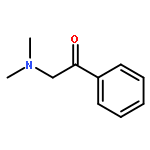
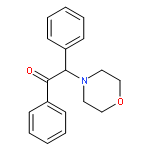
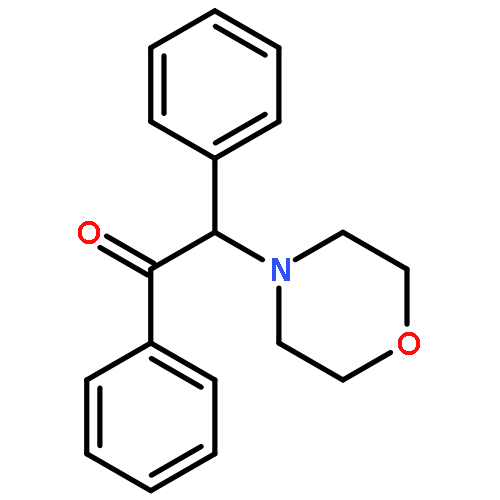
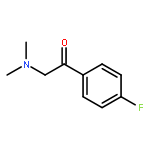
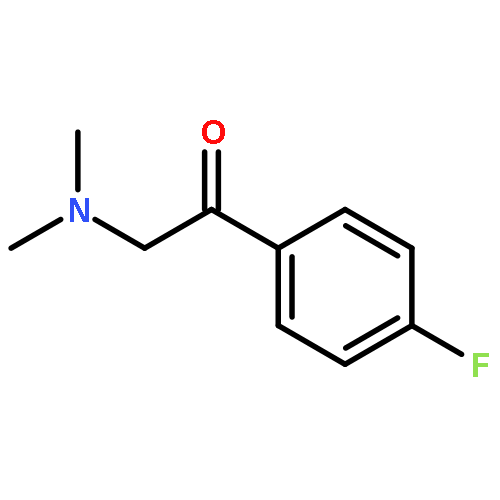
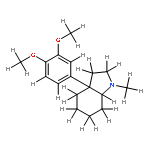
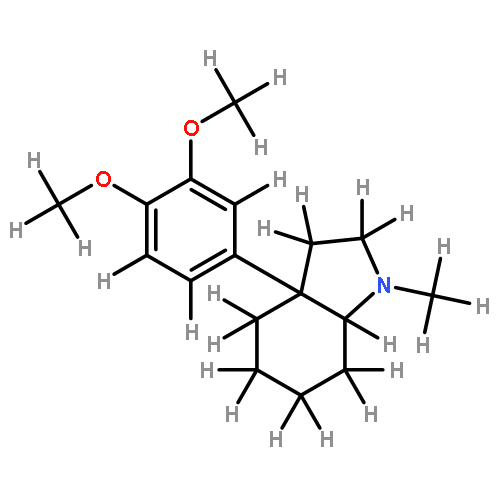
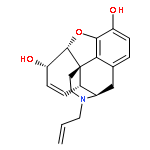
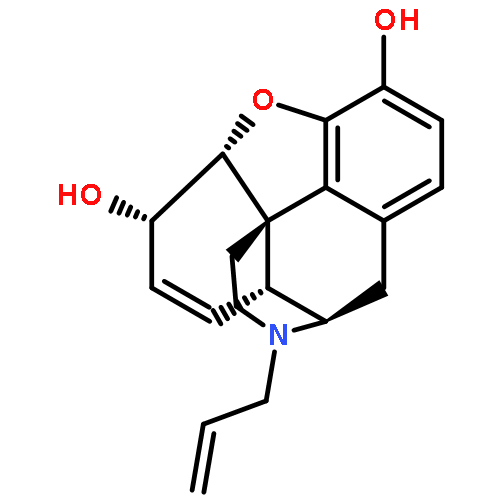
Co-reporter:Yoshikazu Watanabe, Shota Kitazawa, Toru Nemoto, Shigeto Hirayama, Takashi Iwai, Hideaki Fujii, Hiroshi Nagase
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 11) pp:3032-3050
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmc.2013.03.026
Several derivatives with an azabicyclo[2.2.2]octane skeleton having a 7-amide side chain were synthesized. Compounds that had an electron-donating group exhibited high affinity for the μ opioid receptor while those with a bulky substituent at the 8-nitrogen atom had low affinities for all receptor types. High affinities and selectivities for the κ receptor resulted from the introduction of the longer amide side chain at the 7α-position. Our studies indicate that the orientation of the amide side chain at the 7-position within the azabicyclo[2.2.2]octane skeleton is related to selectivity for the κ receptor.
Co-reporter:Toru Nemoto, Naoshi Yamamoto, Naohisa Wada, Yukimasa Harada, Miyuki Tomatsu, Marina Ishihara, Shigeto Hirayama, Takashi Iwai, Hideaki Fujii, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 1) pp:268-272
Publication Date(Web):1 January 2013
DOI:10.1016/j.bmcl.2012.10.100
We have previously reported the essential structure of the opioid κ receptor agonist nalfurafine hydrochloride (TRK-820) for binding to the κ receptor. In the course of this study, we focused on the effect of the substituent at 17-N in nalfurafine on the binding affinity for the κ receptor. The exchange of the 17-N substituent in nalfurafine from cyclopropylmethyl to fluoro-substituted alkyl groups, which are strong electron withdrawing substituents, almost completely diminished the binding affinities for the μ and δ opioid receptors, but the binding affinity for the κ receptor was still maintained. As a result, nalfurafine derivatives with 17-fluoro-substituted alkyl groups showed higher selectivities for the κ receptor than did nalfurafine itself. With regard to the κ agonistic activities, the conversion of the 17-N substituent in nalfurafine from cyclopropylmethyl to fluoro-substituted alkyl groups led to the gradual decrease of the agonistic activities in the order corresponding to their binding affinities for the κ receptor. In contrast, the derivative with the bulky 17-isobutyl group showed lower affinity and agonistic activity for the κ receptor than the derivatives with the smaller functional groups. This research suggested that both the electronic property and the steric characteristics of the 17-N substituent would have a great influence on the binding property for the κ receptor.
