Co-reporter:Lisa M. Doyle, Shane O’Sullivan, Claudia Di Salvo, Michelle McKinney, Patrick McArdle, and Paul V. Murphy
Organic Letters November 3, 2017 Volume 19(Issue 21) pp:5802-5802
Publication Date(Web):October 17, 2017
DOI:10.1021/acs.orglett.7b02760
Glycosyl thiols are widely used in stereoselective S-glycoside synthesis. Their epimerization from 1,2-trans to 1,2-cis thiols (e.g., equatorial to axial epimerization in thioglucopyranose) was attained using TiCl4, while SnCl4 promoted their axial-to-equatorial epimerization. The method included application for stereoselective β-d-manno- and β-l-rhamnopyranosyl thiol formation. Complex formation explains the equatorial preference when using SnCl4, whereas TiCl4 can shift the equilibrium toward the 1,2-cis thiol via 1,3-oxathiolane formation.
Co-reporter:Anthony W. McDonagh, Mary F. Mahon, and Paul V. Murphy
Organic Letters 2016 Volume 18(Issue 3) pp:552-555
Publication Date(Web):January 22, 2016
DOI:10.1021/acs.orglett.5b03591
The TiCl4 induced anomerization of selenium glycosides of galacturonic acid derivatives is reported. The reaction was successful for galacturonic acid when various alkyl, alkenyl, alkynyl, saccharide, steroid, and lipid groups were attached to the anomeric Se atom. An increased amount of TiCl4 and/or higher temperature were needed to ensure completion of the reaction in some cases. Yields were higher for reactions carried out at higher dilution. The reaction was applied to the synthesis of Se-based mimics of the potent immunostimulant α-GalCer (KRN7000).
Co-reporter:Sinclair M. Sweeney, Gemma A. Bullen, Richard B. Gillis, Gary G. Adams, Arthur J. Rowe, Stephen E. Harding, James H.R. Tucker, Anna F.A. Peacock, Paul V. Murphy
Tetrahedron Letters 2016 Volume 57(Issue 13) pp:1414-1417
Publication Date(Web):30 March 2016
DOI:10.1016/j.tetlet.2016.02.005
Scaffold design, synthesis and application are relevant for biomedical research. For example, multivalent interactions, such as those between cell surface glycoproteins and lectins can influence the potency and duration of signalling. The spacing between carbohydrates on their native protein scaffold could be important. Herein, the coiled coil design principle is used to generate synthetic coiled coil type glycoproteins, where three lactose residues are grafted to the coil via N-linkages to asparagine. Molecular modelling indicates that the distance between the galactose anomeric carbon atoms on the neoglycoproteins is ∼30 Å. The inclusion of lactose was accommodated in both the final heptad towards the N-terminus, or more centrally in the penultimate heptad. In either case, neither the helicity nor the assembly to the trimeric form was unduly altered by the presence of the disaccharide.
Co-reporter:Lorna Moynihan, Rekha Chadda, Patrick McArdle, and Paul V. Murphy
Organic Letters 2015 Volume 17(Issue 24) pp:6226-6229
Publication Date(Web):December 9, 2015
DOI:10.1021/acs.orglett.5b03209
Allylic azide rearrangement is used in tandem with intramolecular azide–alkene cycloaddition to give a triazoline that when subsequently decomposed in the presence of a nucleophile gives piperidines. The tandem reaction gives two stereocenters that are generated with high control. The formation of the piperidines required the presence of innate conformational constraint. The applicability of the annulation reaction is demonstrated by the synthesis of iminosugars. A proposal is included to account for the observed stereoselectivity, which is influenced by the precursor structure.
Co-reporter:Jian Zhou, Mairead Reidy, Ciaran O’Reilly, Dilip V. Jarikote, Arvind Negi, Afshin Samali, Eva Szegezdi, and Paul V. Murphy
Organic Letters 2015 Volume 17(Issue 7) pp:1672-1675
Publication Date(Web):March 16, 2015
DOI:10.1021/acs.orglett.5b00404
A build–couple–pair strategy, including double-reductive amination macrocyclization, has been used to generate decorated macrocycles (eannaphanes) with an embedded triazole and monosaccharide. Biological screening led to the identification of an inducer of apoptosis in leukemic cells, which acts at least partially as a 5-HT2 antagonist.
Co-reporter:Sabine André, Shane O'Sullivan, Christiane Koller, Paul V. Murphy and Hans-Joachim Gabius
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 14) pp:4190-4203
Publication Date(Web):17 Feb 2015
DOI:10.1039/C5OB00048C
Emerging insights into the functional spectrum of tissue lectins leads to identification of new targets for the custom-made design of potent inhibitors, providing a challenge for synthetic chemistry. The affinity and selectivity of a carbohydrate ligand for a lectin may immensely be increased by a number of approaches, which includes varying geometrical or topological features. This perspective leads to the design and synthesis of glycoclusters and their testing using assays of physiological relevance. Herein, hydroquinone, resorcinol, benzene-1,3,5-triol and tetra(4-hydroxyphenyl)ethene have been employed as scaffolds and propargyl derivatives obtained. The triazole-containing linker to the α/β-O/S-glycosides of GlcNAc/GalNAc presented on these scaffolds was generated by copper-catalysed azide–alkyne cycloaddition. This strategy was used to give a panel of nine glycoclusters with bi-, tri- and tetravalency. Maintained activity for lectin binding after conjugation was ascertained for both sugars in solid-phase assays with the plant agglutinins WGA (GlcNAc) and DBA (GalNAc). Absence of cross-reactivity excluded any carbohydrate-independent reactivity of the bivalent compounds, allowing us to proceed to further testing with a biomedically relevant lectin specific for GalNAc. Macrophage galactose(-binding C)-type lectin, involved in immune defence by dendritic cells and in virus uptake, was produced as a soluble protein without/with its α-helical coiled-coil stalk region. Binding to ligands presented on a matrix and on cell surfaces was highly susceptible to the presence of the tetravalent inhibitor derived from the tetraphenylethene-containing scaffold, and presentation of GalNAc with an α-thioglycosidic linkage proved favorable. Cross-reactivity of this glycocluster to human galectins-3 and -4, which interact with Tn-antigen-presenting mucins, was rather small. Evidently, the valency and spatial display of α-GalNAc residues is a key factor to design potent and selective inhibitors for this lectin.
Co-reporter:Dr. Daniele LoRe;Dr. Ying Zhou;Dr. Joanna Mucha;Dr. Leigh F. Jones;Lorraine Leahy; Corrado Santocanale; Magdalena Krol; Paul V. Murphy
Chemistry - A European Journal 2015 Volume 21( Issue 50) pp:
Publication Date(Web):
DOI:10.1002/chem.201585001
Co-reporter:Daniele LoRe;Ying Zhou;Joanna Mucha;Leigh F. Jones;Lorraine Leahy;Corrado Santocanale;Magdalena Krol
Chemistry - A European Journal 2015 Volume 21( Issue 50) pp:
Publication Date(Web):
DOI:10.1002/chem.201504167
Abstract
Invited for the cover of this issue are Paul V. Murphy and co-workers at the National University of Ireland Galway (NUI Galway) and Warsaw University. The image depicts MGSTA-6 giving a stop signal to tumour cells that are on the move. Read the full text of the article at 10.1002/chem.201502861.
Co-reporter:Dr. Daniele LoRe;Dr. Ying Zhou;Dr. Joanna Mucha;Dr. Leigh F. Jones;Lorraine Leahy; Corrado Santocanale; Magdalena Krol; Paul V. Murphy
