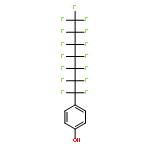
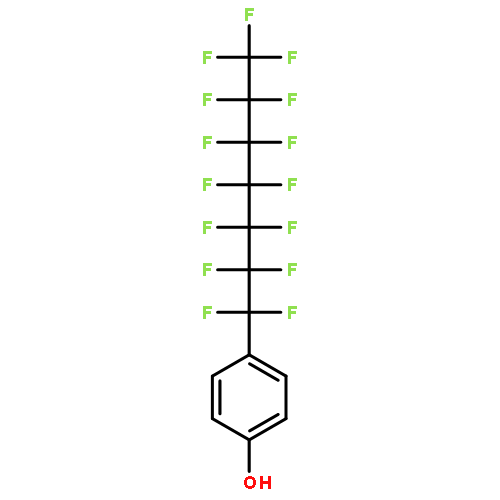
Co-reporter:Dominic J. Pascoe, Kenneth B. Ling, and Scott L. Cockroft
Journal of the American Chemical Society October 25, 2017 Volume 139(Issue 42) pp:15160-15160
Publication Date(Web):October 6, 2017
DOI:10.1021/jacs.7b08511
Favorable molecular interactions between group 16 elements have been implicated in catalysis, biological processes, and materials and medicinal chemistry. Such interactions have since become known as chalcogen bonds by analogy to hydrogen and halogen bonds. Although the prevalence and applications of chalcogen-bonding interactions continues to develop, debate still surrounds the energetic significance and physicochemical origins of this class of σ-hole interaction. Here, synthetic molecular balances were used to perform a quantitative experimental investigation of chalcogen-bonding interactions. Over 160 experimental conformational free energies were measured in 13 different solvents to examine the energetics of O···S, O···Se, S···S, O···HC, and S···HC contacts and the associated substituent and solvent effects. The strongest chalcogen-bonding interactions were found to be at least as strong as conventional H-bonds, but unlike H-bonds, surprisingly independent of the solvent. The independence of the conformational free energies on solvent polarity, polarizability, and H-bonding characteristics showed that electrostatic, solvophobic, and van der Waals dispersion forces did not account for the observed experimental trends. Instead, a quantitative relationship between the experimental conformational free energies and computed molecular orbital energies was consistent with the chalcogen-bonding interactions being dominated by n → σ* orbital delocalization between a lone pair (n) of a (thio)amide donor and the antibonding σ* orbital of an acceptor thiophene or selenophene. Interestingly, stabilization was manifested through the same acceptor molecular orbital irrespective of whether a direct chalcogen···chalcogen or chalcogen···H–C contact was made. Our results underline the importance of often-overlooked orbital delocalization effects in conformational control and molecular recognition phenomena.
Co-reporter:James A. Cooper;Stefan Borsley;Paul J. Lusby
Chemical Science (2010-Present) 2017 vol. 8(Issue 7) pp:5005-5009
Publication Date(Web):2017/06/26
DOI:10.1039/C7SC01940H
Supramolecular chirality may emerge from self-assembly processes to yield architectures that differ only in the topological arrangement of their constituent parts. Since the properties of the resulting enantiomeric assemblies are identical, purification and characterisation can be challenging. Here, we have examined the hypothesis that the intrinsic chirality of a protein nanopore can be exploited to detect supramolecular chirality. Transient blockages in the ion current flowing through a single membrane-spanning α-haemolysin nanopore were shown to discriminate between M4L6 tetrahedral coordination cages of opposing chiralities. The single-molecule nature of the approach facilitated direct access to the rates of association and dissociation with the nanopore, which allowed the concentrations of the enantiomeric supramolecular assemblies to be determined in situ. Thus, we have established that a protein nanopore can be used to discriminate the chiral topologies of supramolecular assemblies, even when they are too large to fully enter the nanopore.
Co-reporter:Scott L. Cockroft
Chem 2017 Volume 3, Issue 3(Volume 3, Issue 3) pp:
Publication Date(Web):14 September 2017
DOI:10.1016/j.chempr.2017.08.008
In this issue of Chem, Flood and co-workers examine solvent effects on chloride anion binding within a neutral macrocyclic host. An inverse relationship was found between the complexation energies and the dielectric constants of aprotic solvents, revealing the limit at which electrostatic interactions are no longer the dominant contributors to binding.