Co-reporter:Yoshihiro Ida, Ayaka Matsubara, Toru Nemoto, Manabu Saito, Shigeto Hirayama, Hideaki Fujii, Hiroshi Nagase
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 19) pp:5810-5831
Publication Date(Web):1 October 2012
DOI:10.1016/j.bmc.2012.08.004
We have reported previously the novel δ opioid agonist KNT-127 which showed high affinity and selectivity for the δ receptor. Moreover, the analgesic effect of subcutaneously administered KNT-127 was more potent than that of a prototypical δ agonist (−)-TAN-67 in the acetic acid writhing test. This study of the structure–activity relationship of KNT-127 derivatives focused on the introduction of substituents onto the 5′-, 6′-, 7′- or 8′-position of the quinoline ring and revealed that many derivatives with 5′- or 8′-substituents showed high affinities and selectivities for the δ receptor. Especially, SYK-153 with an 8′-OH group showed the highest affinity and the most balanced and highest selectivity for the δ receptor among the synthesized compounds.The study of the structure–activity relationship of KNT-127 derivatives showed that the 5′- and 8′-positions on the quinoline ring would be the most appropriate sites for modification to enhance the affinity and selectivity for the δ receptor.
Co-reporter:Yoshihiro Ida, Toru Nemoto, Shigeto Hirayama, Hideaki Fujii, Yumiko Osa, Masayuki Imai, Takashi Nakamura, Toshiyuki Kanemasa, Akira Kato, Hiroshi Nagase
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 2) pp:949-961
Publication Date(Web):15 January 2012
DOI:10.1016/j.bmc.2011.11.047
Co-reporter:Naohisa Wada, Hideaki Fujii, Koji Koyano, Shigeto Hirayama, Takashi Iwai, Toru Nemoto, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 24) pp:7551-7554
Publication Date(Web):15 December 2012
DOI:10.1016/j.bmcl.2012.10.023
Novel double-capped triplet drugs, which have one pharmacophore unit and two epoxymethano or dimethylepoxymethano structures (termed cap or diMe-cap structures, respectively) were synthesized. Key intermediate oxazoline 16 derived from acetone enabled the effective synthesis of double-capped triplets. SYK-134 (7a) and SYK-135 (8a) with N-cyclopropylmethyl substituent and cap structures showed selectivities for the κ opioid receptor. On the other hand, the N-Me series exhibited selectivities for the μ opioid receptor. The double-capped triplet drugs with diMe-cap structures preferred the μ receptor independently of their N-substituents. SYK-385 (19b), one of the μ-selective double-capped triplet drugs, showed the highest selectivity for the μ receptor among the reported μ-selective nonpeptide ligands.
Co-reporter:Hideaki Fujii, Ryo Nakajima, Junko Akiyama, Naoshi Yamamoto, Shigeto Hirayama, Toru Nemoto, Hiroaki Gouda, Shuichi Hirono, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 24) pp:7697-7701
Publication Date(Web):15 December 2012
DOI:10.1016/j.bmcl.2012.09.102
Previously reported propellane derivative KNT-42 preferred the κ receptor and functioned as a message part in the message-address concept, but its affinity for the κ receptor was not high. To improve affinity, we synthesized five pentacyclic propellane derivatives designed for the purpose of fixing the conformation of KNT-42. The etheno- and ethano-bridged derivatives SYK-347 and SYK-393 exhibited high affinity and selectivity for the κ receptor, whereas the other derivatives did not. These results would be due to the different ranges of movement of the basic nitrogens and less basicity of the nitrogens due to the electron withdrawing effect of the introduced hydroxy or keto group. SYK-347 and SYK-393 preferring the κ receptor were expected to be useful for designing selective ligands for opioid receptor types, especially the κ receptor.
Co-reporter:Hideaki Fujii, Satomi Imaide, Shigeto Hirayama, Toru Nemoto, Hiroaki Gouda, Shuichi Hirono, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 24) pp:7711-7714
Publication Date(Web):15 December 2012
DOI:10.1016/j.bmcl.2012.09.101
To clarify the essential structures of an opioid κ receptor selective agonist, nalfurafine, for binding to the κ receptor, we designed and synthesized the decahydro(iminoethano)phenanthrene derivatives with an oxygen functionality at the 3-position. The introduction of a hydroxy group to the derivatives increased the affinity and selectivity to the κ receptor regardless of the configuration at the 3-position. However, their affinities were lower than those of nalfurafine with the phenolic hydroxy group. The results suggested that the acidity of the hydroxy group would play an important role in the interaction with the opioid receptor. The low affinities of the 3-keto derivatives indicated that the 3-hydroxy group may participate in the hydrogen bonding with the receptor site not as a hydrogen acceptor but as a hydrogen donor. This is the first experimental evidence for a role as a hydrogen donor for the 3-hydroxy group in morphinans. Furthermore, the κ selectivities in these derivatives with the 6α-amide side chain were affected by the the 3-hydroxy group. The obtained structure–activity relationship information is expected to be useful for the design of more selective ligands for the κ receptor.
