO2(111) surface indicates that CO adsorption is enhanced due to the strong tin–ceria interactions.
Co-reporter:Bo-Tao Teng, Shi-Yu Jiang, Zong-Xian Yang, Meng-Fei Luo, You-Zhao Lan
Surface Science 2010 Volume 604(Issue 1) pp:68-78
Publication Date(Web):1 January 2010
DOI:10.1016/j.susc.2009.10.024
The effects of different oxygen species and vacancies on the adsorption and oxidation of formaldehyde over CeO2(1 1 1) surface were systematically investigated by using density functional theory (DFT) method. On the stoichiometric CeO2(1 1 1) surface, the C–H bond rupture barriers of chemisorbed formaldehyde are much higher than that of formaldehyde desorption. On the reduced CeO2(1 1 1) surface, the energy barriers of C–H bond ruptures are less than those on the stoichiometric CeO2(1 1 1) surface. If the C–H bond rupture occurs, CO and H2 form quickly with low energy barriers. When O2 adsorbs on the reduced (1 1 1) surface (O2/Ov species), the C–H bond rupture barriers of formaldehyde are greatly reduced in comparison with those on the stoichiometric CeO2(1 1 1) surface. If O2 adsorbs on oxygen vacancy at sub-layer surface, its oxidative roles on formaldehyde are much similar to that of O2/Ov species.
Chemical Physics Letters 2008 Volume 461(1–3) pp:47-52
Publication Date(Web):8 August 2008
DOI:10.1016/j.cplett.2008.06.082
Abstract
Density functional theory (DFT) calculation has been performed to study the adsorption and dissociation of CH2I2 on Ag(1 1 1) surface at different coverages. CH2I2(a) with two iodine atoms bonded to Ag(1 1 1) is the main stable adsorbed species at low coverage, while CH2I2(a) with one iodine atom bonded to Ag(1 1 1) will dominate on the surface at high coverage. The dissociation barriers of CH2I2 to generate CH2(a) species on Ag(1 1 1) also increase with the increase of coverage. Analysis of density of states shows that relatively strong interactions between CH2I2(a) and Ag(1 1 1) surface exist.
Catalysis Communications (5 October 2012) Volume 27() pp:63-68
Publication Date(Web):5 October 2012
DOI:10.1016/j.catcom.2012.06.027
The structures and electronic properties of MxCe1 − xO2 − δ (M = Au, Pd, Pt, and Rh) modeling catalysts were theoretically compared. Two kinds of active oxygen species were obtained by O2 filling into or standing aside surface oxygen vacancy on MxCe1 − xO2 − δ (111). Correspondingly, tridentate and bidentate carbonates form by CO reaction with the two different oxygen species. Finally, the possible oxidative activities of four MxCe1 − xO2 − δ catalysts were evaluated by comparing the formation energy of surface oxygen vacancy, as well as the energy differences between carbonate and CO2 desorption. Our work will provide important information for the design of catalyst with high oxidation performance.Download full-size imageHighlights► Structures and properties of MxCe1 − xO2 − δ (M = Au, Pd, Pt, and Rh) were compared. ► Two oxygen species on MxCe1 − xO2 − δ(111) surface were found. ► Tridentate and bidentate carbonates form by reaction with different oxygen species. ► Activities of MxCe1 − xO2 − δ(M = Au, Pd, Pt, and Rh) were theoretically evaluated.