Co-reporter:Stephen Connelly, David E. Mortenson, Sungwook Choi, Ian A. Wilson, Evan T. Powers, Jeffery W. Kelly, Steven M. Johnson
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmcl.2017.05.080
Rate-limiting dissociation of the tetrameric protein transthyretin (TTR), followed by monomer misfolding and misassembly, appears to cause degenerative diseases in humans known as the transthyretin amyloidoses, based on human genetic, biochemical and pharmacologic evidence. Small molecules that bind to the generally unoccupied thyroxine binding pockets in the native TTR tetramer kinetically stabilize the tetramer, slowing subunit dissociation proportional to the extent that the molecules stabilize the native state over the dissociative transition state—thereby inhibiting amyloidogenesis. Herein, we use previously reported structure-activity relationship data to develop two semi-quantitative algorithms for identifying the structures of potent and selective transthyretin kinetic stabilizers/amyloidogenesis inhibitors. The viability of these prediction algorithms, in particular the more robust in silico docking model, is perhaps best validated by the clinical success of tafamidis, the first-in-class drug approved in Europe, Japan, South America, and elsewhere for treating transthyretin aggregation-associated familial amyloid polyneuropathy. Tafamidis is also being evaluated in a fully-enrolled placebo-controlled clinical trial for its efficacy against TTR cardiomyopathy. These prediction algorithms will be useful for identifying second generation TTR kinetic stabilizers, should these be needed to ameliorate the central nervous system or ophthalmologic pathology caused by TTR aggregation in organs not accessed by oral tafamidis administration.Download high-res image (107KB)Download full-size image
Co-reporter:Wentao Chen; Jiajia Dong; Lars Plate; David E. Mortenson; Gabriel J. Brighty; Suhua Li; Yu Liu; Andrea Galmozzi; Peter S. Lee; Jonathan J. Hulce; Benjamin F. Cravatt; Enrique Saez; Evan T. Powers; Ian A. Wilson; K. Barry Sharpless
Journal of the American Chemical Society 2016 Volume 138(Issue 23) pp:7353-7364
Publication Date(Web):May 18, 2016
DOI:10.1021/jacs.6b02960
Arylfluorosulfates have appeared only rarely in the literature and have not been explored as probes for covalent conjugation to proteins, possibly because they were assumed to possess high reactivity, as with other sulfur(VI) halides. However, we find that arylfluorosulfates become reactive only under certain circumstances, e.g., when fluoride displacement by a nucleophile is facilitated. Herein, we explore the reactivity of structurally simple arylfluorosulfates toward the proteome of human cells. We demonstrate that the protein reactivity of arylfluorosulfates is lower than that of the corresponding aryl sulfonyl fluorides, which are better characterized with regard to proteome reactivity. We discovered that simple hydrophobic arylfluorosulfates selectively react with a few members of the intracellular lipid binding protein (iLBP) family. A central function of iLBPs is to deliver small-molecule ligands to nuclear hormone receptors. Arylfluorosulfate probe 1 reacts with a conserved tyrosine residue in the ligand-binding site of a subset of iLBPs. Arylfluorosulfate probes 3 and 4, featuring a biphenyl core, very selectively and efficiently modify cellular retinoic acid binding protein 2 (CRABP2), both in vitro and in living cells. The X-ray crystal structure of the CRABP2–4 conjugate, when considered together with binding site mutagenesis experiments, provides insight into how CRABP2 might activate arylfluorosulfates toward site-specific reaction. Treatment of breast cancer cells with probe 4 attenuates nuclear hormone receptor activity mediated by retinoic acid, an endogenous client lipid of CRABP2. Our findings demonstrate that arylfluorosulfates can selectively target single iLBPs, making them useful for understanding iLBP function.
Co-reporter:Che-Hsiung Hsu; Sangho Park; David E. Mortenson; B. Lachele Foley; Xiaocong Wang; Robert J. Woods; David A. Case; Evan T. Powers; Chi-Huey Wong; H. Jane Dyson
Journal of the American Chemical Society 2016 Volume 138(Issue 24) pp:7636-7648
Publication Date(Web):June 1, 2016
DOI:10.1021/jacs.6b02879
Interactions between proteins and carbohydrates are ubiquitous in biology. Therefore, understanding the factors that determine their affinity and selectivity are correspondingly important. Herein, we have determined the relative strengths of intramolecular interactions between a series of monosaccharides and an aromatic ring close to the glycosylation site in an N-glycoprotein host. We employed the enhanced aromatic sequon, a structural motif found in the reverse turns of some N-glycoproteins, to facilitate face-to-face monosaccharide–aromatic interactions. A protein host was used because the dependence of the folding energetics on the identity of the monosaccharide can be accurately measured to assess the strength of the carbohydrate–aromatic interaction. Our data demonstrate that the carbohydrate–aromatic interaction strengths are moderately affected by changes in the stereochemistry and identity of the substituents on the pyranose rings of the sugars. Galactose seems to make the weakest and allose the strongest sugar–aromatic interactions, with glucose, N-acetylglucosamine (GlcNAc) and mannose in between. The NMR solution structures of several of the monosaccharide-containing N-glycoproteins were solved to further understand the origins of the similarities and differences between the monosaccharide–aromatic interaction energies. Peracetylation of the monosaccharides substantially increases the strength of the sugar–aromatic interaction in the context of our N-glycoprotein host. Finally, we discuss our results in light of recent literature regarding the contribution of electrostatics to CH−π interactions and speculate on what our observations imply about the absolute conservation of GlcNAc as the monosaccharide through which N-linked glycans are attached to glycoproteins in eukaryotes.
Co-reporter:Aleksandra Baranczak, Jeffery W Kelly
Current Opinion in Chemical Biology 2016 Volume 32() pp:10-21
Publication Date(Web):June 2016
DOI:10.1016/j.cbpa.2016.01.009
•Protein misfolding and misassembly leads to amyloid diseases and tissue degeneration.•Genetic and aggregation insights led to a drug for the transthyretin amyloidoses.•Tafamidis prevents the progression of the otherwise fatal transthyretin amyloidoses.•An understanding of the cellular proteostasis networks is emerging.•Proteostasis regulators should be useful for treating protein misfolding diseases.The list of protein aggregation-associated degenerative diseases is long and growing, while the portfolio of disease-modifying strategies is very small. In this review and perspective, we assess what has worked to slow the progression of an aggregation-associated degenerative disease, covering the underlying mechanism of pharmacologic action and what we have learned about the etiology of the transthyretin amyloid diseases and likely amyloidoses in general. Next, we introduce emerging therapies that should apply more generally to protein misfolding and/or aggregation diseases, approaches that rely on adapting the protein homeostasis or proteostasis network for disease amelioration.
Co-reporter:Wentao Chen, Leopold Kong, Stephen Connelly, Julia M. Dendle, Yu Liu, Ian A. Wilson, Evan T. Powers, and Jeffery W. Kelly
ACS Chemical Biology 2016 Volume 11(Issue 7) pp:1852
Publication Date(Web):April 29, 2016
DOI:10.1021/acschembio.5b01035
Monoclonal antibodies (mAbs) exhibiting highly selective binding to a protein target constitute a large and growing proportion of the therapeutics market. Aggregation of mAbs results in the loss of their therapeutic efficacy and can result in deleterious immune responses. The CH2 domain comprising part of the Fc portion of Immunoglobulin G (IgG) is typically the least stable domain in IgG-type antibodies and therefore influences their aggregation propensity. We stabilized the CH2 domain by engineering an enhanced aromatic sequon (EAS) into the N-glycosylated C′E loop and observed a 4.8 °C increase in the melting temperature of the purified IgG1 Fc fragment. This EAS-stabilized CH2 domain also conferred enhanced stability against thermal and low pH induced aggregation in the context of a full-length monoclonal IgG1 antibody. The crystal structure of the EAS-stabilized (Q295F/Y296A) IgG1 Fc fragment confirms the design principle, i.e., the importance of the GlcNAc1•F295 interaction, and surprisingly reveals that the core fucose attached to GlcNAc1 also engages in an interaction with F295. Inhibition of core fucosylation confirms the contribution of the fucose–Phe interaction to the stabilization. The Q295F/Y296A mutations also modulate the binding affinity of the full-length antibody to Fc receptors by decreasing the binding to low affinity Fc gamma receptors (FcγRIIa, FcγRIIIa, and FcγRIIIb), while maintaining wild-type binding affinity to FcRn and FcγRI. Our results demonstrate that engineering an EAS into the N-glycosylated reverse turn on the C′E loop leads to stabilizing N-glycan–protein interactions in antibodies and that this modification modulates antibody–Fc receptor binding.
Co-reporter:Aleksandra Baranczak; Yu Liu; Stephen Connelly; Wen-Ge Han Du; Erin R. Greiner; Joseph C. Genereux; R. Luke Wiseman; Yvonne S. Eisele; Nadine C. Bradbury; Jiajia Dong; Louis Noodleman; K. Barry Sharpless; Ian A. Wilson; Sandra E. Encalada
Journal of the American Chemical Society 2015 Volume 137(Issue 23) pp:7404-7414
Publication Date(Web):June 8, 2015
DOI:10.1021/jacs.5b03042
Fluorogenic probes, due to their often greater spatial and temporal sensitivity in comparison to permanently fluorescent small molecules, represent powerful tools to study protein localization and function in the context of living systems. Herein, we report fluorogenic probe 4, a 1,3,4-oxadiazole designed to bind selectively to transthyretin (TTR). Probe 4 comprises a fluorosulfate group not previously used in an environment-sensitive fluorophore. The fluorosulfate functional group does not react covalently with TTR on the time scale required for cellular imaging, but does red shift the emission maximum of probe 4 in comparison to its nonfluorosulfated analogue. We demonstrate that probe 4 is dark in aqueous buffers, whereas the TTR·4 complex exhibits a fluorescence emission maximum at 481 nm. The addition of probe 4 to living HEK293T cells allows efficient binding to and imaging of exogenous TTR within intracellular organelles, including the mitochondria and the endoplasmic reticulum. Furthermore, live Caenorhabditis elegans expressing human TTR transgenically and treated with probe 4 display TTR·4 fluorescence in macrophage-like coelomocytes. An analogue of fluorosulfate probe 4 does react selectively with TTR without labeling the remainder of the cellular proteome. Studies on this analogue suggest that certain aryl fluorosulfates, due to their cell and organelle permeability and activatable reactivity, could be considered for the development of protein-selective covalent probes.