Chemistry - A European Journal 2015 Volume 21( Issue 50) pp:18109-18121
Publication Date(Web):
DOI:10.1002/chem.201502861
Abstract
Migrastatin and isomigrastatin analogues have been synthesised in order to contribute to structure–activity studies on tumour cell migration inhibitors. These include macrocycles varying in ring size, functionality and alkene stereochemistry, as well as glucuronides. The synthesis work included application of the Saegusa–Ito reaction for regio- and stereoselective unsaturated macroketone formation, diastereoselective Brown allylation to generate 9-methylmigrastatin analogues and chelation-induced anomerisation to vary glucuronide configuration. Compounds were tested in vitro against both breast and pancreatic cancer cell lines and inhibition of tumour cell migration was observed in both wound-healing (scratch) and Boyden chamber assays. One unsaturated macroketone showed low affinity for a range of secondary drug targets, indicating it is at low risk of displaying adverse side effects.
Co-reporter:Sabine André, Shane O'Sullivan, Hans-Joachim Gabius, Paul V. Murphy
Tetrahedron 2015 Volume 71(Issue 38) pp:6867-6880
Publication Date(Web):23 September 2015
DOI:10.1016/j.tet.2015.07.020
The emerging physiological significance of carbohydrate (glycan)–protein (lectin) recognition engenders the interest to design synthetic inhibitors with a high level of selectivity among natural sugar receptors. Plant agglutinins are common models to determine structure–activity relationships. Focussing on the contribution of valency towards selectivity, copper-catalysed azide (sugar derivative)–alkyne (scaffold) cycloaddition yielded a panel of 10 bi- to tetravalent glycoclusters with N-acetylglucosamine as the bioactive headgroup. They were introduced into assays using (neo)glycoproteins and cell surfaces as platforms to study carbohydrate-dependent lectin binding. The ability of the bivalent compounds, which exhibit a distance profile of the sugar headgroups of about 16–21 Å, for intramolecular bridging of two contact sites from the eight hevein domains of wheat germ agglutinin led to comparatively high enhancements of inhibitory potency relative to a tetrameric leguminous lectin (distance profile of 50–70 Å between sugar-specific sites), especially for a β-S-glycoside. The extent of inhibition at fixed concentrations of the sugar depended on the type of matrix used for the assay. Increases to tri- and tetravalency played a less important role than the anomeric position to keep cross-reactivity low, these tested topologies enabling cross-linking for both lectins. The potential for cis-interactions (intramolecular interactions), with glycoclusters serving as molecular rulers, is suggested to help designing selective blocking reagents.
Co-reporter:Sabine André, Guan-Nan Wang, Hans-Joachim Gabius, Paul V. Murphy
Carbohydrate Research 2014 Volume 389() pp:25-38
Publication Date(Web):7 May 2014
DOI:10.1016/j.carres.2013.12.024
•Synthesis of glycoclusters presenting lactose and 2′-fucosyl lactose on different scaffolds.•Engineering of galectin-4 mutants to probe structure–activity relationships with glycoclusters.•The impact of structural alterations by combining application of glycoclusters and mutants.Complementarity in lectin–glycan interactions in situ is assumed to involve spatial features in both the lectin and the glycan, giving a functional meaning to structural aspects of the lectin beyond its carbohydrate-binding site. In combining protein engineering with glycocluster synthesis, it is shown that the natural linker length of a tandem-repeat-type human lectin (galectin-4) determines binding properties in two binding assays (using surface-presented glycoprotein and cell surface assays). The types of glycocluster tested included bivalent lactosides based on tertiary amides of terephthalic, isophthalic, 2,6-naphthalic and oxalic acids as well as bivalent H(type 2) trisaccharides grafted on secondary/tertiary terephthalamides and two triazole-linker-containing cores. The presented data reveal a marked change in susceptibility to the test compounds when turning the tandem-repeat-type to a proto-type-like display. The testing of glycoclusters is suggested as a general strategy to help to delineate the significance of distinct structural features of lectins beyond their contact sites to the glycan.Graphical abstract
Co-reporter:Stephen Barron and Paul V. Murphy
MedChemComm 2014 vol. 5(Issue 8) pp:1150-1158
Publication Date(Web):20 Mar 2014
DOI:10.1039/C4MD00074A
The synthesis of 1-deoxynojirimycin (DNJ) derivatives presenting a 2-naphthylmethyl and an alkyl amino side chain from L-sorbose is described. The synthetic derivatives were tested for their ability to inhibit the binding of somatostatin-14 to human recombinant somatostatin receptors (hSSTRs). One DNJ derivative showed selective binding for hSSTR5 over hSSTR4. The presence of benzyl groups and acetates on the oxygen atoms of the iminosugar scaffold led to increased affinity for both hSSTR5 and hSSTR4. Ligand-lipophilicity efficiencies (LLEs) are calculated for the iminosugar derivatives. The LLE values are significantly higher for iminosugar derivatives where hydroxyl groups are not protected, as compared to where they are benzylated. This indicates that leaving hydroxyl groups free or avoiding the use of multiple benzyl groups could be important for drug discovery research based on sugar scaffolds.
Co-reporter:Dr. Daniele Lo Re;Dr. Ying Zhou;Max Nobis; Kurt I. Anderson; Paul V. Murphy
ChemBioChem 2014 Volume 15( Issue 10) pp:1459-1464
Publication Date(Web):
DOI:10.1002/cbic.201402061
Abstract
An efficient and scalable synthesis of a key acyclic intermediate used for the preparation of migrastatin and its macroketone analogue is described; Brown alkoxyallylation is the key step for this synthesis. The macroketone was prepared on 100 mg scale by this route. Treatment of invasive pancreatic cancer cells grown on a cell-derived matrix or as subcutaneous tumours in nude mice with the macroketone inhibited E-cadherin dynamics in a manner consistent with increased cell adhesion and reduced invasive potential.
Co-reporter:Anthony W. McDonagh, Paul V. Murphy
Tetrahedron 2014 70(19) pp: 3191-3196
Publication Date(Web):
DOI:10.1016/j.tet.2014.03.029
Co-reporter:Mark Farrell;Dr. Jian Zhou ;Dr. Paul V. Murphy
Chemistry - A European Journal 2013 Volume 19( Issue 44) pp:14836-14851
Publication Date(Web):
DOI:10.1002/chem.201302572
Abstract
Chelation induced anomerisation is promoted when Lewis acids, such as TiCl4 or SnCl4, coordinate to the pyranose ring oxygen atom and another site, giving rise to endocyclic cleavage and isomerisation to the more stable anomer. In this research regiospecific site-directed anomerisation is demonstrated. TiCl4 (2.5 equiv) was employed to induce anomerisation of 15 glycosyl azide and disaccharide substrates of low reactivity, and high yields (>75 %) and stereoselectivies (α/β>9:1) were achieved. The examples included glucopyranuronate, galactopyranuronate and mannopyranuronate as well as N-acetylated glucopyranuronate and galactopyranuronate derivatives. A disaccharide with the α14 linkage found in polygalacturonan was included. The use of benzoylated saccharides was found to be important in disaccharide anomerisation as attempts to isomerise related acetyl protected and 2,3-carbonate protected derivatives were not successful.