Co-reporter:Nicholas Dominelli-Whiteley;Dr. James J. Brown;Dr. Kamila B. Muchowska;Dr. Ioulia K. Mati;Dr. Catherine Adam;Dr. Thomas A. Hubbard;Alex Elmi;Dr. Alisdair J. Brown;Dr. Ian A. W. Bell;Dr. Scott L. Cockroft
Angewandte Chemie International Edition 2017 Volume 56(Issue 26) pp:7658-7662
Publication Date(Web):2017/06/19
DOI:10.1002/anie.201703757
AbstractChains of hydrogen bonds such as those found in water and proteins are often presumed to be more stable than the sum of the individual H bonds. However, the energetics of cooperativity are complicated by solvent effects and the dynamics of intermolecular interactions, meaning that information on cooperativity typically is derived from theory or indirect structural data. Herein, we present direct measurements of energetic cooperativity in an experimental system in which the geometry and the number of H bonds in a chain were systematically controlled. Strikingly, we found that adding a second H-bond donor to form a chain can almost double the strength of the terminal H bond, while further extensions have little effect. The experimental observations add weight to computations which have suggested that strong, but short-range cooperative effects may occur in H-bond chains.
Co-reporter:Thomas A. Hubbard, Alisdair J. Brown, Ian A. W. Bell, and Scott L. Cockroft
Journal of the American Chemical Society 2016 Volume 138(Issue 46) pp:15114-15117
Publication Date(Web):November 6, 2016
DOI:10.1021/jacs.6b09130
Hydrogen bonds are ubiquitous interactions in molecular recognition. The energetics of such processes are governed by the competing influences of pre-organization and flexibility that are often hard to predict. Here we have measured the strength of intramolecular interactions between H-bond donor and acceptor sites separated by a variable linker. A striking distance-dependent threshold was observed in the intramolecular interaction energies. H-bonds were worth less than −1 kJ mol–1 when the interacting groups were separated by ≥6 rotating bonds, but ranged between −5 and −9 kJ mol–1 for ≤5 rotors. Thus, only very strong external H-bond acceptors were able to compete with the stronger internal H-bonds. In addition, a constant energetic penalty per rotor of ∼5–6 kJ mol–1 was observed in less strained situations where the molecule contained ≥4 rotatable bonds.
Co-reporter:Dr. Lixu Yang;Dr. John B. Brazier;Thomas A. Hubbard;Dr. David M. Rogers ;Dr. Scott L. Cockroft
Angewandte Chemie International Edition 2016 Volume 55( Issue 3) pp:912-916
Publication Date(Web):
DOI:10.1002/anie.201508056
Abstract
Experimental support for the dominance of van der Waals dispersion forces in aromatic stacking interactions occurring in organic solution is surprisingly limited. The size-dependence of aromatic stacking in an organic solvent was examined. The interaction energy was found to vary by about 7.5 kJ mol−1 on going from a phenyl–phenyl to an anthracene–pyrene stack. Strikingly, the experimental data were highly correlated with dispersion energies determined using symmetry-adapted perturbation theory (SAPT), while the induction, exchange, electrostatic, and solvation energy components correlated poorly. Both the experimental data and the SAPT-dispersion energies gave high-quality correlations with the change in solvent accessible area upon complexation. Thus, the size-dependence of aromatic stacking interactions is consistent with the dominance of van der Waals dispersion forces even in the presence of a competing polarizable solvent.
Co-reporter:Dr. Lixu Yang;Dr. John B. Brazier;Thomas A. Hubbard;Dr. David M. Rogers ;Dr. Scott L. Cockroft
Angewandte Chemie 2016 Volume 128( Issue 3) pp:924-928
Publication Date(Web):
DOI:10.1002/ange.201508056
Abstract
Experimental support for the dominance of van der Waals dispersion forces in aromatic stacking interactions occurring in organic solution is surprisingly limited. The size-dependence of aromatic stacking in an organic solvent was examined. The interaction energy was found to vary by about 7.5 kJ mol−1 on going from a phenyl–phenyl to an anthracene–pyrene stack. Strikingly, the experimental data were highly correlated with dispersion energies determined using symmetry-adapted perturbation theory (SAPT), while the induction, exchange, electrostatic, and solvation energy components correlated poorly. Both the experimental data and the SAPT-dispersion energies gave high-quality correlations with the change in solvent accessible area upon complexation. Thus, the size-dependence of aromatic stacking interactions is consistent with the dominance of van der Waals dispersion forces even in the presence of a competing polarizable solvent.