Co-reporter:Yoshinori Miyata, Hideaki Fujii, Yuka Uenohara, Seiki Kobayashi, Tsutomu Takeuchi, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 16) pp:5174-5176
Publication Date(Web):15 August 2012
DOI:10.1016/j.bmcl.2012.06.085
Derivatives of 7-benzylidenenaltrexone (BNTX), which was recently reported to be an effective chloroquine (CQ)-resistance reverser, were synthesized and evaluated for their CQ-resistance reversing activities. The synthesized derivatives showed CQ-resistance reversing effects. They also reacted with glutathione (GSH) both enzymatically and chemically, and inhibited glutathione reductase activity. 7-Benzyl derivative, which was obtained by reduction of the olefin group in α,β-unsaturated ketone structure of BNTX, also exhibited CQ-resistance reversing effect, but its potency was significantly lower than that of BNTX. These outcomes suggested that the decrease in GSH level could be one of the mechanisms of CQ-resistance reversing effects induced by BNTX derivatives.
Co-reporter:Yoshikazu Watanabe, Shota Kitazawa, Hideaki Fujii, Toru Nemoto, Shigeto Hirayama, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 8) pp:2689-2692
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmcl.2012.03.001
A novel opioid ligand possessing a stable and cyclic imine 16 and its derivatives with an azabicyclo[2.2.2]octane skeleton were synthesized. The imine 16 showed higher affinity for the μ receptor than compound 21 with an oxabicyclo[2.2.2]octane skeleton. Azabicyclo[2.2.2]octane derivative 18d with a cyclopropylmethyl group on the 8-nitrogen showed the highest affinity for the μ receptor among the synthesized derivatives.
Co-reporter:Hiroshi Nagase, Satomi Imaide, Shigeto Hirayama, Toru Nemoto, Hideaki Fujii
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 15) pp:5071-5074
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmcl.2012.05.122
To clarify the essential structures of an opioid κ receptor selective agonist, nalfurafine, for binding to the κ receptor, we designed and synthesized some nalfurafine derivatives and the decahydro(iminoethano)phenanthrene derivatives with a cyclohexene moiety as a surrogate for the phenol ring. In addition to the 6-amide side chain and the 17-nitrogen substituted by a cyclopropylmethyl group, the 4,5-epoxy ring, phenolic hydroxy group, and angular hydroxy group played important roles in eliciting the binding properties of nalfurafine but these three moieties were not indispensable for binding to the κ receptor. Moreover, the phenol ring was also not essential for the binding to the κ receptor, and the cyclohexene moiety would play an important role in fixing the conformation of decahydro(iminoethano)phenanthrene derivatives to effectively raise the amide side chain, rendering a conformation that resembled the active one of nalfurafine.
Co-reporter:Hiroshi Nagase, Junko Akiyama, Ryo Nakajima, Shigeto Hirayama, Toru Nemoto, Hiroaki Gouda, Shuichi Hirono, Hideaki Fujii
Bioorganic & Medicinal Chemistry Letters 2012 22(8) pp: 2775-2779
Publication Date(Web):
DOI:10.1016/j.bmcl.2012.02.082
Co-reporter:Toru Nemoto, Naoshi Yamamoto, Akio Watanabe, Hideaki Fujii, Ko Hasebe, Mayumi Nakajima, Hidenori Mochizuki, Hiroshi Nagase
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 3) pp:1205-1221
Publication Date(Web):1 February 2011
DOI:10.1016/j.bmc.2010.12.035
A novel 6,14-epoxymorphinan benzamide derivative (NS22) that was previously reported showed opioid κ receptor agonistic activity and analgesic activity. The unsatisfactory κ selectivity of NS22 led us to synthesize its derivatives to improve the opioid κ receptor selectivity and the agonist activity. In the course of SAR of the various derivatives, 17-benzyl-6,14-epoxymorphinan derivatives (KNT-33, 53, 55, 80, 90, 133) were found to show high selectivities and affinities for the opioid κ receptor. In addition, KNT-33, 53, 55 showed dose-dependent analgesic effects in acetic acid writhing tests. Therefore, 17-benzyl substituents may play an important role for developing κ selectivity.We found that 17-benzyl-6,14-epoxymorphinan derivatives showed high selectivities for the opioid κ receptor. The 17-benzyl substituent may play an important role for developing κ selectivity.