Co-reporter:Guan-Nan Wang, Sabine André, Hans-Joachim Gabius and Paul V. Murphy
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 34) pp:6893-6907
Publication Date(Web):29 Jun 2012
DOI:10.1039/C2OB25870F
The emerging functional versatility of cellular glycans makes research on the design of synthetic inhibitors a timely topic. In detail, the combination of ligand (or headgroup or contact site) structure with spatial parameters that depend on topological and geometrical factors underlies the physiological selectivity of glycan-protein (lectin) recognition. We herein tested a panel of bi-, tri- and tetravalent compounds against two plant agglutinins and adhesion/growth-regulatory lectins (galectins). In addition, we examined the impact of headgroup tailoring (converting lactose to 2′-fucosyllactose) in combination with valency increase in two assay types of increasing biorelevance (from solid-phase binding to cell binding). Compounds were prepared using copper-catalysed azide alkyne cycloaddition from peracetylated lactosyl or 2′-fucosyllactosyl azides. Significant inhibition was achieved for the plant toxin with a tetravalent compound. Different levels of sensitivity were noted for the three groups of the galectin family. The headgroup extension to 2′-fucosyllactose led to a selectivity gain, especially for the chimera-type galectin-3. Valency increase established discrimination against the homodimeric proteins, whereas the combination of valency with the headgroup extension led to discrimination against the tandem-repeat-type galectin-8 for chicken galectins but not human galectins-3 and -4. Thus, detailed structure–activity profiling of glycoclusters combined with suitably modifying the contact site for the targeted lectin will help minimize cross-reactivity among this class of closely related proteins.
Co-reporter:Sabine André, Dilip V. Jarikote, Dandan Yan, Lisa Vincenz, Guan-Nan Wang, Herbert Kaltner, Paul V. Murphy, Hans-Joachim Gabius
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 1) pp:313-318
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmcl.2011.11.010
The synthesis of nine bivalent lactosides (based on ditriazoles, diamides, a glycocyclophane and an acyclic analogue of the glycocyclophane) and one monovalent lactosyl triazole facilitated the assessment of the sensitivity of plant/animal lectins to this type of ligand display. The inhibitory potency of the compounds was determined in two assays of increasing biorelevance. These were solid-phase and cell binding set-ups. Hereby, the ability of the compounds to inhibit the binding of two plant agglutinins and the entire set of adhesion/growth-regulatory galectins from one organism (chicken) to a glycoprotein or to cell surfaces was systematically evaluated. Differential sensitivities were detected between plant and animal lectins and also between distinct galectin forms within the chicken series. Two of the bivalent probes can be considered as sensors for interlectin differences. Most pronounced were the selectivities of N-glycosyl 1,2,3-triazole derivatives for the chimera-type galectin and its proteolytically truncated version.Synthesis of bivalent lactosides generates selective tools to interfere with cell binding of adhesion/growth-regulatory lectins, with the compound shown being the most potent.
Co-reporter:Carmela Napolitano, Viraja R. Palwai, Leif A. Eriksson, Paul V. Murphy
Tetrahedron 2012 68(27–28) pp: 5533-5540
Publication Date(Web):
DOI:10.1016/j.tet.2012.04.082
Co-reporter:Dandan Yan, Julie Naughton, Marguerite Clyne, Paul V. Murphy
Carbohydrate Research 2012 360() pp: 1-7
Publication Date(Web):
DOI:10.1016/j.carres.2012.07.011
Co-reporter:Dilip V. Jarikote, Wei Li, Tao Jiang, Leif A. Eriksson, Paul V. Murphy
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 2) pp:826-835
Publication Date(Web):15 January 2011
DOI:10.1016/j.bmc.2010.12.009
Echinomycin is a natural depsipeptide, which is a bisintercalator, inserting quinoxaline units preferentially adjacent to CG base pairs of DNA. Herein the design and synthesis of echinomycin mimetics based on grafting of two quinoxaline residues onto a macrocyclic scaffold (glycophane) is addressed. Binding of the compounds to calf-thymus DNA was studied using UV–vis and steady state fluorescence spectroscopy, as well as thermal denaturation. An interesting observation was enhancement of fluorescence emission for the peptidomimetics on binding to DNA, which contrasted with observations for echinomycin. Molecular dynamics simulations were exploited to explore in more detail if bis-intercalation to DNA was possible for one of the glycophanes. Bis-intercalating echinomycin complexes with DNA were found to be stable during 20 ns simulations at 298 K. However, the MD simulations of a glycophane complexed with a DNA octamer displayed very different behaviour to echinomycin and its quinoxaline units were found to rapidly migrate out from the intercalation site. Release of bis-intercalation strain occurred with only one of the quinoxaline chromophores remaining intercalated throughout the simulation. The distance between the quinoxaline residues in the glycophane at the end of the MD simulation was 7.3–7.5 Å, whereas in echinomycin, the distance between the residues was ∼11 Å, suggesting that longer glycophane scaffolds would be required to generate bis-intercalating echinomycin mimetics.
Co-reporter:Yunxue Zhao, Min Liu, Vincent Chagnault, Juying Wang, Xiumei Zhang, Paul V. Murphy
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 2) pp:824-828
Publication Date(Web):15 January 2011
DOI:10.1016/j.bmcl.2010.11.088
Previously the synthesis of novel somatostatin mimetic from 1-deoxynojirimycin (DNJ) led to identification of a compound with affinity for human somatostatin receptor subtypes 4 and 5 (hSSTR4 and hSSTR5). Here we examined the properties of this peptidomimetic in a human umbilical vein endothelial cell (HUVEC) based assays. The peptidomimetic prevented capillary tube formation based on HUVECs. It also inhibited HUVEC proliferation by inducing G1 phase cell cycle arrest and apoptosis. Stress fiber assembly and cell migration in HUVECs was markedly suppressed by the somatostatin receptor ligand.
Co-reporter:Carmela Napolitano, Alessandro Natoni, Corrado Santocanale, Lasse Evensen, James B. Lorens, Paul V. Murphy
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 4) pp:1167-1170
Publication Date(Web):15 February 2011
DOI:10.1016/j.bmcl.2010.12.100
The synthesis of a small library of resorcylic acid lactones and evaluation of their biological properties as kinase inhibitors is described. Within the series E-enones were found more active than corresponding Z-enones as inhibitors of a subset of kinases containing a conserved cysteine. Replacement of the enone moiety with a β-haloketone group led to compounds with an interesting kinase selectivity profile and also antiproliferative activity against Jurkat cells. An E-enone derivative also showed activity against capillary tube formation based on a co-culture of primary human umbilical cord endothelial cells (HUVECs) and vascular smooth muscle cells (vSMCs).
Co-reporter:Dilip Venkatrao Jarikote
European Journal of Organic Chemistry 2010 Volume 2010( Issue 26) pp:4959-4970
Publication Date(Web):