Co-reporter:Yu Liu; Xin Zhang; Wentao Chen; Yun Lei Tan
Journal of the American Chemical Society 2015 Volume 137(Issue 35) pp:11303-11311
Publication Date(Web):August 25, 2015
DOI:10.1021/jacs.5b04366
Proteome misfolding and/or aggregation, caused by a thermal perturbation or a related stress, transiently challenges the cellular protein homeostasis (proteostasis) network capacity of cells by consuming chaperone/chaperonin pathway and degradation pathway capacity. Developing protein client-based probes to quantify the cellular proteostasis network capacity in real time is highly desirable. Herein we introduce a small-molecule-regulated fluorescent protein folding sensor based on a thermo-labile mutant of the de novo designed retroaldolase (RA) enzyme. Since RA enzyme activity is not present in any cell, the protein folding sensor is bioorthogonal. The fluorogenic small molecule was designed to become fluorescent when it binds to and covalently reacts with folded and functional RA. Thus, in the first experimental paradigm, cellular proteostasis network capacity and its dynamics are reflected by RA–small molecule conjugate fluorescence, which correlates with the amount of folded and functional RA present, provided that pharmacologic chaperoning is minimized. In the second experimental scenario, the RA–fluorogenic probe conjugate is pre-formed in a cell by simply adding the fluorogenic probe to the cell culture media. Unreacted probe is then washed away before a proteome misfolding stress is applied in a pulse-chase-type experiment. Insufficient proteostasis network capacity is reflected by aggregate formation of the fluorescent RA–fluorogenic probe conjugate. Removal of the stress results in apparent RA–fluorogenic probe conjugate re-folding, mediated in part by the heat-shock response transcriptional program augmenting cytosolic proteostasis network capacity, and in part by time-dependent RA–fluorogenic probe conjugate degradation by cellular proteolysis.
Co-reporter:Amber N. Murray, Wentao Chen, Aristotelis Antonopoulos, Sarah R. Hanson, R. Luke Wiseman, Anne Dell, Stuart M. Haslam, David L. Powers, Evan T. Powers, Jeffery W. Kelly
Chemistry & Biology 2015 Volume 22(Issue 8) pp:1052-1062
Publication Date(Web):20 August 2015
DOI:10.1016/j.chembiol.2015.06.017
•Aromatic amino acids two residues before sequons increase glycosylation efficiency•Aromatic amino acids two residues before sequons decrease N-glycan heterogeneity•Increased glycan occupancy results from oligosaccharyltransferase preferences•Decreased N-glycan heterogeneity results from suppressed Golgi glycan remodelingN-Glycosylation plays an important role in protein folding and function. Previous studies demonstrate that a phenylalanine residue introduced at the n-2 position relative to an Asn-Xxx-Thr/Ser N-glycosylation sequon increases the glycan occupancy of the sequon in insect cells. Here, we show that any aromatic residue at n-2 increases glycan occupancy in human cells and that this effect is dependent upon oligosaccharyltransferase substrate preferences rather than differences in other cellular processing events such as degradation or trafficking. Moreover, aromatic residues at n-2 alter glycan processing in the Golgi, producing proteins with less complex N-glycan structures. These results demonstrate that manipulating the sequence space surrounding N-glycosylation sequons is useful both for controlling glycosylation efficiency, thus enhancing glycan occupancy, and for influencing the N-glycan structures produced.Figure optionsDownload full-size imageDownload high-quality image (170 K)Download as PowerPoint slide
Co-reporter:Yu Liu ; Xin Zhang ; Yun Lei Tan ; Gira Bhabha ; Damian C. Ekiert ; Yakov Kipnis ; Sinisa Bjelic ; David Baker
Journal of the American Chemical Society 2014 Volume 136(Issue 38) pp:13102-13105
Publication Date(Web):September 11, 2014
DOI:10.1021/ja5056356
Enzyme-based tags attached to a protein-of-interest (POI) that react with a small molecule, rendering the conjugate fluorescent, are very useful for studying the POI in living cells. These tags are typically based on endogenous enzymes, so protein engineering is required to ensure that the small-molecule probe does not react with the endogenous enzyme in the cell of interest. Here we demonstrate that de novo-designed enzymes can be used as tags to attach to POIs. The inherent bioorthogonality of the de novo-designed enzyme–small-molecule probe reaction circumvents the need for protein engineering, since these enzyme activities are not present in living organisms. Herein, we transform a family of de novo-designed retroaldolases into variable-molecular-weight tags exhibiting fluorescence imaging, reporter, and electrophoresis applications that are regulated by tailored, reactive small-molecule fluorophores.
Co-reporter:Yun Lei Tan, Joseph C. Genereux, Sandra Pankow, Johannes M.F.G. Aerts, John R. Yates III, Jeffery W. Kelly
Chemistry & Biology 2014 Volume 21(Issue 8) pp:967-976
Publication Date(Web):14 August 2014
DOI:10.1016/j.chembiol.2014.06.008
•Immunoprecipitation and mass spectrometry identify ERdj3 as a GCase interactor•ERdj3 depletion enhances mutant GCase folding, trafficking, and lysosomal function•The ERdj3 degradation factor competes with profolding calnexin for mutant GCase•ERdj3 depletion and increased calnexin function synergize to rescue mutant GCaseGaucher’s disease (GD) is caused by mutations that compromise β-glucocerebrosidase (GCase) folding in the endoplasmic reticulum (ER), leading to excessive degradation instead of trafficking, which results in insufficient lysosomal function. We hypothesized that ER GCase interacting proteins play critical roles in making quality control decisions, i.e., facilitating ER-associated degradation (ERAD) instead of folding and trafficking. Utilizing GCase immunoprecipitation followed by mass-spectrometry-based proteomics, we identified endogenous HeLa cell GCase protein interactors, including ERdj3, an ER resident Hsp40 not previously established to interact with GCase. Depleting ERdj3 reduced the rate of mutant GCase degradation in patient-derived fibroblasts, while increasing folding, trafficking, and function by directing GCase to the profolding ER calnexin pathway. Inhibiting ERdj3-mediated mutant GCase degradation while simultaneously enhancing calnexin-associated folding, by way of a diltiazem-mediated increase in ER Ca2+ levels, yields a synergistic rescue of L444P GCase lysosomal function. Our findings suggest a combination therapeutic strategy for ameliorating GD.
Co-reporter:Xin Zhang, Yu Liu, Joseph C. Genereux, Chandler Nolan, Meha Singh, and Jeffery W. Kelly
ACS Chemical Biology 2014 Volume 9(Issue 9) pp:1945
Publication Date(Web):July 22, 2014
DOI:10.1021/cb5004477
The biosynthesis of soluble, properly folded recombinant proteins in large quantities from Escherichia coli is desirable for academic research and industrial protein production. The basal E. coli protein homeostasis (proteostasis) network capacity is often insufficient to efficiently fold overexpressed proteins. Herein we demonstrate that a transcriptionally reprogrammed E. coli proteostasis network is generally superior for producing soluble, folded, and functional recombinant proteins. Reprogramming is accomplished by overexpressing a negative feedback deficient heat-shock response transcription factor before and during overexpression of the protein-of-interest. The advantage of transcriptional reprogramming versus simply overexpressing select proteostasis network components (e.g., chaperones and co-chaperones, which has been explored previously) is that a large number of proteostasis network components are upregulated at their evolved stoichiometry, thus maintaining the system capabilities of the proteostasis network that are currently incompletely understood. Transcriptional proteostasis network reprogramming mediated by stress-responsive signaling in the absence of stress should also be useful for protein production in other cells.
Co-reporter:Irit Rappley, Cecília Monteiro, Marta Novais, Aleksandra Baranczak, Gregory Solis, R. Luke Wiseman, Stephen Helmke, Mathew S. Maurer, Teresa Coelho, Evan T. Powers, and Jeffery W. Kelly
Biochemistry 2014 Volume 53(Issue 12) pp:
Publication Date(Web):March 12, 2014
DOI:10.1021/bi500171j
The transthyretin (TTR) amyloidoses are a group of degenerative diseases caused by TTR aggregation, requiring rate-limiting tetramer dissociation. Kinetic stabilization of TTR, by preferential binding of a drug to the native tetramer over the dissociative transition state, dramatically slows the progression of familial amyloid polyneuropathy. An established method for quantifying the kinetic stability of recombinant TTR tetramers in buffer is subunit exchange, in which tagged TTR homotetramers are added to untagged homotetramers at equal concentrations to measure the rate at which the subunits exchange. Herein, we report a subunit exchange method for quantifying the kinetic stability of endogenous TTR in human plasma. The subunit exchange reaction is initiated by the addition of a substoichiometric quantity of FLAG-tagged TTR homotetramers to endogenous TTR in plasma. Aliquots of the subunit exchange reaction, taken as a function of time, are then added to an excess of a fluorogenic small molecule, which immediately arrests further subunit exchange. After binding, the small molecule reacts with the TTR tetramers, rendering them fluorescent and detectable in human plasma after subsequent ion exchange chromatography. The ability to report on the extent of TTR kinetic stabilization resulting from treatment with oral tafamidis is important, especially for selection of the appropriate dose for patients carrying rare mutations. This method could also serve as a surrogate biomarker for the prediction of the clinical outcome. Subunit exchange was used to quantify the stabilization of WT TTR from senile systemic amyloidosis patients currently being treated with tafamidis (20 mg orally, once daily). TTR kinetic stability correlated with the tafamidis plasma concentration.