Co-reporter:Dr. Matthew A. Watson ;Dr. Scott L. Cockroft
Angewandte Chemie 2016 Volume 128( Issue 4) pp:1367-1371
Publication Date(Web):
DOI:10.1002/ange.201508845
Abstract
Biological molecular machines operate far from equilibrium by coupling chemical potential to repeated cycles of dissipative nanomechanical motion. This principle has been exploited in supramolecular systems that exhibit true machine behavior in solution and on surfaces. However, designed membrane-spanning assemblies developed to date have been limited to simple switches or stochastic shuttles, and true machine behavior has remained elusive. Herein, we present a transmembrane nanoactuator that turns over chemical fuel to drive autonomous reciprocating (back-and-forth) nanomechanical motion. Ratcheted reciprocating motion of a DNA/PEG copolymer threaded through a single α-hemolysin pore was induced by a combination of DNA strand displacement processes and enzyme-catalyzed reactions. Ion-current recordings revealed saw-tooth patterns, indicating that the assemblies operated in autonomous, asymmetric cycles of conformational change at rates of up to one cycle per minute.
Co-reporter:Dr. Matthew A. Watson ;Dr. Scott L. Cockroft
Angewandte Chemie International Edition 2016 Volume 55( Issue 4) pp:1345-1349
Publication Date(Web):
DOI:10.1002/anie.201508845
Abstract
Biological molecular machines operate far from equilibrium by coupling chemical potential to repeated cycles of dissipative nanomechanical motion. This principle has been exploited in supramolecular systems that exhibit true machine behavior in solution and on surfaces. However, designed membrane-spanning assemblies developed to date have been limited to simple switches or stochastic shuttles, and true machine behavior has remained elusive. Herein, we present a transmembrane nanoactuator that turns over chemical fuel to drive autonomous reciprocating (back-and-forth) nanomechanical motion. Ratcheted reciprocating motion of a DNA/PEG copolymer threaded through a single α-hemolysin pore was induced by a combination of DNA strand displacement processes and enzyme-catalyzed reactions. Ion-current recordings revealed saw-tooth patterns, indicating that the assemblies operated in autonomous, asymmetric cycles of conformational change at rates of up to one cycle per minute.
Co-reporter:Lixu Yang; Catherine Adam
Journal of the American Chemical Society 2015 Volume 137(Issue 32) pp:10084-10087
Publication Date(Web):July 10, 2015
DOI:10.1021/jacs.5b05736
The hydrophobic effect plays a central role in determining the structure, activity, and properties of biomolecules and materials. In contrast, the general manifestation of this phenomenon in other solvents—the solvophobic effect—although widely invoked, is currently poorly defined because of the lack of a universally accepted descriptor. Here we have used synthetic molecular balances to measure solvent effects on aromatic, aliphatic, and fluorous nonpolar interactions. Our solvent screening data combined with independent experimental measurements of supramolecular association, single-molecule folding, and bulk phase transfer energies were all found to correlate well with the cohesive energy density (ced) of the solvent. Meanwhile, other measures of solvent cohesion, such as surface tension and internal pressure, gave inferior correlations. Thus, we establish ced as a readily accessible, quantitative descriptor of solvophobic association in a range of chemical contexts.
Co-reporter:Matthew A. Watson and Scott L. Cockroft
Chemical Communications 2015 vol. 51(Issue 61) pp:12243-12246
Publication Date(Web):23 Jun 2015
DOI:10.1039/C5CC01563D
Here we investigate the modulation of solvent isotope effects by the entry of DNA molecules into individual α-haemolysin nanopores. Solvent isotope effects in D2O versus H2O were enhanced (kH/kD ≈ 1.6) compared to the bulk (kH/kD ≈ 1.2), except when the pore was most blocked (kH/kD ≤ 1.1).
Co-reporter:Dr. Catherine Adam;Dr. Lixu Yang ;Dr. Scott L. Cockroft
Angewandte Chemie 2015 Volume 127( Issue 4) pp:1180-1183
Publication Date(Web):
DOI:10.1002/ange.201408982
Abstract
Fluorocarbons often have distinct miscibility properties compared to their nonfluorinated analogues. These differences may be attributed to van der Waals dispersion forces or solvophobic effects, but their contributions are notoriously difficult to separate in molecular recognition processes. Here, molecular torsion balances were used to compare cohesive alkyl and perfluoroalkyl interactions in a range of solvents. A simple linear regression enabled the energetic partitioning of solvophobic and van der Waals forces in the self-association of apolar chains. The contributions of dispersion interactions in apolar cohesion were found to be strongly attenuated in solution compared to the gas phase, but still play a major role in fluorous and organic solvents. In contrast, solvophobic effects were found to be dominant in driving the association of apolar chains in aqueous solution. The results are expected to assist the computational modelling of van der Waals forces in solution.