Co-reporter:Naoshi Yamamoto, Hideaki Fujii, Toru Nemoto, Ryo Nakajima, Shinobu Momen, Naoki Izumimoto, Ko Hasebe, Hidenori Mochizuki, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 13) pp:4104-4107
Publication Date(Web):1 July 2011
DOI:10.1016/j.bmcl.2011.04.147
Co-reporter:Hiroshi Nagase, Akio Watanabe, Toru Nemoto, Mayumi Nakajima, Ko Hasebe, Hidenori Mochizuki, Hideaki Fujii
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 13) pp:4023-4026
Publication Date(Web):1 July 2011
DOI:10.1016/j.bmcl.2011.04.134
We synthesized symmetrical and nonsymmetrical triplet drugs with 1,3,5-trioxazatriquinane skeletons. The isolation of key intermediates, oxazoline dimers, made it possible to effectively produce nonsymmetrical triplets. Among the synthesized triplets, KNT-93, composed of three identical opioid μ receptor agonists, showed dose-dependent antinociception via the μ receptor. The effect was 56-fold more potent than that of morphine, a representative μ agonist. The profound analgesic effect induced by KNT-93 might result from simultaneous occupation of three μ opioid receptors.Triplet drug KNT-93 induced profound antinociception; the effect was 56-fold more potent than that of morphine.
Co-reporter:Yoshinori Miyata, Hideaki Fujii, Yumiko Osa, Seiki Kobayashi, Tsutomu Takeuchi, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 16) pp:4710-4712
Publication Date(Web):15 August 2011
DOI:10.1016/j.bmcl.2011.06.085
We evaluated antimalarial and/or chloroquine-resistance reversing effects of five opioid receptor antagonists. Although none of the evaluated compounds showed antimalarial effects, some of them, especially the δ1 receptor antagonist, 7-benzylidenenaltrexone (BNTX) exhibited potent chloroquine-resistance reversing effects in Plasmodium chabaudi.Opioid δ1 receptor antagonist 7-benzylidenenaltrexone (BNTX) exhibited profound resistance reversing effects for chloroquine-resistant Plasmodium chabaudi.
Co-reporter:Hiroshi Nagase, Koji Koyano, Naohisa Wada, Shigeto Hirayama, Akio Watanabe, Toru Nemoto, Mayumi Nakajima, Kaoru Nakao, Hidenori Mochizuki, Hideaki Fujii
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 20) pp:6198-6202
Publication Date(Web):15 October 2011
DOI:10.1016/j.bmcl.2011.07.065
An improved synthetic method for triplet drugs with the 1,3,5-trioxazatriquinane skeleton was developed that used p-toluenesulfonylmethyl isocyanide (TosMIC) instead of 1,3-dithiane. Using the improved method, we synthesized compounds with two identical pharmacophore units and an epoxymethano group, that is, capped homotriplets. Among the synthesized capped homotriplets, KNT-123 showed high selectivity for the μ receptor over the κ receptor, and the μ selectivity was the highest among the reported μ selective nonpeptide ligands. KNT-123 administered subcutaneously induced a dose-dependent analgesic effect in the acetic acid writhing assay, and its potency was 11-fold more potent than that of morphine. KNT-123 may serve as a useful tool for the study of the pharmacological actions mediated specifically via the μ receptor.Capped homotriplet KNT-123 with high selectivity for the μ receptor over the κ receptor induced profound antinociception; the effect was 11-fold more potent than that of morphine in the acetic acid writhing assay.
Co-reporter:Kohei Hayashida, Hideaki Fujii, Shigeto Hirayama, Toru Nemoto, Hiroshi Nagase
Tetrahedron 2011 67(35) pp: 6682-6688
Publication Date(Web):
DOI:10.1016/j.tet.2011.04.097
Co-reporter:Naoshi Yamamoto, Hideaki Fujii, Satomi Imaide, Shigeto Hirayama, Toru Nemoto, Junji Inokoshi, Hiroshi Tomoda, and Hiroshi Nagase
The Journal of Organic Chemistry 2011 Volume 76(Issue 7) pp:2257-2260
Publication Date(Web):March 7, 2011
DOI:10.1021/jo1022487
Acetylcholinesterase inhibitor (−)-homogalanthamine 3 was synthesized from μ opioid antagonist naltrexone (2) in 16% total yield. The synthesis features Grob fragmentation as a key reaction, which was especially accelerated in the presence of 15-crown-5.
Co-reporter:Hiroshi Nagase, Satomi Imaide, Miyuki Tomatsu, Toru Nemoto, Mayumi Nakajima, Kaoru Nakao, Hidenori Mochizuki, Hideaki Fujii
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 12) pp:3726-3729
Publication Date(Web):15 June 2010
DOI:10.1016/j.bmcl.2010.04.080
We synthesized novel 15–16 nornaltrexone derivatives 9, 11 and 22 to examine the importance of the cavity in the Beckett–Casy model, which was proposed to interact with the 15–16 ethylene moiety in the morphine structure. All the synthesized compounds showed lower affinities for the opioid receptor than did the naltrexone (10). The binding affinities of 14-OH derivatives 11, in which the rotation of the 9–17 bond would be restricted by an intramolecular hydrogen bond, was improved compared to the corresponding 14-H derivatives 9. Compound 22 whose 9–17 bond was strictly fixed by the ethylene bridge hardly bound to the opioid receptor. Compound 26 also showed very weak binding affinity in spite of the existence of the 15–16 ethylene unit. We proposed an important role for the orientation of the lone electron pair on the 17-nitrogen rather than the significance of the cavity in the Beckett–Casy model.Novel 15–16 nornaltrexone derivatives 2 were synthesized using a double decarboxylation reaction as the key reaction to examine the Beckett–Casy model.