Co-reporter:Lisa M. Ryno;Christina B. Cooley;Lars Plate;Gareth J. Morgan;John D. Hulleman;R. Luke Wiseman
PNAS 2014 Volume 111 (Issue 36 ) pp:13046-13051
Publication Date(Web):2014-09-09
DOI:10.1073/pnas.1406050111
Light-chain amyloidosis (AL) is a degenerative disease characterized by the extracellular aggregation of a destabilized amyloidogenic
Ig light chain (LC) secreted from a clonally expanded plasma cell. Current treatments for AL revolve around ablating the cancer
plasma cell population using chemotherapy regimens. Unfortunately, this approach is limited to the ∼70% of patients who do
not exhibit significant organ proteotoxicity and can tolerate chemotherapy. Thus, identifying new therapeutic strategies to
alleviate LC organ proteotoxicity should allow AL patients with significant cardiac and/or renal involvement to subsequently
tolerate established chemotherapy treatments. Using a small-molecule screening approach, the unfolded protein response (UPR)
was identified as a cellular signaling pathway whose activation selectively attenuates secretion of amyloidogenic LC, while
not affecting secretion of a nonamyloidogenic LC. Activation of the UPR-associated transcription factors XBP1s and/or ATF6
in the absence of stress recapitulates the selective decrease in amyloidogenic LC secretion by remodeling the endoplasmic
reticulum proteostasis network. Stress-independent activation of XBP1s, or especially ATF6, also attenuates extracellular
aggregation of amyloidogenic LC into soluble aggregates. Collectively, our results show that stress-independent activation
of these adaptive UPR transcription factors offers a therapeutic strategy to reduce proteotoxicity associated with LC aggregation.
Co-reporter:Aleksra Baranczak;Stephen Connelly;Yu Liu;Sungwook Choi;Neil P. Grimster;Evan T. Powers;Ian A. Wilson
Biopolymers 2014 Volume 101( Issue 5) pp:484-495
Publication Date(Web):
DOI:10.1002/bip.22407
ABSTRACT
We seek fluorogenic small molecules that generate a fluorescent conjugate signal if and only if they react with a given protein-of-interest (i.e., small molecules for which noncovalent binding to the protein-of-interest is insufficient to generate fluorescence). Consequently, it is the new chemical entity afforded by the generally irreversible reaction between the small molecule and the protein-of-interest that enables the energy of an electron occupying the lowest unoccupied molecular orbital (LUMO) of the chromophore to be given off as a photon instead of being dissipated by nonradiative mechanisms in complex biological environments. This category of fluorogenic small molecules is created by starting with environmentally sensitive fluorophores that are modified by an essential functional group that efficiently quenches the fluorescence until a chemoselective reaction between that functional group and the protein-of-interest occurs, yielding the fluorescent conjugate. Fluorogenic small molecules are envisioned to be useful for a wide variety of applications, including live cell imaging without the requirement for washing steps and pulse-chase kinetic analyses of protein synthesis, trafficking, degradation, etc. © 2013 Wiley Periodicals, Inc. Biopolymers 101: 484–495, 2014.
Co-reporter:Yu Liu;Yun Lei Tan;Xin Zhang;Gira Bhabha;Damian C. Ekiert;Joseph C. Genereux;Younhee Cho;Yakov Kipnis;Sinisa Bjelic;David Baker
PNAS 2014 111 (12 ) pp:4449-4454
Publication Date(Web):2014-03-25
DOI:10.1073/pnas.1323268111
Although much is known about protein folding in buffers, it remains unclear how the cellular protein homeostasis network functions
as a system to partition client proteins between folded and functional, soluble and misfolded, and aggregated conformations.
Herein, we develop small molecule folding probes that specifically react with the folded and functional fraction of the protein
of interest, enabling fluorescence-based quantification of this fraction in cell lysate at a time point of interest. Importantly,
these probes minimally perturb a protein’s folding equilibria within cells during and after cell lysis, because sufficient
cellular chaperone/chaperonin holdase activity is created by rapid ATP depletion during cell lysis. The folding probe strategy
and the faithful quantification of a particular protein’s functional fraction are exemplified with retroaldolase, a de novo
designed enzyme, and transthyretin, a nonenzyme protein. Our findings challenge the often invoked assumption that the soluble
fraction of a client protein is fully folded in the cell. Moreover, our results reveal that the partitioning of destabilized
retroaldolase and transthyretin mutants between the aforementioned conformational states is strongly influenced by cytosolic
proteostasis network perturbations. Overall, our results suggest that applying a chemical folding probe strategy to other
client proteins offers opportunities to reveal how the proteostasis network functions as a system to regulate the folding
and function of individual client proteins in vivo.
Co-reporter:Wentao Chen ; Sebastian Enck ; Joshua L. Price ; David L. Powers ; Evan T. Powers ; Chi-Huey Wong ; H. Jane Dyson
Journal of the American Chemical Society 2013 Volume 135(Issue 26) pp:9877-9884
Publication Date(Web):June 7, 2013
DOI:10.1021/ja4040472
Carbohydrate–aromatic interactions mediate many biological processes. However, the structure–energy relationships underpinning direct carbohydrate–aromatic packing interactions in aqueous solution have been difficult to assess experimentally and remain elusive. Here, we determine the structures and folding energetics of chemically synthesized glycoproteins to quantify the contributions of the hydrophobic effect and CH−π interactions to carbohydrate–aromatic packing interactions in proteins. We find that the hydrophobic effect contributes significantly to protein–carbohydrate interactions. Interactions between carbohydrates and aromatic amino acid side chains, however, are supplemented by CH−π interactions. The strengths of experimentally determined carbohydrate CH−π interactions do not correlate with the electrostatic properties of the involved aromatic residues, suggesting that the electrostatic component of CH−π interactions in aqueous solution is small. Thus, tight binding of carbohydrates and aromatic residues is driven by the hydrophobic effect and CH−π interactions featuring a dominating dispersive component.
Co-reporter:Eul Hyun Suh ; Yu Liu ; Stephen Connelly ; Joseph C. Genereux ; Ian A. Wilson
Journal of the American Chemical Society 2013 Volume 135(Issue 47) pp:17869-17880
Publication Date(Web):November 1, 2013
DOI:10.1021/ja408230k
Small molecules that react selectively with a specific non-enzyme drug-target protein in a complex biological environment without displacement of a leaving group (tracelessly) are rare and highly desirable. Herein we describe the development of a family of fluorogenic stilbene-based vinyl amides and vinyl sulfonamides that covalently modify transthyretin (TTR) tracelessly. These small molecules bind selectively to TTR in complex biological environments and then undergo a rapid and chemoselective 1,4-Michael addition with the pKa-perturbed Lys-15 ε-amino group of TTR. Replacing the vinyl amide in 2 with the more reactive vinyl sulfonamide in 4 hastens the conjugation kinetics. X-ray cocrystallography verified the formation of the secondary amine bond mediating the conjugation in the case of 2 and 4 and confirmed the expected orientation of the stilbene within the TTR binding sites. Vinyl amide 2 and vinyl sulfonamide 4 potently inhibit TTR dissociation and amyloid fibril formation in vitro. The TTR binding selectivity, modification yield, and reaction chemoselectivity of vinyl sulfonamide 4 are good enough in human plasma to serve as a starting point for medicinal chemistry efforts. Moreover, vinyl sulfonamide 4 is fluorogenic: it exhibits minimal background fluorescence in complex biological environments, remains dark upon binding to TTR, and becomes fluorescent only upon reaction with TTR. The fluorogenicity of 4 was utilized to accurately quantify the native TTR concentration in Escherichia coli lysate using a fluorescence plate reader.
Co-reporter:Neil P. Grimster ; Stephen Connelly ; Aleksandra Baranczak ; Jiajia Dong ; Larissa B. Krasnova ; K. Barry Sharpless ; Evan T. Powers ; Ian A. Wilson
Journal of the American Chemical Society 2013 Volume 135(Issue 15) pp:5656-5668
Publication Date(Web):January 25, 2013
DOI:10.1021/ja311729d
Molecules that bind selectively to a given protein and then undergo a rapid chemoselective reaction to form a covalent conjugate have utility in drug development. Herein a library of 1,3,4-oxadiazoles substituted at the 2 position with an aryl sulfonyl fluoride and at the 5 position with a substituted aryl known to have high affinity for the inner thyroxine binding subsite of transthyretin (TTR) was conceived of by structure-based design principles and was chemically synthesized. When bound in the thyroxine binding site, most of the aryl sulfonyl fluorides react rapidly and chemoselectively with the pKa-perturbed K15 residue, kinetically stabilizing TTR and thus preventing amyloid fibril formation, known to cause polyneuropathy. Conjugation t50s range from 1 to 4 min, ∼1400 times faster than the hydrolysis reaction outside the thyroxine binding site. X-ray crystallography confirms the anticipated binding orientation and sheds light on the sulfonyl fluoride activation leading to the sulfonamide linkage to TTR. A few of the aryl sulfonyl fluorides efficiently form conjugates with TTR in plasma. Eleven of the TTR covalent kinetic stabilizers synthesized exhibit fluorescence upon conjugation and therefore could have imaging applications as a consequence of the environment sensitive fluorescence of the chromophore.