DOI:10.1002/ejoc.201000491
Abstract
Macrocycles containing embedded carbohydrates are found in nature. Tricholorin A and G, woodrosin, sophorolipid lactone, cycloviracin B1, glucolipsin A and ipomoeassins A–F are natural products with interesting biological properties. They have inspired synthesis of non-natural macrocyclic structures which contain embedded carbohydrates. Glycophanes (hybrids of carbohydrates and cyclophanes) were prepared which show potential in host-guest chemistry and which have been applied as rigid scaffolds for the synthesis of bivalent inhibitors of lectin binding to tumour cells. Macrocyclic neoglycoconjugates have also been prepared. These include macrocyclic neooligoaminodeoxysaccharides which bind to RNA regions containing either asymmetric internal loop or hairpin loop-stem junctions, and vaccine candidates. Non-natural macrolides having antibacterial and antifungal activity have been synthesised. Metathesis using Grubbs and Hoyveda-Grubbs catalysts has been key to the successful generation of these compounds. Herein we review the recent application of metathesis to the synthesis of ipomoeassins A, B and F, glycophanes, neooligoaminodeoxysaccharides,macrolides and other macrocycles with embedded carbohydrates.
Co-reporter:Yunxue Zhao;Ying Zhou;Kathy M. O’ Boyle
Chemical Biology & Drug Design 2010 Volume 75( Issue 6) pp:570-577
Publication Date(Web):
DOI:10.1111/j.1747-0285.2010.00968.x
The α-glucosidase inhibitors N-methyl-1-deoxynojirimycin (MDNJ) and castanospermine have been shown to inhibit angiogenesis. A hybrid of 1-deoxynojirimycin (DNJ) and an aryl-1,2,3-triazole, which inhibits both an α-glucosidase and methionine aminopeptidase-2 (MetAP2), displayed properties associated with inhibition of angiogenesis (Bioorg. Med. Chem., 16, 2008, 6333–7). The biological evaluation of a structural analogue N-(8-(3-ethynylphenoxy)octyl-1-deoxynojirimycin is described herein. Although this alkyne derivative did not inhibit MetAP2, it inhibited a bacterial α-glucosidase, altered bovine aortic endothelial cell (BAEC) surface oligosaccharide expression and inhibited BAEC proliferation by inducing G1 phase cell cycle arrest. Experiments showed G1 arrest was attributable to the α-glucosidase inhibitor inducing an increase in p27Kip1 expression and high phosphorylation of ERK1/2 without a reduction in cyclin D1. The DNJ derivative (0.1 mm) prevented capillary tube formation from bovine aortic endothelial cells, whereas DNJ or other analogues were unable to inhibit tube formation at the same concentration. Stress fiber assembly in bovine aortic endothelial cells was abolished, and BAEC migration was inhibited indicating the inhibition of tube formation by this derivative is partially a result of a reduction in cell motility. The agent also caused a reduction in secretion of MMP-2 from bovine aortic endothelial cells. Therefore, the new α-glucosidase inhibitor has a different mechanism by which it inhibits angiogenesis in vitro when compared with deoxynojirimycin, the deoxynojirimycin -triazole hybrid, N-methyl-1-deoxynojirimycin and castanospermine.
Co-reporter:Dilip V. Jarikote, Ciaran O’Reilly, Paul V. Murphy
Tetrahedron Letters 2010 Volume 51(Issue 51) pp:6776-6778
Publication Date(Web):22 December 2010
DOI:10.1016/j.tetlet.2010.10.113
A significant rate enhancement was observed in the preparation of allyl and allenyl-C-glycosides from glycosyl acetate or methyl O-glycoside precursors when ultrasound irradiation was employed as an energy source. The C-glycosides were obtained in 77–96% yields in <20 min using TMSOTf as promoter. These results show that sonication provides rapid and efficient access to useful C-glycoside-based building blocks.
Co-reporter:Ying Zhou, Paul V. Murphy
Tetrahedron Letters 2010 Volume 51(Issue 40) pp:5262-5264
Publication Date(Web):6 October 2010
DOI:10.1016/j.tetlet.2010.07.141
Co-reporter:Wayne Pilgrim and Paul V. Murphy
The Journal of Organic Chemistry 2010 Volume 75(Issue 20) pp:6747-6755
Publication Date(Web):September 13, 2010
DOI:10.1021/jo101090f
The quantification of factors that influence both rates and stereoselectivity of anomerization reactions catalyzed by SnCl4 and TiCl4 and how this has informed the synthesis of α-O- and α-S-glycolipids is discussed. The SnCl4-catalyzed anomerization reactions of β-S- and β-O-glycosides of 18 substrates followed first order equilibrium kinetics and kf + kr values were obtained, where kf is the rate constant for the forward reaction (β → α) and kr is the rate constant for the reverse reaction (α → β). Comparison of the kf + kr values showed that reactions of glucuronic acid or galacturonic acid derivatives were ∼10 to 3000 times faster than those of related glucoside and galactopyranoside counterparts and α:β ratios were generally also higher. Stereoelectronic effects contributed from galacto-configured compounds were up to 2-fold faster than those of corresponding glucosides. The introduction of groups, including protecting groups, which are increasingly electron releasing generally led to rate enhancements. The anomerization of S-glycosides was consistently faster than that of corresponding O-glycosides. Reactions were generally faster for reactions with TiCl4 than those with SnCl4. Anomeric ratios depended on the Lewis acid, the number equivalents of the Lewis acid, temperature, and substrate. Very high ratios of α-products for both O- and S-glucuronides were observed for reactions promoted by TiCl4; for these substrates TiCl4 was superior to SnCl4. Anomeric ratios from anomerization of S-glucosides were higher with SnCl4 than with TiCl4. The dependence of equilibrium ratio on Lewis acid and the number of equivalents of Lewis acid indicated that the equilibrium ratio is determined by a complex of the saccharide residue bound to the Lewis acid and not the free glycoside. The high α:β ratios observed for anomerization of both O- and S-glycuronic acids can be explained by coordination of the C-1 heteroatom and C-6 carbonyl group of the product to the Lewis acid, which would enhance the anomeric effect by increasing the electron-withdrawing ability of the anomeric substituent and lead to an increase in the proportion of the α-anomer. Such an observation would argue against the existence of a reverse anomeric effect. Support for a chelation-induced endocyclic cleavage mechanism for the anomerization is provided by the trapping of a key intermediate. The data herein will help predict the tendency of β-glycosides to undergo anomerization; this includes cases where 1,2-trans glycosides are initial products of glycosidation reactions catalyzed by TiCl4 or SnCl4.
Co-reporter:Sabine André, Trinidad Velasco-Torrijos, Rosaria Leyden, Sebastien Gouin, Manuela Tosin, Paul V. Murphy and Hans-Joachim Gabius
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 22) pp:4715-4725
Publication Date(Web):23 Sep 2009
DOI:10.1039/B913010A