Co-reporter:Satomi Imaide, Hideaki Fujii, Akio Watanabe, Toru Nemoto, Mayumi Nakajima, Kaoru Nakao, Hidenori Mochizuki, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 3) pp:1055-1058
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmcl.2009.12.039
Novel 16,17-seco-naltrexone derivatives 3 were synthesized using a 16–17 bond cleavage reaction of naltrexone as the key reaction to examine the Beckett–Casy model. All the prepared 16,17-seco-naltrexone derivatives 3 showed lower affinities for opioid receptors than naltrexone. Although the results of binding assay seem to support the existence of a cavity in the model, further investigation using 15,16-nornaltrexone derivatives 26 will be needed to confirm the model.Novel 16,17-seco-naltrexone derivatives 3 were synthesized using 16–17 bond cleavage reaction of naltrexone as the key reaction to examine the Beckett–Casy model.
Co-reporter:Hideaki Fujii, Yoshihiro Ida, Shinichi Hanamura, Yumiko Osa, Toru Nemoto, Mayumi Nakajima, Ko Hasebe, Kaoru Nakao, Hidenori Mochizuki, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 17) pp:5035-5038
Publication Date(Web):1 September 2010
DOI:10.1016/j.bmcl.2010.07.035
We synthesized pyrrolomorphinan derivatives 6, 7, and 9 to examine whether the pyrrole ring would be an accessory site in the κ opioid receptor selective antagonist, nor-binaltorphimine. Derivative 6 had an α,β-unsaturated ketone substituent that strongly bound to the κ receptor. The compound with the highest κ receptor selectivity, 6e, produced a dose-dependent antinociceptive effect in the mouse acetic acid writhing test. However, derivatives 7 and 9, which did not have α,β-unsaturated ketone substituents, showed less κ receptor selectivity than compound 6. Based on structure–activity relationships, we proposed that these compounds adopted active structures for κ selective agonist activity. The pyrrole ring would not function as an accessory site, but the ability of the side chain on the pyrrole ring to localize above the C-ring appeared to confer κ selective agonist activity. These results will promote the design of novel κ agonists.Pyrrolomorphinans 1 were synthesized to examine whether the pyrrole ring might be an accessory site in the κ receptor selective antagonist, nor-binaltorphimine.
Co-reporter:Hiroshi Nagase, Akio Watanabe, Toru Nemoto, Noriyuki Yamaotsu, Kohei Hayashida, Mayumi Nakajima, Ko Hasebe, Kaoru Nakao, Hidenori Mochizuki, Shuichi Hirono, Hideaki Fujii
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 1) pp:121-124
Publication Date(Web):1 January 2010
DOI:10.1016/j.bmcl.2009.11.027
A conformational analysis of κ opioid receptor agonists, TRK-820 and U-50,488H indicated an active conformation of TRK-820 in which the C-ring was in the boat form with the 14-OH interacting with the amide nitrogen. Based on the obtained active conformation of TRK-820, we designed and synthesized a novel κ agonist KNT-63 with oxabicyclo[2.2.2]octane skeleton. KNT-63 showed profound antinociceptive effects via the κ receptor which were as potent as that of TRK-820.KNT-63 designed on the basis of an active conformation of selective κ agonist TRK-820 showed strong antinociceptive effects via the κ receptor.
Co-reporter:Hiroshi Nagase, Satomi Imaide, Miyuki Tomatsu, Shigeto Hirayama, Toru Nemoto, Noriko Sato, Mayumi Nakajima, Kaoru Nakao, Hidenori Mochizuki, Hiroaki Gouda, Shuichi Hirono, Hideaki Fujii
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 12) pp:3801-3804
Publication Date(Web):15 June 2010
DOI:10.1016/j.bmcl.2010.04.044
Novel naltrexone derivatives 7 and 8 with contracted and expanded D-rings were synthesized to investigate the importance of orientation of lone electron pair on the nitrogen for binding abilities to the opioid receptor. Compound 7 showed almost no binding affinity, whereas compound 8 was comparable to naltrexone (6) in binding affinity. Conformational analyses and NOE experiments in D2O of compounds 6–8 suggested that the lone electron pairs of compounds 6 and 8 with respective six- and seven-membered D-rings would project in the pseudo-axial orientation, whereas compound 7 with five-membered D-ring would have the lone electron pair directing in pseudo-equatorial position. These results strongly supported the proposal that the axial orientation of the lone electron pair on nitrogen would provide sufficient binding abilities to the opioid receptor and that the 15–16 ethylene moiety in the morphine structure would play a role in fixation of the lone electron pair in the axial direction rather than interaction with the putative cavity in the Beckett–Casy model.Novel naltrexone derivatives 7 and 8 were synthesized to examine the Beckett–Casy model.
Co-reporter:Hiroshi Nagase, Toru Nemoto, Ayaka Matsubara, Manabu Saito, Naoshi Yamamoto, Yumiko Osa, Shigeto Hirayama, Mayumi Nakajima, Kaoru Nakao, Hidenori Mochizuki, Hideaki Fujii
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 21) pp:6302-6305
Publication Date(Web):1 November 2010
DOI:10.1016/j.bmcl.2010.08.083
We have reported previously the novel δ-opioid agonist, SN-28, which was more potent in in vitro assays than the prototype δ-agonists, TAN-67 and SNC-80. However, when administered by subcutaneous injection, this compound showed no analgesic effect at dosages greater than 30 mg/kg in the acetic acid writhing test. We speculated that SN-28 was not effective in the test because the presence of the charged ammonium groups prevented its penetration through the blood–brain barrier. On the basis of our proposal, we designed the novel δ-agonist, KNT-127, which was effective with systemic administration.We designed novel δ-agonists, effective in systemic administration. One of the agonists, KNT-127, is expected to be a useful tool to clarify the real pharmacological effects mediated via the δ-receptor including analgesia and addiction.