Co-reporter:Lisa M Ryno, R Luke Wiseman, Jeffery W Kelly
Current Opinion in Chemical Biology 2013 Volume 17(Issue 3) pp:346-352
Publication Date(Web):June 2013
DOI:10.1016/j.cbpa.2013.04.009
•The UPR adapts ER proteostasis through translational attenuation and transcriptional remodeling.•Activating distinct arms of the UPR differentially influences ER proteostasis.•Arm-selective UPR activation shows promise to ameliorate protein misfolding diseases.•Small molecules can manipulate independent arms of the UPR to change the fate of misfolded proteins.Protein homeostasis (or proteostasis) within the endoplasmic reticulum (ER) is regulated by the unfolded protein response (UPR). The UPR consists of three integrated signaling pathways activated by the accumulation of misfolded proteins within the ER lumen. Activation of the UPR alters ER proteostasis through translational attenuation of new protein synthesis and transcriptional remodeling of ER proteostasis pathways, providing a mechanism to adapt ER proteostasis in response to cellular stress. The capacity of the UPR to alter ER proteostasis suggests that exogenous manipulation of UPR signaling pathways offers therapeutic promise to alter the fate of pathologic proteins associated with human protein misfolding diseases. Here, we discuss the therapeutic potential of exogenous UPR activation to treat human disease and highlight specific small molecule approaches for regulating UPR signaling that could be beneficial to treat protein misfolding diseases.
Co-reporter:Derrick Sek Tong Ong, Ya-Juan Wang, Yun Lei Tan, John R. Yates III, Ting-Wei Mu, Jeffery W. Kelly
Chemistry & Biology 2013 Volume 20(Issue 3) pp:403-415
Publication Date(Web):21 March 2013
DOI:10.1016/j.chembiol.2012.11.014
Lysosomal storage diseases (LSDs) are often caused by mutations compromising lysosomal enzyme folding in the endoplasmic reticulum (ER), leading to degradation and loss of function. Mass spectrometry analysis of Gaucher fibroblasts treated with mechanistically distinct molecules that increase LSD enzyme folding, trafficking, and function resulted in the identification of nine commonly downregulated and two jointly upregulated proteins, which we hypothesized would be critical proteostasis network components for ameliorating loss-of-function diseases. LIMP-2 and FK506 binding protein 10 (FKBP10) were validated as such herein. Increased FKBP10 levels accelerated mutant glucocerebrosidase degradation over folding and trafficking, whereas decreased ER FKBP10 concentration led to more LSD enzyme partitioning into the calnexin profolding pathway, enhancing folding and activity to levels thought to ameliorate LSDs. Thus, targeting FKBP10 appears to be a heretofore unrecognized therapeutic strategy to ameliorate LSDs.Highlights► Whole-cell proteomics identifies FKBP10 as key to lysosomal enzyme proteostasis ► FKBP10 knockdown increases mutant lysosomal enzyme folding, trafficking, and function ► FKBP10 overexpression accelerates ERAD of mutant lysosomal enzymes ► FKBP10 appears to act as an ER degradation versus folding partitioning factor
Co-reporter:Joshua L. Price, Evan T. Powers, and Jeffery W. Kelly
ACS Chemical Biology 2011 Volume 6(Issue 11) pp:1188
Publication Date(Web):September 22, 2011
DOI:10.1021/cb200277u
The intrinsic stabilization of therapeutic proteins by N-glycosylation can endow them with increased shelf and serum half-lives owing to lower populations of misfolded and unfolded states, which are susceptible to aggregation and proteolysis. Conjugation of poly(ethylene glycol) (PEG) oligomers to nucleophilic groups on the surfaces of folded proteins (i.e., PEGylation) is a chemical alternative to N-glycosylation, in that it can also enhance the pharmacologic attributes of therapeutic proteins. However, the energetic consequences of PEGylation are currently not predictable. We find that PEGylation of an Asn residue in reverse turn 1 of the Pin WW domain is intrinsically stabilizing in several sequence contexts, unlike N-glycosylation, which is only stabilizing in a particular sequence context. Our thermodynamic data are consistent with the hypothesis that PEGylation destabilizes the protein denatured state ensemble via an excluded volume effect, whereas N-glycosylation-associated stabilization results primarily from native state interactions between the N-glycan and the protein.
Co-reporter:Deguo Du, Amber N. Murray, Ehud Cohen, Hyun-Eui Kim, Ryan Simkovsky, Andrew Dillin, and Jeffery W. Kelly
Biochemistry 2011 Volume 50(Issue 10) pp:1607-1617
Publication Date(Web):January 26, 2011
DOI:10.1021/bi1013744
The process of amyloid-β (Aβ) fibril formation is genetically and pathologically linked to Alzheimer’s disease (AD). Thus, a selective and sensitive method for quantifying Aβ fibrils in complex biological samples allows a variety of hypotheses to be tested. Herein, we report the basis for a quantitative in vitro kinetic aggregation assay that detects seeding-competent Aβ aggregates in mammalian cell culture media, in Caenorhabditis elegans lysate, and in mouse brain homogenate. Sonicated, proteinase K-treated Aβ fibril-containing tissue homogenates or cell culture media were added to an initially monomeric Aβ1−40 reporter peptide to seed an in vitro nucleated aggregation reaction. The reduction in the half-time (t50) of the amyloid growth phase is proportional to the quantity of seeding-competent Aβ aggregates present in the biological sample. An ion-exchange resin amyloid isolation strategy from complex biological samples is demonstrated as an alternative for improving the sensitivity and linearity of the kinetic aggregation assay.
Co-reporter:James P. Solomon, Steve Bourgault, Evan T. Powers, and Jeffery W. Kelly
Biochemistry 2011 Volume 50(Issue 13) pp:
Publication Date(Web):February 24, 2011
DOI:10.1021/bi101905n
Glycosaminoglycans (GAGs) are highly sulfated linear polysaccharides prevalent in the extracellular matrix, and they associate with virtually all amyloid deposits in vivo. GAGs accelerate the aggregation of many amyloidogenic peptides in vitro, but little mechanistic evidence is available to explain why. Herein, spectroscopic methods demonstrate that GAGs do not affect the secondary structure of the monomeric 8 kDa amyloidogenic fragment of human plasma gelsolin. Moreover, monomerized 8 kDa gelsolin does not bind to heparin under physiological conditions. In contrast, 8 kDa gelsolin cross-β-sheet oligomers and amyloid fibrils bind strongly to heparin, apparently because of electrostatic interactions between the negatively charged polysaccharide and a positively charged region of the 8 kDa gelsolin assemblies. Our observations are consistent with a scaffolding mechanism whereby cross-β-sheet oligomers, upon formation, bind to GAGs, accelerating the fibril extension phase of amyloidogenesis, possibly by concentrating and orienting the oligomers to more efficiently form amyloid fibrils. Notably, heparin decreases the 8 kDa gelsolin concentration necessary for amyloid fibril formation, likely a consequence of fibril stabilization through heparin binding. Because GAG overexpression, which is common in amyloidosis, may represent a strategy for minimizing cross-β-sheet oligomer toxicity by transforming them into amyloid fibrils, the mechanism described herein for GAG-mediated acceleration of 8 kDa gelsolin amyloidogenesis provides a starting point for therapeutic strategy development. The addition of GAG mimetics, small molecule sulfonates shown to reduce the amyloid load in animal models of amyloidosis, to a heparin-accelerated 8 kDa gelsolin aggregation reaction mixture neither significantly alters the rate of amyloidogenesis nor prevents oligomers from binding to GAGs, calling into question their commonly accepted mechanism.
Co-reporter:
Biochemistry 2011 Volume 50(Issue 6) pp:1001-1015
Publication Date(Web):December 31, 2010
DOI:10.1021/bi101822y
Glycosaminoglycans (GAGs), which are found in association with all extracellular amyloid deposits in humans, are known to accelerate the aggregation of various amyloidogenic proteins in vitro. However, the precise molecular mechanism(s) by which GAGs accelerate amyloidogenesis remains elusive. Herein, we show that sulfated GAGs, especially heparin, accelerate transthyretin (TTR) amyloidogenesis by quaternary structural conversion. The clustering of sulfate groups on heparin and its polymeric nature are essential features for accelerating TTR amyloidogenesis. Heparin does not influence TTR tetramer stability or TTR dissociation kinetics, nor does it alter the folded monomer−misfolded monomer equilibrium directly. Instead, heparin accelerates the conversion of preformed TTR oligomers into larger aggregates. The more rapid disappearance of monomeric TTR in the presence of heparin likely reflects the fact that the monomer−misfolded amyloidogenic monomer−oligomer−TTR fibril equilibria are all linked, a hypothesis that is strongly supported by the light scattering data. TTR aggregates prepared in the presence of heparin exhibit a higher resistance to trypsin and proteinase K proteolysis and a lower exposure of hydrophobic side chains comprising hydrophobic clusters, suggesting an active role for heparin in amyloidogenesis. Our data suggest that heparin accelerates TTR aggregation by a scaffold-based mechanism, in which the sulfate groups comprising GAGs interact primarily with TTR oligomers through electrostatic interactions, concentrating and orienting the oligomers, facilitating the formation of higher molecular weight aggregates. This model raises the possibility that GAGs may play a protective role in human amyloid diseases by interacting with proteotoxic oligomers and promoting their association into less toxic amyloid fibrils.
Co-reporter:Sungwook Choi, Jeffery W. Kelly
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 4) pp:1505-1514
Publication Date(Web):15 February 2011
DOI:10.1016/j.bmc.2010.12.050
Herein we demonstrate that competition between candidate kinetic stabilizer binding to transthyretin (TTR) and stilbene binding to and reaction with the same thyroxine sites within TTR can be utilized to discover potent and highly selective non-covalent TTR amyloidogenesis inhibitors. We report two stilbenes, S1 and S2, for use in distinct competition assays. Each bind selectively to TTR and then chemoselectively react to form an amide bond with the Lys-15 residue of TTR, creating a fluorescent conjugate. We used 28 TTR kinetic stabilizers exhibiting a known spectrum of plasma TTR binding selectivities and TTR amyloid fibril inhibition efficacies to validate the ‘TTR fluorescence conjugate competition assay’. The kinetic stabilizers competed with S1 for binding to recombinant TTR in buffer and with S2 for binding to endogenous levels of TTR in human blood serum. In both assay scenarios, we demonstrate that the lower the TTR–stilbene conjugate fluorescence after a 3 h competition, the greater the binding selectivity and potency of the candidate TTR kinetic stabilizer. These assays, particularly the assay utilizing S2 in human serum, replace two assays previously utilized to gather the same information. While not the focus of this manuscript, it is clear that the ‘TTR fluorescence conjugate competition assay’ could be adapted for high throughput screening applications.