The conjugation of carbohydrates to synthetic scaffolds has the goal of preparing potent inhibitors of lectin binding. We herein report the synthesis of a panel of bivalent compounds (cyclophane and terephthalamide-derivatives) then used to establish the influence of scaffold flexibility on respective inhibitory potency in a medically relevant test system. Synthetic routes to two phenylenediamine-based glycocyclophanes involving Ugi reactions of glucuronic acid derivatives and subsequent ring closing metathesis are described, as are improvements for producing terephthalamide-based carbohydrate carriers. Assays were performed with human tumour cells measuring quantitatively the influence of the test compounds on fluorescent surface staining by labelled lectins. Biological evaluation using two different lines of cancer cells as well as cells with known alterations in the glycomic profile (cells treated with an inhibitor of glycan processing and a glycosylation mutant) reduced the risk of generating premature generalizations regarding inhibitor potency. Bioactivity relative to free mannose was invariably determined for the synthetic compounds. A clear trend for enhanced inhibitory properties for macrocyclic compounds compared to non-macrocyclic derivatives was discerned for one type of glycocyclophane. Herein we also document the impact of altering the spacing between the mannose residues, altering cell surface ligand density and cell-type reactivity. The applied strategy for the cell assays is proposed to be of general importance in the quest to identify medically relevant lectin inhibitors.
Co-reporter:Ciaran O’Reilly, Colin O’Brien, Paul V. Murphy
Tetrahedron Letters 2009 50(31) pp: 4427-4429
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.05.060
Co-reporter:Sabine André, Shane O'Sullivan, Christiane Koller, Paul V. Murphy and Hans-Joachim Gabius
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 14) pp:NaN4203-4203
Publication Date(Web):2015/02/17
DOI:10.1039/C5OB00048C
Emerging insights into the functional spectrum of tissue lectins leads to identification of new targets for the custom-made design of potent inhibitors, providing a challenge for synthetic chemistry. The affinity and selectivity of a carbohydrate ligand for a lectin may immensely be increased by a number of approaches, which includes varying geometrical or topological features. This perspective leads to the design and synthesis of glycoclusters and their testing using assays of physiological relevance. Herein, hydroquinone, resorcinol, benzene-1,3,5-triol and tetra(4-hydroxyphenyl)ethene have been employed as scaffolds and propargyl derivatives obtained. The triazole-containing linker to the α/β-O/S-glycosides of GlcNAc/GalNAc presented on these scaffolds was generated by copper-catalysed azide–alkyne cycloaddition. This strategy was used to give a panel of nine glycoclusters with bi-, tri- and tetravalency. Maintained activity for lectin binding after conjugation was ascertained for both sugars in solid-phase assays with the plant agglutinins WGA (GlcNAc) and DBA (GalNAc). Absence of cross-reactivity excluded any carbohydrate-independent reactivity of the bivalent compounds, allowing us to proceed to further testing with a biomedically relevant lectin specific for GalNAc. Macrophage galactose(-binding C)-type lectin, involved in immune defence by dendritic cells and in virus uptake, was produced as a soluble protein without/with its α-helical coiled-coil stalk region. Binding to ligands presented on a matrix and on cell surfaces was highly susceptible to the presence of the tetravalent inhibitor derived from the tetraphenylethene-containing scaffold, and presentation of GalNAc with an α-thioglycosidic linkage proved favorable. Cross-reactivity of this glycocluster to human galectins-3 and -4, which interact with Tn-antigen-presenting mucins, was rather small. Evidently, the valency and spatial display of α-GalNAc residues is a key factor to design potent and selective inhibitors for this lectin.
Co-reporter:Guan-Nan Wang, Sabine André, Hans-Joachim Gabius and Paul V. Murphy
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 34) pp:NaN6907-6907
Publication Date(Web):2012/06/29
DOI:10.1039/C2OB25870F
The emerging functional versatility of cellular glycans makes research on the design of synthetic inhibitors a timely topic. In detail, the combination of ligand (or headgroup or contact site) structure with spatial parameters that depend on topological and geometrical factors underlies the physiological selectivity of glycan-protein (lectin) recognition. We herein tested a panel of bi-, tri- and tetravalent compounds against two plant agglutinins and adhesion/growth-regulatory lectins (galectins). In addition, we examined the impact of headgroup tailoring (converting lactose to 2′-fucosyllactose) in combination with valency increase in two assay types of increasing biorelevance (from solid-phase binding to cell binding). Compounds were prepared using copper-catalysed azide alkyne cycloaddition from peracetylated lactosyl or 2′-fucosyllactosyl azides. Significant inhibition was achieved for the plant toxin with a tetravalent compound. Different levels of sensitivity were noted for the three groups of the galectin family. The headgroup extension to 2′-fucosyllactose led to a selectivity gain, especially for the chimera-type galectin-3. Valency increase established discrimination against the homodimeric proteins, whereas the combination of valency with the headgroup extension led to discrimination against the tandem-repeat-type galectin-8 for chicken galectins but not human galectins-3 and -4. Thus, detailed structure–activity profiling of glycoclusters combined with suitably modifying the contact site for the targeted lectin will help minimize cross-reactivity among this class of closely related proteins.
Co-reporter:Sabine André, Trinidad Velasco-Torrijos, Rosaria Leyden, Sebastien Gouin, Manuela Tosin, Paul V. Murphy and Hans-Joachim Gabius
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 22) pp:NaN4725-4725
Publication Date(Web):2009/09/23
DOI:10.1039/B913010A
The conjugation of carbohydrates to synthetic scaffolds has the goal of preparing potent inhibitors of lectin binding. We herein report the synthesis of a panel of bivalent compounds (cyclophane and terephthalamide-derivatives) then used to establish the influence of scaffold flexibility on respective inhibitory potency in a medically relevant test system. Synthetic routes to two phenylenediamine-based glycocyclophanes involving Ugi reactions of glucuronic acid derivatives and subsequent ring closing metathesis are described, as are improvements for producing terephthalamide-based carbohydrate carriers. Assays were performed with human tumour cells measuring quantitatively the influence of the test compounds on fluorescent surface staining by labelled lectins. Biological evaluation using two different lines of cancer cells as well as cells with known alterations in the glycomic profile (cells treated with an inhibitor of glycan processing and a glycosylation mutant) reduced the risk of generating premature generalizations regarding inhibitor potency. Bioactivity relative to free mannose was invariably determined for the synthetic compounds. A clear trend for enhanced inhibitory properties for macrocyclic compounds compared to non-macrocyclic derivatives was discerned for one type of glycocyclophane. Herein we also document the impact of altering the spacing between the mannose residues, altering cell surface ligand density and cell-type reactivity. The applied strategy for the cell assays is proposed to be of general importance in the quest to identify medically relevant lectin inhibitors.