Co-reporter:Hideaki Fujii, Ryo Ogawa, Kenji Ohata, Toru Nemoto, Mayumi Nakajima, Ko Hasebe, Hidenori Mochizuki, Hiroshi Nagase
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 16) pp:5983-5988
Publication Date(Web):15 August 2009
DOI:10.1016/j.bmc.2009.06.067
Aerobic oxidation of indolomorphinan 1 without a 4,5-epoxy bridge proceeded in the presence of platinum catalyst to give indoleninomorphinan 2 or quinolono-C-normorphinan 5. The 4-hydroxy group would play an important role in deciding the course of the reaction. Treatment of 2a with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) gave spiroindolinonyl-C-normorphinan 3a whose structure resembles that of δ opioid receptor agonist spiroindanyloxymorphone (SIOM). Boron trichloride was effective for the reverse reaction from 3a to 2a without side reaction. This practical interconversion method between hydroxyindolenine and spiroindolinone would be useful for the design and construction of drug-like compound libraries. Although the compound 3b was expected to show the selectivity for δ opioid receptor because of the structural resemblance to SIOM, it was rather selective for μ opioid receptor (μ: Ki = 0.75 nM; δ: Ki = 2.90 nM; κ: Ki = 13.4 nM). The result suggests that the slight difference of the spatial location of the benzene rings in these compounds may definitively affect the binding affinity for δ opioid receptor.Indolomorphinan without 4,5-epoxy bridge 1a was easily oxidized to give indoleninomorphinan 2a. Practical interconversion between indoleninomorphinan 2a and spiroindolinonyl-C-normorphinan 3a became possible.
Co-reporter:Hiroshi Nagase, Yumiko Osa, Toru Nemoto, Hideaki Fujii, Masayuki Imai, Takashi Nakamura, Toshiyuki Kanemasa, Akira Kato, Hiroaki Gouda, Shuichi Hirono
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 10) pp:2792-2795
Publication Date(Web):15 May 2009
DOI:10.1016/j.bmcl.2009.03.099
We re-examined the accessory site of the 4,5-epoxymorphinan skeleton by camdas conformational analysis in an effort to deign novel δ opioid receptor antagonists. We synthesized three novel compounds (SN-11, 23 and 28) with a 10-methylene bridge and without a 4,5-epoxy ring. Among them, compounds SN-23 (17-isobutyl derivative) and SN-28 (17-methyl derivative) showed very strong agonist activity (over 10 times more than TAN-67). SN-28 also showed high δ selectivity. The δ agonist activity of SN-23 was weaker than that of SN-28, but in terms of the δ selectivity, SN-23 was higher than that of SN-28. These unexpected results indicated that the 4,5-epoxy ring, but not the 10-methylene bridge, was an accessory site in δ opioid receptor agonists.An accessory site of NTI would be only 4,5-epoxy ring. SN-23 and 28 with 10-methylene bridge showed satisfactory δ selectivities and potent agonist activities.
Co-reporter:Akio Watanabe, Hideaki Fujii, Mayumi Nakajima, Ko Hasebe, Hidenori Mochizuki, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 9) pp:2416-2419
Publication Date(Web):1 May 2009
DOI:10.1016/j.bmcl.2009.03.068
An attempt to prepare a trimer having the 1,3,5-trioxazatriquinane skeleton led to discovery of a novel rearrangement reaction that afforded a compound with an oxabicyclo[3.2.1]octane skeleton whose reaction mechanism was proposed. On the basis of this mechanism, we synthesized the rearranged product from a dimethyl acetal intermediate in excellent yield. The compound with an oxabicyclo[3.2.1]octane skeleton showed high affinity for μ and κ but not δ opioid receptor types. The compound expected to be a key intermediate for novel κ selective ligands.Novel morphinan derivative having oxabicyclo[3.2.1]octane skeleton was prepared from dimethyl acetal derivative. The novel compound showed strong affinity for μ and κ opioid receptor types.