Co-reporter:Elizabeth K. Culyba;Joshua L. Price;Sarah R. Hanson;Apratim Dhar;Chi-Huey Wong;Martin Gruebele;Evan T. Powers
Science 2011 Volume 331(Issue 6017) pp:571-575
Publication Date(Web):04 Feb 2011
DOI:10.1126/science.1198461
Protein reverse turns that interact with a phenlyalanine group allow stable introduction of glycan groups at asparagine residues.
Co-reporter:Joshua L. Price ; Dalit Shental-Bechor ; Apratim Dhar ; Maurice J. Turner ; Evan T. Powers ; Martin Gruebele ; Yaakov Levy
Journal of the American Chemical Society 2010 Volume 132(Issue 43) pp:15359-15367
Publication Date(Web):October 11, 2010
DOI:10.1021/ja106896t
Asparagine glycosylation is one of the most common and important post-translational modifications of proteins in eukaryotic cells. N-Glycosylation occurs when a triantennary glycan precursor is transferred en bloc to a nascent polypeptide (harboring the N−X−T/S sequon) as the peptide is cotranslationally translocated into the endoplasmic reticulum (ER). In addition to facilitating binding interactions with components of the ER proteostasis network, N-glycans can also have intrinsic effects on protein folding by directly altering the folding energy landscape. Previous work from our laboratories (Hanson et al. Proc. Natl. Acad. Sci. U.S.A. 2009, 109, 3131−3136; Shental-Bechor, D.; Levy, Y. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 8256−8261) suggested that the three sugar residues closest to the protein are sufficient for accelerating protein folding and stabilizing the resulting structure in vitro; even a monosaccharide can have a dramatic effect. The highly conserved nature of these three proximal sugars in N-glycans led us to speculate that introducing an N-glycosylation site into a protein that is not normally glycosylated would stabilize the protein and increase its folding rate in a manner that does not depend on the presence of specific stabilizing protein−saccharide interactions. Here, we test this hypothesis experimentally and computationally by incorporating an N-linked GlcNAc residue at various positions within the Pin WW domain, a small β-sheet-rich protein. The results show that an increased folding rate and enhanced thermodynamic stability are not general, context-independent consequences of N-glycosylation. Comparison between computational predictions and experimental observations suggests that generic glycan-based excluded volume effects are responsible for the destabilizing effect of glycosylation at highly structured positions. However, this reasoning does not adequately explain the observed destabilizing effect of glycosylation within flexible loops. Our data are consistent with the hypothesis that specific, evolved protein−glycan contacts must also play an important role in mediating the beneficial energetic effects on protein folding that glycosylation can confer.
Co-reporter:Sungwook Choi ; Derrick Sek Tong Ong
Journal of the American Chemical Society 2010 Volume 132(Issue 45) pp:16043-16051
Publication Date(Web):October 21, 2010
DOI:10.1021/ja104999v
We describe a non-fluorescent, second generation stilbene that very selectively binds to transthyretin in complex biological environments and remains dark until it chemoselectively reacts with the pKa-perturbed Lys-15 ε-amino group of transthyretin to form a bright blue fluorescent conjugate. Stilbene A2 is mechanistically unusual in that it remains non-fluorescent in cell lysates lacking transthyretin, even though there is likely some proteome binding. Thus, it is especially useful for cellular imaging, as background fluorescence is undetectable until A2 reacts with transthyretin. The mechanistic basis for the effective lack of environment-sensitive fluorescence of A2 when bound to, but before reacting with, transthyretin is reported. Stilbene A2 exhibits sufficiently rapid transthyretin conjugation kinetics at 37 °C to enable pulse−chase experiments to be performed, in this case demonstrating that transthyretin is secreted from HeLa cells. As the chase compound, we employed C1, a cell-permeable, highly selective, non-covalent, transthyretin-binding dihydrostilbene that cannot become fluorescent. The progress reported is viewed as a first and necessary step toward our long-term goal of creating a one-chain, one-binding-site transthyretin tag, whose fluorescence can be regulated by adding A2 or an analogous molecule. Fusing proteins of interest to a one-chain, one-binding-site transthyretin tag regulated by A2 should be useful for studying folding, trafficking, and degradation in the cellular secretory pathway, utilizing pulse−chase experiments. Immediate applications of A2 include utilizing its conjugate fluorescence to quantify transthyretin concentration in human plasma, reflecting nutritional status, and determining the binding stoichiometry of kinetic stabilizer drugs to transthyretin in plasma.
Co-reporter:Michelle R. Bunagan ; Jianmin Gao ; Jeffery W. Kelly ;Feng Gai
Journal of the American Chemical Society 2009 Volume 131(Issue 21) pp:7470-7476
Publication Date(Web):May 8, 2009
DOI:10.1021/ja901860f
Backbone−backbone hydrogen bonds are a common feature of native protein structures, yet their thermodynamic and kinetic influence on folding has long been debated. This is reflected by the disparity between current protein folding models, which place hydrogen bond formation at different stages along the folding trajectory. For example, previous studies have suggested that the denatured state of the villin headpiece subdomain contains a residual helical structure that may provide a bias toward the folded state by confining the conformational search associated with its folding. Although helical hydrogen bonds clearly stabilize the folded state, here we show, using an amide-to-ester mutation strategy, that the formation of backbone hydrogen bonds within helices is not rate-limiting in the folding of the subdomain, thereby suggesting that such hydrogen bonds are unlikely to be formed en route from the denatured to the transition state. On the other hand, elimination of hydrogen bonds within the turn region elicits a slower folding rate, consistent with the hypothesis that these residues are involved in the formation of a folding nucleus. While illustrating a potentially conserved aspect of helix-turn-helix folding, our results further underscore the inherent importance of turns in protein supersecondary structure formation.
Co-reporter:Steven M. Johnson ; Stephen Connelly ; Ian A. Wilson
Journal of Medicinal Chemistry 2009 Volume 52(Issue 4) pp:1115-1125
Publication Date(Web):February 3, 2009
DOI:10.1021/jm801347s
Transthyretin (TTR) amyloidogenesis inhibitors are typically composed of two aromatic rings and a linker. We have previously established optimal structures for one aromatic ring and the linker. Herein, we employ a suboptimal linker and an optimal aryl-X substructure to rank order the desirability of aryl-Z substructures—using a library of 56 N-(3,5-dibromo-4-hydroxyphenyl)benzamides. Coconsideration of amyloid inhibition potency and ex vivo plasma TTR binding selectivity data reveal that 2,6, 2,5, 2, 3,4,5, and 3,5 substituted aryls bearing small substituents generate the most potent and selective inhibitors, in descending order. These benzamides generally lack undesirable thyroid hormone receptor binding and COX-1 inhibition activity. Three high-resolution TTR·inhibitor crystal structures (1.31−1.35 Å) provide insight into why these inhibitors are potent and selective, enabling future structure-based design of TTR kinetic stabilizers.
Co-reporter:James P. Solomon, Isaac T. Yonemoto, Amber N. Murray, Joshua L. Price, Evan T. Powers, William E. Balch and Jeffery W. Kelly
Biochemistry 2009 Volume 48(Issue 48) pp:
Publication Date(Web):November 11, 2009
DOI:10.1021/bi901368e
Familial amyloidosis of Finnish type (FAF), or gelsolin amyloidosis, is a systemic amyloid disease caused by a mutation (D187N/Y) in domain 2 of human plasma gelsolin, resulting in domain 2 misfolding within the secretory pathway. When D187N/Y gelsolin passes through the Golgi, furin endoproteolysis within domain 2 occurs as a consequence of the abnormal conformations that enable furin to bind and cleave, resulting in the secretion of a 68 kDa C-terminal fragment (amino acids 173−755, C68). The C68 fragment is cleaved upon secretion from the cell by membrane type 1 matrix metalloprotease (MT1-MMP), affording the 8 and 5 kDa fragments (amino acids 173−242 and 173−225, respectively) comprising the amyloid fibrils in FAF patients. Herein, we show that the 8 and 5 kDa gelsolin fragments form amyloid fibrils by a nucleated polymerization mechanism. In addition to demonstrating the expected concentration dependence of a nucleated polymerization reaction, the addition of preformed amyloid fibrils, or “seeds”, was shown to bypass the requirement for the formation of a high-energy nucleus, accelerating 8 and 5 kDa D187N gelsolin amyloidogenesis. The C68 fragment can form small oligomers, but not amyloid fibrils, even when seeded with preformed 8 kDa fragment plasma gelsolin fibrils. Because the 68 kDa fragment of gelsolin does not form amyloid fibrils in vitro or in a recently published transgenic mouse model of FAF, we propose that administration of an MT1-MMP inhibitor could be an effective strategy for the treatment of FAF.
Co-reporter:Sarah R. Hanson;Elizabeth K. Culyba;Tsui-Ling Hsu;Chi-Huey Wong;Evan T. Powers
PNAS 2009 Volume 106 (Issue 9 ) pp:3131-3136
Publication Date(Web):2009-03-03
DOI:10.1073/pnas.0810318105
The folding energetics of the mono-N-glycosylated adhesion domain of the human immune cell receptor cluster of differentiation
2 (hCD2ad) were studied systematically to understand the influence of the N-glycan on the folding energy landscape. Fully
elaborated N-glycan structures accelerate folding by 4-fold and stabilize the β-sandwich structure by 3.1 kcal/mol, relative
to the nonglycosylated protein. The N-glycan's first saccharide unit accounts for the entire acceleration of folding and for
2/3 of the native state stabilization. The remaining third of the stabilization is derived from the next 2 saccharide units.