![(3aR,4S,6S,7R,7aR)-4-(iodomethyl)-6-methoxy-2,2-dimethyltetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-7-ol](http://img.cochemist.com/ccimg/71800/71732-12-2.png)
![(3aR,4S,6S,7R,7aR)-4-(iodomethyl)-6-methoxy-2,2-dimethyltetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-7-ol](http://img.cochemist.com/ccimg/71800/71732-12-2_b.png)




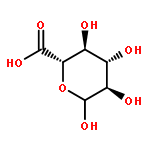
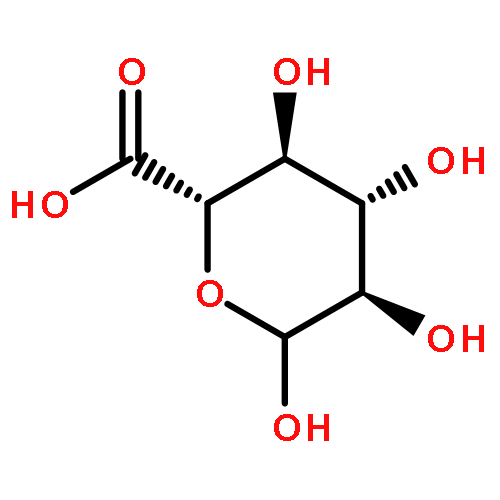