Co-reporter:Dr. Catherine Adam;Dr. Lixu Yang ;Dr. Scott L. Cockroft
Angewandte Chemie International Edition 2015 Volume 54( Issue 4) pp:1164-1167
Publication Date(Web):
DOI:10.1002/anie.201408982
Abstract
Fluorocarbons often have distinct miscibility properties compared to their nonfluorinated analogues. These differences may be attributed to van der Waals dispersion forces or solvophobic effects, but their contributions are notoriously difficult to separate in molecular recognition processes. Here, molecular torsion balances were used to compare cohesive alkyl and perfluoroalkyl interactions in a range of solvents. A simple linear regression enabled the energetic partitioning of solvophobic and van der Waals forces in the self-association of apolar chains. The contributions of dispersion interactions in apolar cohesion were found to be strongly attenuated in solution compared to the gas phase, but still play a major role in fluorous and organic solvents. In contrast, solvophobic effects were found to be dominant in driving the association of apolar chains in aqueous solution. The results are expected to assist the computational modelling of van der Waals forces in solution.
Co-reporter:Lixu Yang, Thomas A. Hubbard and Scott L. Cockroft
Chemical Communications 2014 vol. 50(Issue 40) pp:5212-5214
Publication Date(Web):22 Oct 2013
DOI:10.1039/C3CC46048G
We examine an unusual case where a neutral hydrogen atom acts as a hydrogen-bond acceptor. The association constant between trihexylsilane and perfluoro-tert-butanol was measured as ∼0.8 M−1 in cyclohexane. Computations and experimental NMR data are consistent with a weak, but favourable Si–H⋯HO interaction.
Co-reporter:Kamila B. Muchowska ; Catherine Adam ; Ioulia K. Mati
Journal of the American Chemical Society 2013 Volume 135(Issue 27) pp:9976-9979
Publication Date(Web):June 24, 2013
DOI:10.1021/ja402566w
Solvent effects are implicated as playing a major role in modulating electrostatic interactions via through-space and polarization effects, but these phenomena are often hard to dissect. By using synthetic molecular torsion balances and a simple explicit solvation model, we demonstrate that the solvation of substituents substantially affects the electrostatic potential of aromatic rings. Although polarization effects are important, we show that a simple additive through-space model also provides a reasonable account of the experimental data. The results deliver insights into solvent structure and might contribute to the development of computationally inexpensive solvent models.
Co-reporter:Ioulia K. Mati, Catherine Adam and Scott L. Cockroft
Chemical Science 2013 vol. 4(Issue 10) pp:3965-3972
Publication Date(Web):23 Jul 2013
DOI:10.1039/C3SC51764K
The study of molecular interactions is often complicated by solvent effects. Here we have used a series of 11 synthetic molecular balances to measure solvent and substituent effects on the positions of conformational equilibria in 13 different solvents. Despite the simplicity of the model system, surprisingly complicated behaviour was seen to emerge from the interplay of conformational, intramolecular and solvent effects. Nonetheless, 138 experimental conformational free energies were analysed using a simple solvent model, which was able to account for both the major and more unusual patterns observed. The success of the solvent model can be attributed to its ability to facilitate consideration of individual intramolecular and solute–solvent interactions, as confirmed by comparison with NMR chemical shifts and DFT calculations. The approach provides a means of dissecting electrostatic and solvent effects to reveal pseudo gas-phase behaviour from experimental data obtained in solution. For example, the method facilitated the identification of an unexpected, but highly favourable CO⋯NO2 interaction worth up to 3.6 kJ mol−1, which was shown not to be driven by solvent effects.
Co-reporter:James J. Brown and Scott L. Cockroft
Chemical Science 2013 vol. 4(Issue 4) pp:1772-1780
Publication Date(Web):11 Feb 2013
DOI:10.1039/C3SC50309G
The prediction of reactivity is one of the long-standing objectives of chemistry. We have extracted reactivity patterns observed in aromatic molecules spanning 150 years of synthetic developments and used the data to test the predictive capacity of popular reactivity models. This systematic analysis has exposed numerous regioselectivities that are not predicted by resonance theory, electrostatic potentials or frontier molecular orbital theory. In contrast, calculated local ionisation energy surfaces are shown to consistently reveal the most nucleophilic sites in aromatic molecules even where established reactivity models fail. Furthermore, these local ionisation energy minima are found to correlate with experimentally determined reactivity parameters. Since ionisation energy surfaces are simple to interpret and are provided as standard in popular computational chemistry software, the approach serves as a readily accessible tool for visualising the fundamental factors governing the reactivity of aromatic molecules.