Thus, the conserved N-linked triose core, ManGlcNAc2, improves both the kinetics and the thermodynamics of protein folding. The native state stabilization and decreased activation
barrier for folding conferred by N-glycosylation provide a powerful and potentially general mechanism for enhancing folding
in the secretory pathway.
Co-reporter:Kenji Usui;John D. Hulleman;Johan F. Paulsson;Sarah J. Siegel;Evan T. Powers
PNAS 2009 Volume 106 (Issue 44 ) pp:18563-18568
Publication Date(Web):2009-11-03
DOI:10.1073/pnas.0804758106
Accumulation of amyloid β-peptide (Aβ) and tau aggregates, possibly linked to age-associated deficiencies in protein homeostasis,
appear to cause Alzheimer's disease. Schiff-base formation between Aβ and the aldehyde-bearing cholesterol oxidation product
3-β-hydroxy-5-oxo-5,6-secocholestan-6-al is known to increase Aβ amyloidogenicity. Here, we synthesized Aβ variants site-specifically modified with
the cholesterol aldehyde at Asp-1, Lys-16, or Lys-28, rather than studying mixtures. These distinct modifications have a similar
effect on the thermodynamic propensity for aggregation, enabling aggregation at low concentrations. In contrast, the modification
site differentially influences the aggregation kinetics; Lys-16-modified Aβ formed amorphous aggregates fastest and at the
lowest concentration (within 2 h at a concentration of 20 nM), followed by the Lys-28 and Asp-1 conjugates. Also, the aggregates
resulting from Aβ Lys-16 cholesterol aldehyde conjugation were more toxic to primary rat cortical neurons than treatment with
unmodified Aβ under identical conditions and at the same concentration. Our results show that Aβ modification by cholesterol
derivatives, especially at Lys-16, renders it kinetically and thermodynamically competent to form neurotoxic aggregates at
concentrations approaching the physiologic concentration of Aβ.
Co-reporter:Lesley J. Page;Ji Young Suk;Lyudmila Bazhenova;Sheila M. Fleming;Malcolm Wood;Yun Jiang;Ling T. Guo;Andrew P. Mizisin;Robert Kisilevsky;G. Diane Shelton;William E. Balch;
Proceedings of the National Academy of Sciences 2009 106(27) pp:11125-11130
Publication Date(Web):June 19, 2009
DOI:10.1073/pnas.0811753106
Familial amyloidosis of Finnish type (FAF) is a systemic amyloid disease associated with the deposition of proteolytic fragments
of mutant (D187N/Y) plasma gelsolin. We report a mouse model of FAF featuring a muscle-specific promoter to drive D187N gelsolin
synthesis. This model recapitulates the aberrant endoproteolytic cascade and the aging-associated extracellular amyloid deposition
of FAF. Amyloidogenesis is observed only in tissues synthesizing human D187N gelsolin, despite the presence of full-length
D187N gelsolin and its 68-kDa cleavage product in blood—demonstrating the importance of local synthesis in FAF. Loss of muscle
strength was progressive in homozygous D187N gelsolin mice. The presence of misfolding-prone D187N gelsolin appears to exacerbate
the age-associated decline in cellular protein homeostasis (proteostasis), reflected by the intracellular deposition of numerous
proteins, a characteristic of the most common degenerative muscle disease of aging humans, sporadic inclusion body myositis.
Co-reporter:Amelia A. Fuller;Deguo Du;Feng Liu;Jennifer E. Davoren;Gira Bhabha;Gerard Kroon;David A. Case;H. Jane Dyson;Evan T. Powers;Peter Wipf;Martin Gruebele;
Proceedings of the National Academy of Sciences 2009 106(27) pp:11067-11072
Publication Date(Web):June 18, 2009
DOI:10.1073/pnas.0813012106
β-Turns are common conformations that enable proteins to adopt globular structures, and their formation is often rate limiting
for folding. β-Turn mimics, molecules that replace the i + 1 and i + 2 amino acid residues of a β-turn, are envisioned to act as folding nucleators by preorganizing the pendant polypeptide
chains, thereby lowering the activation barrier for β-sheet formation. However, the crucial kinetic experiments to demonstrate
that β-turn mimics can act as strong nucleators in the context of a cooperatively folding protein have not been reported.
We have incorporated 6 β-turn mimics simulating varied β-turn types in place of 2 residues in an engineered β-turn 1 or β-bulge
turn 1 of the Pin 1 WW domain, a three-stranded β-sheet protein. We present 2 lines of kinetic evidence that the inclusion
of β-turn mimics alters β-sheet folding rates, enabling us to classify β-turn mimics into 3 categories: those that are weak
nucleators but permit Pin WW folding, native-like nucleators, and strong nucleators. Strong nucleators accelerate folding
relative to WW domains incorporating all α-amino acid sequences. A solution NMR structure reveals that the native Pin WW β-sheet
structure is retained upon incorporating a strong E-olefin nucleator. These β-turn mimics can now be used to interrogate protein folding transition state structures and the
2 kinetic analyses presented can be used to assess the nucleation capacity of other β-turn mimics.
Co-reporter:Steven M. Johnson ; Stephen Connelly ; Ian A. Wilson
Journal of Medicinal Chemistry 2008 Volume 51(Issue 2) pp:260-270
Publication Date(Web):December 21, 2007
DOI:10.1021/jm0708735
To develop potent transthyretin (TTR) amyloidogenesis inhibitors that also display high binding selectivity in blood, it proves useful to systematically optimize each of the three substructural elements that comprise a typical inhibitor: the two aryl rings and the linker joining them. In the first study, described herein, structural modifications to one aryl ring were evaluated by screening a library of 2-arylbenzoxazoles bearing thyroid hormone-like aryl substituents on the 2-aryl ring. Several potent and highly selective amyloidogenesis inhibitors were identified that exhibit minimal thyroid hormone nuclear receptor and COX-1 binding. High resolution crystal structures (1.3–1.5 Å) of three inhibitors (2f, 4f, and 4d) in complex with TTR were obtained to characterize their binding orientation. Collectively, the results demonstrate that thyroid hormone-like substitution patterns on one aryl ring lead to potent and highly selective TTR amyloidogenesis inhibitors that lack undesirable thyroid hormone receptor or COX-1 binding.
Co-reporter:Steven M. Johnson ; Stephen Connelly ; Ian A. Wilson
Journal of Medicinal Chemistry 2008 Volume 51(Issue 20) pp:6348-6358
Publication Date(Web):September 24, 2008
DOI:10.1021/jm800435s
To develop potent and highly selective transthyretin (TTR) amyloidogenesis inhibitors, it is useful to systematically optimize the three substructural elements that compose a typical TTR kinetic stabilizer: the two aryl rings and the linker joining them. Herein, we evaluated 40 bisaryl molecules based on 10 unique linker substructures to determine how these linkages influence inhibitor potency and selectivity. These linkers connect one unsubstituted aromatic ring to either a 3,5-X2 or a 3,5-X2-4-OH phenyl substructure (X = Br or CH3). Coconsideration of amyloid inhibition and ex vivo plasma TTR binding selectivity data reveal that direct connection of the two aryls or linkage through nonpolar E-olefin or −CH2CH2− substructures generates the most potent and selective TTR amyloidogenesis inhibitors exhibiting minimal undesirable binding to the thyroid hormone nuclear receptor or the COX-1 enzyme. Five high-resolution TTR·inhibitor crystal structures (1.4−1.8 Å) provide insight into why such linkers afford inhibitors with greater potency and selectivity.
Co-reporter:Amy R. Hurshman Babbes, Evan T. Powers and Jeffery W. Kelly
Biochemistry 2008 Volume 47(Issue 26) pp:
Publication Date(Web):June 7, 2008
DOI:10.1021/bi800636q
Urea denaturation studies were carried out as a function of transthyretin (TTR) concentration to quantify the thermodynamically linked quaternary and tertiary structural stability and to improve our understanding of the relationship between mutant folding energetics and amyloid disease phenotype. Urea denaturation of TTR involves at least two equilibria: dissociation of tetramers into folded monomers and monomer unfolding. To deal with the thermodynamic linkage of these equilibria, we analyzed concentration-dependent denaturation data by globally fitting them to an equation that simultaneously accounts for the two-step denaturation process. Using this method, the quaternary and tertiary structural stabilities of well-behaved TTR sequences, wild-type (WT) TTR and the disease-associated variant V122I, were scrutinized. The V122I variant is linked to late onset familial amyloid cardiomyopathy, the most common familial TTR amyloid disease. V122I TTR exhibits a destabilized quaternary structure and a stable tertiary structure relative to those of WT TTR. Three other variants of TTR were also examined, L55P, V30M, and A25T TTR. The L55P mutation is associated with the most aggressive familial TTR amyloid disease. L55P TTR has a complicated denaturation pathway that includes dimers and trimers, so globally fitting its concentration-dependent urea denaturation data yielded error-laden estimates of stability parameters. Nevertheless, it is clear that L55P TTR is substantially less stable than WT TTR, primarily because its tertiary structure is unstable, although its quaternary structure is destabilized as well. V30M is the most common mutation associated with neuropathic forms of TTR amyloid disease. V30M TTR is certainly destabilized relative to WT TTR, but like L55P TTR, it has a complex denaturation pathway that cannot be fit to the aforementioned two-step denaturation model. Literature data suggest that V30M TTR has stable quaternary structure but unstable tertiary structure. The A25T mutant, associated with central nervous system amyloidosis, is highly aggregation-prone and exhibits drastically reduced quaternary and tertiary structural stabilities. The observed differences in stability among the disease-associated TTR variants highlight the complexity and heterogeneity of TTR amyloid disease, an observation that has important implications for the treatment of these maladies.