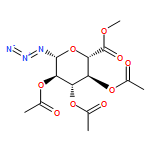





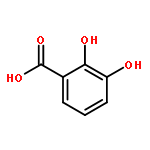

![[(2R,3R,4S,5S,6R)-3,4,5-triacetyloxy-6-azidooxan-2-yl]methyl acetate](http://img.cochemist.com/ccimg/65900/65864-60-0.png)
![[(2R,3R,4S,5S,6R)-3,4,5-triacetyloxy-6-azidooxan-2-yl]methyl acetate](http://img.cochemist.com/ccimg/65900/65864-60-0_b.png)








![[(sulfonatoperoxy)sulfonyl]oxidanide](http://img.cochemist.com/ccimg/15100/15092-81-6.png)
![[(sulfonatoperoxy)sulfonyl]oxidanide](http://img.cochemist.com/ccimg/15100/15092-81-6_b.png)












![2,7-Octadienal,5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-methoxy-2,4-dimethyl-,(2Z,4R,5S,6S)-](http://img.cochemist.com/ccimg/545400/545339-14-8.png)
![2,7-Octadienal,5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-methoxy-2,4-dimethyl-,(2Z,4R,5S,6S)-](http://img.cochemist.com/ccimg/545400/545339-14-8_b.png)
![Benzene, 1-methoxy-4-[(2-propynyloxy)methyl]-](http://img.cochemist.com/ccimg/4100/4039-83-2.png)
![Benzene, 1-methoxy-4-[(2-propynyloxy)methyl]-](http://img.cochemist.com/ccimg/4100/4039-83-2_b.png)

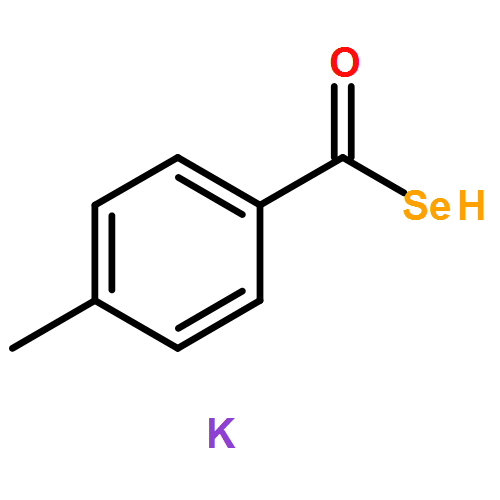


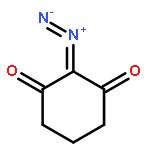
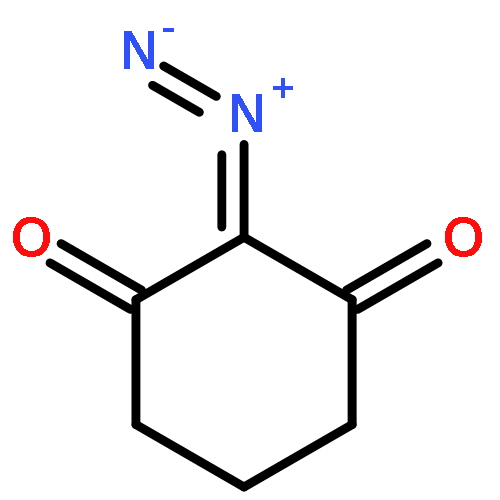






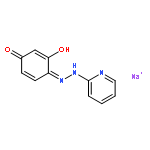


![1,10,15-Hexadecatrien-7-one,13-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-14-methoxy-10,12-dimethyl-,(10Z,12R,13S,14S)-](http://img.cochemist.com/ccimg/663700/663613-05-6.png)
![1,10,15-Hexadecatrien-7-one,13-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-14-methoxy-10,12-dimethyl-,(10Z,12R,13S,14S)-](http://img.cochemist.com/ccimg/663700/663613-05-6_b.png)


![1-Butanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3R)-](http://img.cochemist.com/ccimg/109800/109715-47-1.png)
![1-Butanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3R)-](http://img.cochemist.com/ccimg/109800/109715-47-1_b.png)
















![Butanoic acid, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester, (3R)-](/data/chemimg/1042900/104524-19-8.png)
![Butanoic acid, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester, (3R)-](/data/chemimg/1042900/104524-19-8_b.png)




