Co-reporter:Ioulia K. Mati and Scott L. Cockroft
Chemical Society Reviews 2010 vol. 39(Issue 11) pp:4195-4205
Publication Date(Web):16 Sep 2010
DOI:10.1039/B822665M
Molecular interactions underlie the whole of chemistry and biology. This tutorial review illustrates the use of rotameric folding molecules, topoisomers, atropoisomers, and tautomers as molecular balances for quantifying non-covalent interactions. This intramolecular approach enables a wide variety of interactions to be examined with a degree of geometric control that is difficult to achieve in supramolecular complexes. Synthetic variation of molecular balances allows the fundamental physicochemical origins of molecular recognition to be systematically examined by providing insights into the interplay of geometry and solvation on non-covalent interactions.
Co-reporter:Long Ma Dr.
ChemBioChem 2010 Volume 11( Issue 1) pp:25-34
Publication Date(Web):
DOI:10.1002/cbic.200900526
Co-reporter:Scott L. Cockroft and Christopher A. Hunter
Chemical Communications 2009 (Issue 26) pp:3961-3963
Publication Date(Web):27 May 2009
DOI:10.1039/B902351H
Experimental measurements of aromatic edge-to-face interaction energies in both molecular torsion balances and supramolecular zipper complexes can be reliably estimated using a simple electrostatic solvation model and α/βH-bond constants.
Co-reporter:Matthew A. Watson and Scott L. Cockroft
Chemical Society Reviews 2016 - vol. 45(Issue 22) pp:NaN6129-6129
Publication Date(Web):2016/03/02
DOI:10.1039/C5CS00874C
Nature's molecular machines are a constant source of inspiration to the chemist. Many of these molecular machines function within lipid membranes, allowing them to exploit potential gradients between spatially close, but chemically distinct environments to fuel their work cycle. Indeed, the realisation of such principles in synthetic transmembrane systems remains a tantalising goal. This tutorial review opens by highlighting seminal examples of synthetic molecular machines. We illustrate the importance of surfaces for facilitating the extraction of work from molecular switches and motors. We chart the development of man-made transmembrane systems; from passive to machine-like stimuli-responsive channels, to fully autonomous transmembrane molecular machines. Finally, we highlight higher-order compartmentalised systems that exhibit emergent properties. We suggest that such higher-order architectures could serve as platforms for sophisticated devices that co-ordinate the activity of numerous transmembrane molecular machines.
Co-reporter:James J. Brown and Scott L. Cockroft
Chemical Science (2010-Present) 2013 - vol. 4(Issue 4) pp:NaN1780-1780
Publication Date(Web):2013/02/11
DOI:10.1039/C3SC50309G
The prediction of reactivity is one of the long-standing objectives of chemistry. We have extracted reactivity patterns observed in aromatic molecules spanning 150 years of synthetic developments and used the data to test the predictive capacity of popular reactivity models. This systematic analysis has exposed numerous regioselectivities that are not predicted by resonance theory, electrostatic potentials or frontier molecular orbital theory. In contrast, calculated local ionisation energy surfaces are shown to consistently reveal the most nucleophilic sites in aromatic molecules even where established reactivity models fail. Furthermore, these local ionisation energy minima are found to correlate with experimentally determined reactivity parameters. Since ionisation energy surfaces are simple to interpret and are provided as standard in popular computational chemistry software, the approach serves as a readily accessible tool for visualising the fundamental factors governing the reactivity of aromatic molecules.
Co-reporter:Ioulia K. Mati, Catherine Adam and Scott L. Cockroft
Chemical Science (2010-Present) 2013 - vol. 4(Issue 10) pp:NaN3972-3972
Publication Date(Web):2013/07/23
DOI:10.1039/C3SC51764K
The study of molecular interactions is often complicated by solvent effects. Here we have used a series of 11 synthetic molecular balances to measure solvent and substituent effects on the positions of conformational equilibria in 13 different solvents. Despite the simplicity of the model system, surprisingly complicated behaviour was seen to emerge from the interplay of conformational, intramolecular and solvent effects. Nonetheless, 138 experimental conformational free energies were analysed using a simple solvent model, which was able to account for both the major and more unusual patterns observed. The success of the solvent model can be attributed to its ability to facilitate consideration of individual intramolecular and solute–solvent interactions, as confirmed by comparison with NMR chemical shifts and DFT calculations. The approach provides a means of dissecting electrostatic and solvent effects to reveal pseudo gas-phase behaviour from experimental data obtained in solution. For example, the method facilitated the identification of an unexpected, but highly favourable CO⋯NO2 interaction worth up to 3.6 kJ mol−1, which was shown not to be driven by solvent effects.