Co-reporter:Isaac T. Yonemoto, Gerard J. A. Kroon, H. Jane Dyson, William E. Balch and Jeffery W. Kelly
Biochemistry 2008 Volume 47(Issue 37) pp:
Publication Date(Web):August 19, 2008
DOI:10.1021/bi800828u
Human amylin, or islet amyloid polypeptide, is a peptide cosecreted with insulin by the beta cells of the pancreatic islets of Langerhans. The 37-residue, C-terminally amidated human amylin peptide derives from a proprotein that undergoes disulfide bond formation in the endoplasmic reticulum and is then subjected to four enzymatic processing events in the immature secretory granule. Human amylin forms both intracellular and extracellular amyloid deposits in the pancreas of most type II diabetic subjects, likely reflecting compromised secretory cell function. In addition, amylin processing intermediates, postulated to initiate intracellular amyloidogenesis, have been reported as components of intracellular amyloid in beta cells. We investigated the amyloidogenicity of amylin and its processing intermediates in vitro. Chaotrope-denatured amylin and amylin processing intermediates were subjected to size exclusion chromatography, affording high concentrations of monomeric peptides. NMR studies reveal that human amylin samples helical conformations. Under conditions mimicking the immature secretory granule (37 °C, pH 6), amylin forms amyloid aggregates more rapidly than its processing intermediates, and more rapidly than its reduced counterparts. Our studies also show that the amyloidogenicity of amylin and its processing intermediates is negatively correlated with net charge and charge at the C-terminus. Although our conditions may not precisely reflect those of amyloidogenesis in vivo, the lower amyloidogenicity of the processing intermediates relative to amylin suggests their presence in intracellular amyloid deposits in the increasingly stressed beta cells of diabetic subjects may be a consequence of general defects in protein homeostasis control known to occur in diabetes rather than serving as amyloid initiators.
Co-reporter:William E. Balch;Andrew Dillin;Richard I. Morimoto
Science 2008 Volume 319(Issue 5865) pp:916-919
Publication Date(Web):15 Feb 2008
DOI:10.1126/science.1141448
Abstract
The protein components of eukaryotic cells face acute and chronic challenges to their integrity. Eukaryotic protein homeostasis, or proteostasis, enables healthy cell and organismal development and aging and protects against disease. Here, we describe the proteostasis network, a set of interacting activities that maintain the health of proteome and the organism. Deficiencies in proteostasis lead to many metabolic, oncological, neurodegenerative, and cardiovascular disorders. Small-molecule or biological proteostasis regulators that manipulate the concentration, conformation, quaternary structure, and/or the location of protein(s) have the potential to ameliorate some of the most challenging diseases of our era.
Co-reporter:Martin Gruebele;Joseph P. Noel;Maria Dendle;Marianne E. Bowman;Marcus Jäger;Jan Bieschke;Houbi Nguyen;Yan Zhang
PNAS 2006 Volume 103 (Issue 28 ) pp:10648-10653
Publication Date(Web):2006-07-11
DOI:10.1073/pnas.0600511103
Protein folding barriers result from a combination of factors including unavoidable energetic frustration from nonnative interactions,
natural variation and selection of the amino acid sequence for function, and/or selection pressure against aggregation. The
rate-limiting step for human Pin1 WW domain folding is the formation of the loop 1 substructure. The native conformation of
this six-residue loop positions side chains that are important for mediating protein–protein interactions through the binding
of Pro-rich sequences. Replacement of the wild-type loop 1 primary structure by shorter sequences with a high propensity to
fold into a type-I′ β-turn conformation or the statistically preferred type-I G1 bulge conformation accelerates WW domain
folding by almost an order of magnitude and increases thermodynamic stability. However, loop engineering to optimize folding
energetics has a significant downside: it effectively eliminates WW domain function according to ligand-binding studies. The
energetic contribution of loop 1 to ligand binding appears to have evolved at the expense of fast folding and additional protein
stability. Thus, the two-state barrier exhibited by the wild-type human Pin1 WW domain principally results from functional
requirements, rather than from physical constraints inherent to even the most efficient loop formation process.
Co-reporter:Sean R Miller, Yoshiki Sekijima and Jeffery W Kelly
Laboratory Investigation 2004 84(5) pp:545-552
Publication Date(Web):February 16, 2004
DOI:10.1038/labinvest.3700059
Transthyretin (TTR) tetramer dissociation and misfolding affords a monomeric amyloidogenic intermediate that misassembles into aggregates including amyloid fibrils. Amyloidogenesis of wild-type (WT) TTR causes senile systemic amyloidosis (SSA), whereas fibril formation from one of the more than 80 TTR variants leads to familial amyloidosis, typically with earlier onset than SSA. Several nonsteroidal anti-inflammatory drugs (NSAIDs) stabilize the native tetramer, strongly inhibiting TTR amyloid fibril formation in vitro. Structure-based designed NSAID analogs are even more potent amyloid inhibitors. The effectiveness of several NSAIDs, including diclofenac, diflunisal, and flufenamic acid, as well as the diclofenac analog, 2–[(3,5-dichlorophenyl) amino] benzoic acid (inhibitor 1), has been demonstrated against WT TTR amyloidogenesis. Herein, the efficacy of these compounds at preventing acid-induced fibril formation and urea-induced tetramer dissociation of the most common disease-associated TTR variants (V30M, V122I, T60A, L58H, and I84S) was evaluated. Homotetramers of these variants were employed for the studies within, realizing that the tetramers in compound heterozygote patients are normally composed of a mixture of WT and variant subunits. The most common familial TTR variants were stabilized substantially by flufenamic acid and inhibitor 1, and to a lesser extent by diflunisal, against acid-mediated fibril formation and chaotrope denaturation, suggesting that this chemotherapeutic option is viable for patients with familial transthyretin amyloidosis.
Co-reporter:Qinghai Zhang;Evan T. Powers;Jorge Nieva;Mary E. Huff;Maria A. Dendle;Jan Bieschke;Charles G. Glabe;Albert Eschenmoser;Paul Wentworth, Jr.;Richard A. Lerner;
Proceedings of the National Academy of Sciences 2004 101(14) pp:4752-4757
Publication Date(Web):March 19, 2004
DOI:10.1073/pnas.0400924101
Anfinsen showed that a protein's fold is specified by its sequence. Although it is clear why mutant proteins form amyloid,
it is harder to rationalize why a wild-type protein adopts a native conformation in most individuals, but it misfolds in a
minority of others, in what should be a common extracellular environment. This discrepancy suggests that another event likely
triggers misfolding in sporadic amyloid disease. One possibility is that an abnormal metabolite, generated only in some individuals,
covalently modifies the protein or peptide and causes it to misfold, but evidence for this is sparse. Candidate metabolites
are suggested by the recently appreciated links between Alzheimer's disease (AD) and atherosclerosis, known chronic inflammatory
metabolites, and the newly discovered generation of ozone during inflammation. Here we report detection of cholesterol ozonolysis
products in human brains. These products and a related, lipid-derived aldehyde covalently modify Aβ, dramatically accelerating
its amyloidogenesis in vitro, providing a possible chemical link between hypercholesterolemia, inflammation, atherosclerosis, and sporadic AD.
Co-reporter:Shu-Li You Dr.;Jeffery W. Kelly Dr.
Chemistry - A European Journal 2004 Volume 10(Issue 1) pp:
Publication Date(Web):19 DEC 2003
DOI:10.1002/chem.200305504
The interesting biological activities of heterocycle-containing cyclic peptide-derived natural products, isolated from marine organisms over the past twenty years, have attracted the interest of many synthetic and natural products chemists. Bistratamides E–J, members of this class of natural products that were isolated very recently from Lissoclinum bistratum, exhibited cytotoxic activity against a human colon tumor (HCT-116) cell line. Here we report the first total syntheses of bistratamides E (1) and J (2) in overall yields of 19 and 34 %, respectively. The thiazole substructures have been synthesized by oxidation of their corresponding thiazoline substructures, which were obtained from cysteine containing peptides using a novel biomimetic approach wherein Val-Cys dipeptide units were converted to thiazolines by a bisphosphonium salt. The final macrocyclization was promoted efficiently using the combination of PyBOP and DMAP. This approach allows the use of readily available Fmoc-protected amino acids to make complex thiazole and oxazoline-containing natural products.
Co-reporter:Songpon Deechongkit,
Houbi Nguyen,
Evan T. Powers,
Philip E. Dawson,
Martin Gruebele
and
Jeffery W. Kelly
Nature 2004 430(6995) pp:101
Publication Date(Web):
DOI:10.1038/nature02611
Co-reporter:Hossein Razavi Dr.;Satheesh K. Palaninathan Dr.;Evan T. Powers ;R. Luke Wiseman;Hans E. Purkey Dr.;Nilofar N. Mohamedmohaideen Dr.;Songpon Deechongkit;Kyle P. Chiang;Maria T. A. Dendle;James C. Sacchettini
Angewandte Chemie International Edition 2003 Volume 42(Issue 24) pp:
Publication Date(Web):17 JUN 2003
DOI:10.1002/anie.200351179
Benzoxazoles pevent misfolding: Benzoxazole-based inhibitors of transthyretin (TTR) amyloid fibril formation are among the most effective found to date. They stabilize TTR against both acid-mediated misfolding and urea denaturation by raising the activation barrier to tetramer dissociation, the rate-limiting step for amyloid formation. The figure depicts the cocrystal structure of one of the better benzoxazole inhibitors bound to TTR.
Co-reporter:Shu-Li You;Hossein Razavi
Angewandte Chemie 2003 Volume 115(Issue 1) pp:
Publication Date(Web):10 JAN 2003
DOI:10.1002/ange.200390029
Chemiker synthetisieren Thiazoline zumeist mit Serinsubstraten, die Natur dagegen mit Cysteinresten. Eine biomimetische Methode nutzt [(Ph3P+)2O](OTf−)2 zur Abspaltung von Triphenylmethyl(Tr)-Schutzgruppen von Cystein-N-amiden und In-situ-Dehydrierung/Cyclisierung, die in ausgezeichneten Ausbeuten und Enantiomerenüberschüssen zu Thiazolinen führt (siehe Schema). Diese milde Methode wurde unter anderem auf die Synthese von Thiazolinen ausgehend von Cystein enthaltenden Dipeptiden ausgedehnt, die in hohen Ausbeuten und ohne nennenswerte Chiralitätseinbußen am C2-Exomethin-C-Atom verläuft.