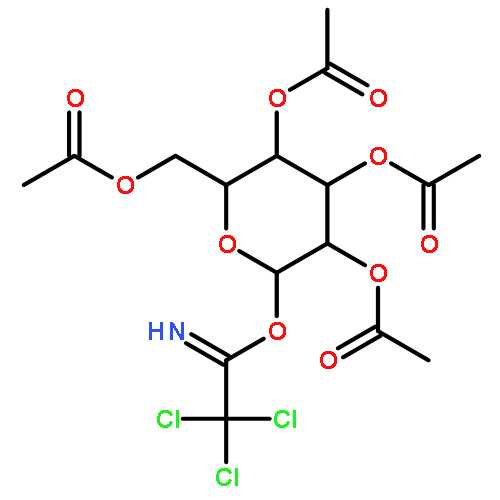










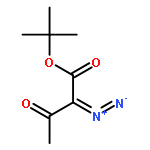
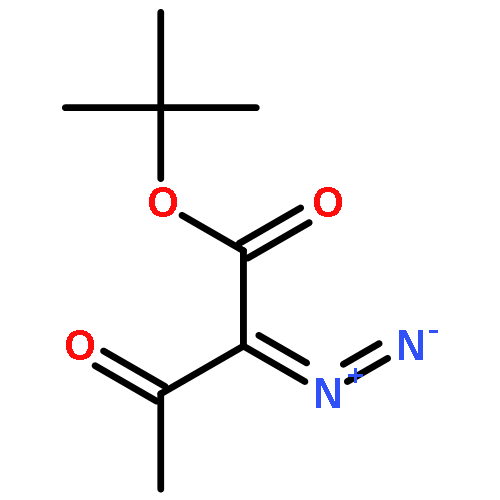








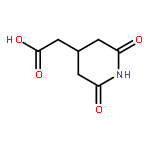



METHANONE](http://img.cochemist.com/ccimg/3100/3006-48-2.png)
METHANONE](http://img.cochemist.com/ccimg/3100/3006-48-2_b.png)










![2-(3,5-DIMETHOXYPHENYL)-6-ETHYL-5-METHYLPYRAZOLO[1,5-A]PYRIMIDIN-7-OL](http://img.cochemist.com/ccimg/149800/149707-75-5.png)
![2-(3,5-DIMETHOXYPHENYL)-6-ETHYL-5-METHYLPYRAZOLO[1,5-A]PYRIMIDIN-7-OL](http://img.cochemist.com/ccimg/149800/149707-75-5_b.png)










![[(2r,3r,4s,5r,6r)-4,5-diacetyloxy-6-azido-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxyoxan-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/30900/30854-62-7.png)
![[(2r,3r,4s,5r,6r)-4,5-diacetyloxy-6-azido-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxyoxan-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/30900/30854-62-7_b.png)
![(3as,4r,6s,7r,7ar)-4-(hydroxymethyl)-6-methoxy-2,2-dimethyl-4,6,7,7a-tetrahydro-3ah-[1,3]dioxolo[4,5-c]pyran-7-ol](http://img.cochemist.com/ccimg/40300/40269-01-0.png)
![(3as,4r,6s,7r,7ar)-4-(hydroxymethyl)-6-methoxy-2,2-dimethyl-4,6,7,7a-tetrahydro-3ah-[1,3]dioxolo[4,5-c]pyran-7-ol](http://img.cochemist.com/ccimg/40300/40269-01-0_b.png)


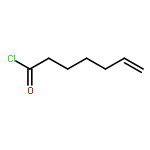
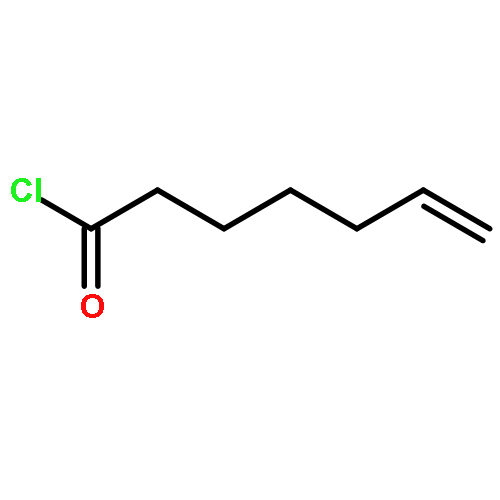














![Hexacosanamide,N-[(1S,2S,3R)-1-[(a-D-galactopyranosyloxy)methyl]-2,3-dihydroxyheptadecyl]-](http://img.cochemist.com/ccimg/158100/158021-47-7.png)
![Hexacosanamide,N-[(1S,2S,3R)-1-[(a-D-galactopyranosyloxy)methyl]-2,3-dihydroxyheptadecyl]-](http://img.cochemist.com/ccimg/158100/158021-47-7_b.png)






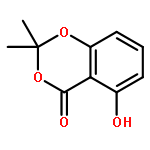
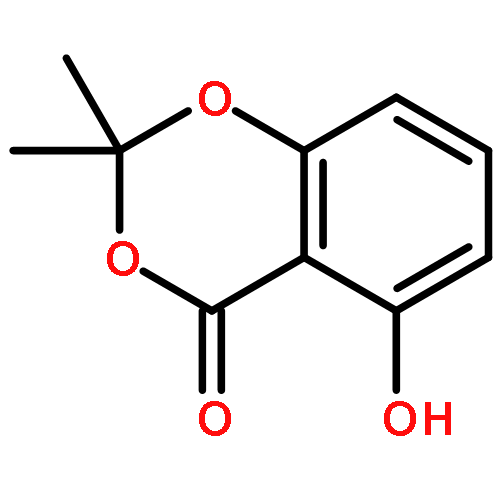








![Benzene, 1,3-bis[[3,5-bis(2-propyn-1-yloxy)phenyl]methoxy]-5-(bromomethyl)-](/data/chemimg/106200/852511-56-9.png)
![Benzene, 1,3-bis[[3,5-bis(2-propyn-1-yloxy)phenyl]methoxy]-5-(bromomethyl)-](/data/chemimg/106200/852511-56-9_b.png)