Co-reporter:James A. Cooper, Stefan Borsley, Paul J. Lusby and Scott L. Cockroft
Chemical Science (2010-Present) 2017 - vol. 8(Issue 7) pp:NaN5009-5009
Publication Date(Web):2017/05/11
DOI:10.1039/C7SC01940H
Supramolecular chirality may emerge from self-assembly processes to yield architectures that differ only in the topological arrangement of their constituent parts. Since the properties of the resulting enantiomeric assemblies are identical, purification and characterisation can be challenging. Here, we have examined the hypothesis that the intrinsic chirality of a protein nanopore can be exploited to detect supramolecular chirality. Transient blockages in the ion current flowing through a single membrane-spanning α-haemolysin nanopore were shown to discriminate between M4L6 tetrahedral coordination cages of opposing chiralities. The single-molecule nature of the approach facilitated direct access to the rates of association and dissociation with the nanopore, which allowed the concentrations of the enantiomeric supramolecular assemblies to be determined in situ. Thus, we have established that a protein nanopore can be used to discriminate the chiral topologies of supramolecular assemblies, even when they are too large to fully enter the nanopore.
Co-reporter:Ioulia K. Mati and Scott L. Cockroft
Chemical Society Reviews 2010 - vol. 39(Issue 11) pp:NaN4205-4205
Publication Date(Web):2010/09/16
DOI:10.1039/B822665M
Molecular interactions underlie the whole of chemistry and biology. This tutorial review illustrates the use of rotameric folding molecules, topoisomers, atropoisomers, and tautomers as molecular balances for quantifying non-covalent interactions. This intramolecular approach enables a wide variety of interactions to be examined with a degree of geometric control that is difficult to achieve in supramolecular complexes. Synthetic variation of molecular balances allows the fundamental physicochemical origins of molecular recognition to be systematically examined by providing insights into the interplay of geometry and solvation on non-covalent interactions.
Co-reporter:Scott L. Cockroft and Christopher A. Hunter
Chemical Communications 2009(Issue 26) pp:NaN3963-3963
Publication Date(Web):2009/05/27
DOI:10.1039/B902351H
Experimental measurements of aromatic edge-to-face interaction energies in both molecular torsion balances and supramolecular zipper complexes can be reliably estimated using a simple electrostatic solvation model and α/βH-bond constants.
Co-reporter:Matthew A. Watson and Scott L. Cockroft
Chemical Communications 2015 - vol. 51(Issue 61) pp:NaN12246-12246
Publication Date(Web):2015/06/23
DOI:10.1039/C5CC01563D
Here we investigate the modulation of solvent isotope effects by the entry of DNA molecules into individual α-haemolysin nanopores. Solvent isotope effects in D2O versus H2O were enhanced (kH/kD ≈ 1.6) compared to the bulk (kH/kD ≈ 1.2), except when the pore was most blocked (kH/kD ≤ 1.1).
Co-reporter:Lixu Yang, Thomas A. Hubbard and Scott L. Cockroft
Chemical Communications 2014 - vol. 50(Issue 40) pp:NaN5214-5214
Publication Date(Web):2013/10/22
DOI:10.1039/C3CC46048G
We examine an unusual case where a neutral hydrogen atom acts as a hydrogen-bond acceptor. The association constant between trihexylsilane and perfluoro-tert-butanol was measured as ∼0.8 M−1 in cyclohexane. Computations and experimental NMR data are consistent with a weak, but favourable Si–H⋯HO interaction.














![Ethanol, 2,2'-[1,5-naphthalenediylbis(oxy-2,1-ethanediyloxy-2,1-ethanediyloxy-2,1-ethanediyloxy)]bis-](/data/chemimg/103300/168211-64-1.png)
![Ethanol, 2,2'-[1,5-naphthalenediylbis(oxy-2,1-ethanediyloxy-2,1-ethanediyloxy-2,1-ethanediyloxy)]bis-](/data/chemimg/103300/168211-64-1_b.png)