Co-reporter:Hideaki Fujii, Akio Watanabe, Toru Nemoto, Minoru Narita, Kan Miyoshi, Atsushi Nakamura, Tsutomu Suzuki, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2009 19(2) pp: 438-441
Publication Date(Web):
DOI:10.1016/j.bmcl.2008.11.050
Co-reporter:Hideaki Fujii, Yoshikazu Watanabe, Yumiko Osa, Toru Nemoto, Noriko Sato, Hiroshi Nagase
Tetrahedron 2009 65(25) pp: 4808-4813
Publication Date(Web):
DOI:10.1016/j.tet.2009.04.055
Co-reporter:Hideaki Fujii, Kayoko Okada, Marina Ishihara, Shinichi Hanamura, Yumiko Osa, Toru Nemoto, Hiroshi Nagase
Tetrahedron 2009 65(51) pp: 10623-10630
Publication Date(Web):
DOI:10.1016/j.tet.2009.10.064
Co-reporter:Satoshi Sakami ; Masayuki Maeda ; Koji Kawai ; Takumi Aoki ; Kuniaki Kawamura ; Hideaki Fujii ; Ko Hasebe ; Mayumi Nakajima ; Takashi Endo ; Shinya Ueno ; Tsuyoshi Ito ; Junzo Kamei
Journal of Medicinal Chemistry 2008 Volume 51(Issue 15) pp:4404-4411
Publication Date(Web):July 19, 2008
DOI:10.1021/jm701440h
We have previously reported antitussive effects of naltrindole (NTI), a typical δ opioid receptor antagonist, in a rat model. The ED50 values of NTI by intraperitoneal and peroral injections were 104 μg/kg and 1840 μg/kg, respectively, comparable to those of codeine. Codeine, one of the most reliable centrally acting antitussive drugs, has μ agonist activity and thus the same side effects as morphine, e.g., constipation, dependency, and respiratory depression. Because NTI is a δ opioid antagonist, its derivatives have potential as highly potent antitussives, free from the μ opioid agonist side effects. We attempted to optimize the NTI derivatives to develop novel antitussive agents. On the basis of the studies of structure−antitussive activity relationships of alkyl substituted NTI derivatives, we designed NTI derivatives with extra ring fused structures. As a clinical candidate, we identified a highly potent new compound, (5R,9R,13S,14S)-17-cyclopropylmethyl-6,7-didehydro-4,5-epoxy-5′,6′-dihydro-3-methoxy-4′H-pyrrolo[3,2,1-ij]quinolino[2′,1′:6,7]morphinan-14-ol (5b) methanesulfonate (TRK-850) which was effective even by oral administration (ED50 6.40 μg/kg).
Co-reporter:Toru Nemoto, Hideaki Fujii, Minoru Narita, Kan Miyoshi, Atsushi Nakamura, Tsutomu Suzuki, Hiroshi Nagase
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 8) pp:4304-4312
Publication Date(Web):15 April 2008
DOI:10.1016/j.bmc.2008.02.082
A modification of the message site in the skeleton of naltrexone was carried out to improve the potency and selectivity of the compound for an opioid receptor subtype. In the course of conversion, we synthesized 7-membered ring ether derivatives, which had an inserted OCH2 group between 4- and 6-positions of morphinan skeleton. One of the 7-membered ring ether derivatives possessed more potent antagonistic activity than naltrexone for the μ opioid receptor. Another compound possessing 17-methyl group derived from noroxycodone may be a μ opioid receptor partial agonist and showed analgesic activity. We are currently examining the subtype selectivity of these compounds.We succeeded in syntheses of 4,6′-epoxymorphinan derivatives. The 4,6′-epoxymorphinan skeleton could be an alternative message site.
Co-reporter:Hideaki Fujii, Yumiko Osa, Marina Ishihara, Shinichi Hanamura, Toru Nemoto, Mayumi Nakajima, Ko Hasebe, Hidenori Mochizuki, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 18) pp:4978-4981
Publication Date(Web):15 September 2008
DOI:10.1016/j.bmcl.2008.08.019
Selective ring opening reaction of the N-cyclopropylmethyl group in naltrexone (1d) was effected in the presence of platinum (IV) oxide and hydrobromic acid under a hydrogen atmosphere at rt to selectively afford N-isobutyl derivative 10. The binding affinity of N-i-Bu derivative 10 for opioid receptors was 11–17 times less than that of the corresponding N-CPM compound, naltrexone (1d). However, compound 10 showed dose-dependent analgesic effects. Contrary to expectations based on previous structure–activity relationship studies for a series of N-substituted naltrexone derivatives that compound 10 would be an opioid antagonist, 10 showed dose-dependent analgesia in the mouse acetic acid writhing test (ED50: 5.05 mg/kg, sc), indicating it was an opioid agonist. This finding may have a great influence on the drug design of opioid agonists.
Co-reporter:Toru Nemoto, Hideaki Fujii, Minoru Narita, Kan Miyoshi, Atsushi Nakamura, Tsutomu Suzuki, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 24) pp:6398-6401
Publication Date(Web):15 December 2008
DOI:10.1016/j.bmcl.2008.10.081
A novel 6,14-epoxymorphinan benzamide derivative (NS22) was synthesized, which showed opioid κ receptor agonistic activity in the [35S]GTPγS binding assay. The antinociceptive effect of NS22 was evaluated in the tail-flick and the hot-plate test. This compound showed a potent antinociceptive activity in mice by sc administration, which was attenuated with nor-BNI (selective opioid κ receptor antagonist).The antinociceptive effect of NS22 was evaluated in the tail-flick and the hot-plate test. NS22 showed a potent antinociceptive activity in mice (sc), which was attenuated with nor-BNI.

![[2-(4-METHOXY-PHENYL)-2-OXO-ETHYL]-CARBAMIC ACID TERT-BUTYL ESTER](http://img.cochemist.com/ccimg/778700/778617-61-1.png)
![[2-(4-METHOXY-PHENYL)-2-OXO-ETHYL]-CARBAMIC ACID TERT-BUTYL ESTER](http://img.cochemist.com/ccimg/778700/778617-61-1_b.png)
![Carbamic acid, [2-(4-methoxyphenyl)-2-oxoethyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/178900/178888-27-2.png)
![Carbamic acid, [2-(4-methoxyphenyl)-2-oxoethyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/178900/178888-27-2_b.png)
![Ethanone, 2-[bis(phenylmethyl)amino]-1-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/140500/140420-07-1.png)
![Ethanone, 2-[bis(phenylmethyl)amino]-1-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/140500/140420-07-1_b.png)


![2-[benzyl(methyl)amino]-1-(4-methoxyphenyl)ethanone](http://img.cochemist.com/ccimg/109000/108976-12-1.png)
![2-[benzyl(methyl)amino]-1-(4-methoxyphenyl)ethanone](http://img.cochemist.com/ccimg/109000/108976-12-1_b.png)
![1H-Indole, 3-acetyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/104200/104142-24-7.png)
![1H-Indole, 3-acetyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/104200/104142-24-7_b.png)
![4-(2-CHLORO-11H-DIBENZO[B,E][1,4]OXATHIEPIN-11-YL)-1-METHYLPIPERI<WBR />DINE (2E)-2-BUTENEDIOATE (1:1)](http://img.cochemist.com/ccimg/72800/72782-05-9.png)
![4-(2-CHLORO-11H-DIBENZO[B,E][1,4]OXATHIEPIN-11-YL)-1-METHYLPIPERI<WBR />DINE (2E)-2-BUTENEDIOATE (1:1)](http://img.cochemist.com/ccimg/72800/72782-05-9_b.png)


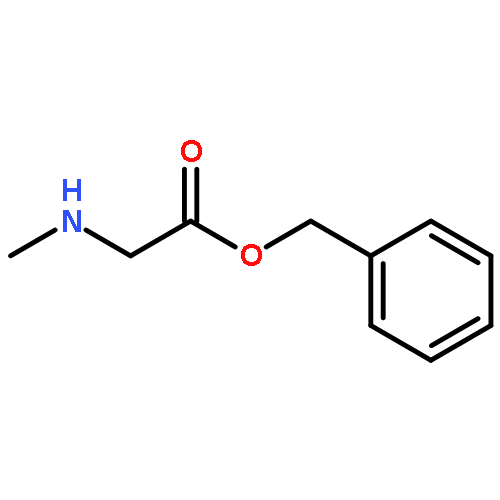
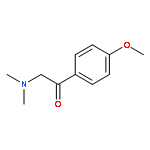
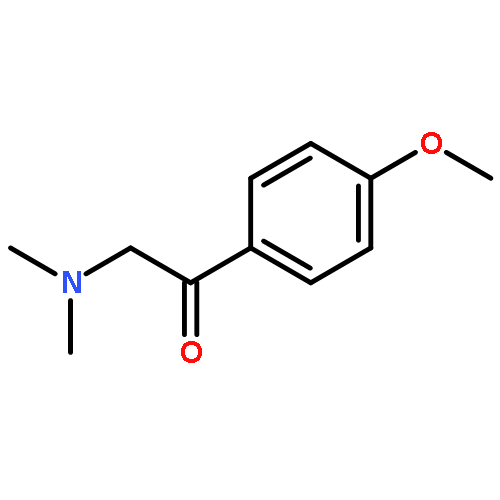
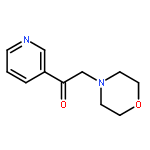
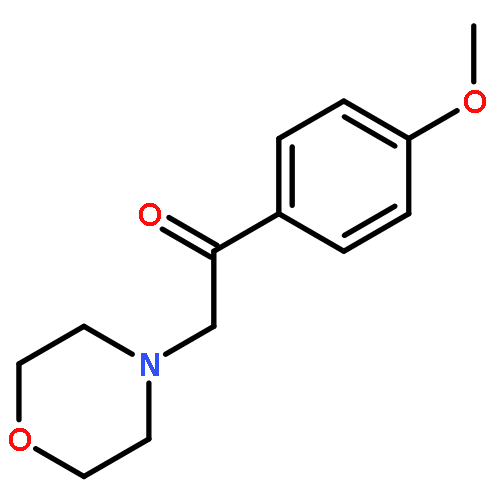
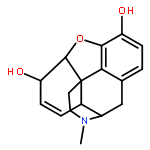
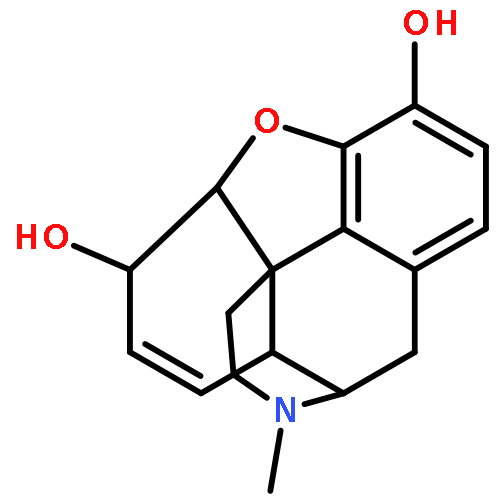
![4,5-epoxy-3-methoxy-17-methyl-[9,16-14C]morphin-7-en-6-ol](http://img.cochemist.com/ccimg/16300/16206-70-5.png)
![4,5-epoxy-3-methoxy-17-methyl-[9,16-14C]morphin-7-en-6-ol](http://img.cochemist.com/ccimg/16300/16206-70-5_b.png)




![N-[2-(4-METHOXYPHENYL)-2-OXOETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/3800/3755-89-3.png)
![N-[2-(4-METHOXYPHENYL)-2-OXOETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/3800/3755-89-3_b.png)