Co-reporter:Hossein Razavi Dr.;Satheesh K. Palaninathan Dr.;Evan T. Powers ;R. Luke Wiseman;Hans E. Purkey Dr.;Nilofar N. Mohamedmohaideen Dr.;Songpon Deechongkit;Kyle P. Chiang;Maria T. A. Dendle;James C. Sacchettini
Angewandte Chemie 2003 Volume 115(Issue 24) pp:
Publication Date(Web):17 JUN 2003
DOI:10.1002/ange.200351179
Fester Zusammenhalt: Wirkstoffe auf Benzoxazol-Basis zählen zu den effektivsten Inhibitoren der Bildung von Amyloidfibrillen aus Transthyretin (TTR). Zum einen stabilisieren sie TTR gegen Fehlfaltungen infolge Säureeinwirkung, zum anderen vergrößern sie die Aktivierungsbarriere für die Dissoziation der TTR-Tetramere, den geschwindigkeitsbestimmenden Schritt der Amyloidbildung. Unser Bild zeigt die Struktur eines Komplexes von TTR mit einem Benzoxazolinhibitor im Kristall.
Co-reporter:Shu-Li You;Hossein Razavi
Angewandte Chemie International Edition 2003 Volume 42(Issue 1) pp:
Publication Date(Web):14 JAN 2003
DOI:10.1002/anie.200390059
Most chemical syntheses of thiazolines use serine residues, whereas nature employs cysteine residues. In a biomimetic approach, [(Ph3P+)2O](OTf−)2 was used to promote triphenylmethyl (Tr) deprotection and dehydrocyclization of simple cysteine N-amides to give thiazolines in excellent chemical and optical yields (see scheme). This mild method was extended to the synthesis of, for example, thiazolines from cysteine-containing dipeptides in high yield and without significant loss of chirality at the C2-exomethine carbon atom. TF=trifluoromethanesulfonyl, Cbz=benzyloxycarbonyl
Co-reporter:Jeffery W. Kelly;Martin Gruebele;Houbi Nguyen;Alessandro Moretto;Marcus Jäger
PNAS 2003 Volume 100 (Issue 7 ) pp:3948-3953
Publication Date(Web):2003-04-01
DOI:10.1073/pnas.0538054100
The equilibrium unfolding of the Formin binding protein 28 (FBP) WW domain, a stable three-stranded β-sheet protein, can be
described as reversible apparent two-state folding. Kinetics studied by laser temperature jump reveal a third state at temperatures
below the midpoint of unfolding. The FBP free-energy surface can be tuned between three-state and two-state kinetics by changing
the temperature, by truncation of the C terminus, or by selected point mutations. FBP WW domain is the smallest three-state
folder studied to date and the only one that can be freely tuned between three-state and apparent two-state folding by several
methods (temperature, truncation, and mutation). Its small size (28–37 residues), the availability of a quantitative reaction
coordinate (φT), the fast folding time scale (10s of μs), and the tunability of the folding routes by small temperature or sequence changes
make this system the ideal prototype for studying more subtle features of the folding free-energy landscape by simulations
or analytical theory.
Co-reporter:Wei-Chieh Cheng;Anu R. Sawkar;Ernest Beutler;William E. Balch;Chi-Huey Wong
PNAS 2002 Volume 99 (Issue 24 ) pp:15428-15433
Publication Date(Web):2002-11-26
DOI:10.1073/pnas.192582899
Gaucher disease is a lysosomal storage disorder caused by deficient lysosomal β-glucosidase (β-Glu) activity. A marked decrease
in enzyme activity results in progressive accumulation of the substrate (glucosylceramide) in macrophages, leading to hepatosplenomegaly,
anemia, skeletal lesions, and sometimes CNS involvement. Enzyme replacement therapy for Gaucher disease is costly and relatively
ineffective for CNS involvement. Chemical chaperones have been shown to stabilize various proteins against misfolding, increasing
proper trafficking from the endoplasmic reticulum. We report herein that the addition of subinhibitory concentrations (10
μM) of N-(n-nonyl)deoxynojirimycin (NN-DNJ) to a fibroblast culture medium for 9 days leads to a 2-fold increase in the activity of N370S
β-Glu, the most common mutation causing Gaucher disease. Moreover, the increased activity persists for at least 6 days after
the withdrawal of the putative chaperone. The NN-DNJ chaperone also increases WT β-Glu activity, but not that of L444P, a
less prevalent Gaucher disease variant. Incubation of isolated soluble WT enzyme with NN-DNJ reveals that β-Glu is stabilized
against heat denaturation in a dose-dependent fashion. We propose that NN-DNJ chaperones β-Glu folding at neutral pH, thus
allowing the stabilized enzyme to transit from the endoplasmic reticulum to the Golgi, enabling proper trafficking to the
lysosome. Clinical data suggest that a modest increase in β-Glu activity may be sufficient to achieve a therapeutic effect.
Co-reporter:Evan T. Powers ;Sung Ik Yang Dr.;Charles M. Lieber
Angewandte Chemie 2002 Volume 114(Issue 1) pp:
Publication Date(Web):4 JAN 2002
DOI:10.1002/1521-3757(20020104)114:1<135::AID-ANGE135>3.0.CO;2-1
Die Peptidlänge beeinflusst die Form der mit Kraftmikroskopie (AFM) aufgenommenen Grate der Langmuir-Blodgett-Filme aus amphiphilen Peptiden: Wohlgeordnete LB-Filme (links) können aus einem 14-gliedrigen amphiphilen Peptid, weniger geordnete LB-Filme mit einem breiteren Gitter (rechts) aus einem 18-gliedrigen Peptid hergestellt werden. Die gezeigten Bilder mit einer Auflösung von 100×100 nm wurden mit AFM unter Verwendung von Spitzen aus Kohlenstoff-Nanoröhren erhalten.
Co-reporter:Jennifer A. Kowalski;Kai Liu
Biopolymers 2002 Volume 63(Issue 2) pp:
Publication Date(Web):2 JAN 2002
DOI:10.1002/bip.10020
The NMR solution structure of the isolated Apo Pin1 WW domain (6–39) reveals that it adopts a twisted three-stranded antiparallel β-sheet conformation, very similar to the structure exhibited by the crystal of this domain in the context of the two domain Pin1 protein. While the B factors in the apo x-ray crystal structure indicate that loop 1 and loop 2 are conformationally well defined, the solution NMR data suggest that loop 1 is quite flexible, at least in the absence of the ligand. The NMR chemical shift and nuclear Overhauser effect pattern exhibited by the 6–39 Pin1 WW domain has proven to be diagnostic for demonstrating that single site variants of this domain adopt a normally folded structure. Knowledge of this type is critical before embarking on time-consuming kinetic and thermodynamic studies required for a detailed understanding of β-sheet folding. © 2002 John Wiley & Sons, Inc. Biopolymers 63: 111–121, 2002
Co-reporter:Evan T. Powers ;Sung Ik Yang Dr.;Charles M. Lieber
Angewandte Chemie International Edition 2002 Volume 41(Issue 1) pp:
Publication Date(Web):2 JAN 2002
DOI:10.1002/1521-3773(20020104)41:1<127::AID-ANIE127>3.0.CO;2-F
Peptide length affects the size of the ridges observed in the atomic force microscopy (AFM) images of the Langmuir–Blodgett films of amphiphilic peptides: Well-ordered LB films can be prepared from a 14-residue amphiphilic peptide (left), while ordered LB films with a wider lattice (right) are obtained from an 18-residue peptide. The 100 nm×100 nm images pictured were obtained by AFM using carbon nanotube tips.
Co-reporter:
Nature Structural and Molecular Biology 2002 9(5) pp:323 - 325
Publication Date(Web):
DOI:10.1038/nsb0502-323
Co-reporter:Jeffery W. Kelly;Michael I. Dorrell;Hans E. Purkey
PNAS 2001 Volume 98 (Issue 10 ) pp:5566-5571
Publication Date(Web):2001-05-08
DOI:10.1073/pnas.091431798
Transthyretin (TTR) tetramer dissociation and misfolding facilitate
assembly into amyloid fibrils that putatively cause senile systemic
amyloidosis and familial amyloid polyneuropathy. We have previously
discovered more than 50 small molecules that bind to and stabilize
tetrameric TTR, inhibiting amyloid fibril formation in
vitro. A method is presented here to evaluate the binding
selectivity of these inhibitors to TTR in human plasma, a complex
biological fluid composed of more than 60 proteins and numerous small
molecules. Our immunoprecipitation approach isolates TTR and bound
small molecules from a biological fluid such as plasma, and quantifies
the amount of small molecules bound to the protein by HPLC analysis.
This approach demonstrates that only a small subset of the inhibitors
that saturate the TTR binding sites in vitro do so in
plasma. These selective inhibitors can now be tested in animal models
of TTR amyloid disease to probe the validity of the amyloid hypothesis.
This method could be easily extended to evaluate small molecule binding
selectivity to any protein in a given biological fluid without the
necessity of determining or guessing which other protein components may
be competitors. This is a central issue to understanding the
distribution, metabolism, activity, and toxicity of potential drugs.
Co-reporter:Joleen T. White
PNAS 2001 Volume 98 (Issue 23 ) pp:13019-13024
Publication Date(Web):2001-11-06
DOI:10.1073/pnas.241406698
The amyloidoses are a large group of protein misfolding diseases. Genetic and biochemical evidence support the hypothesis
that amyloid formation from wild-type or 1 of 80 sequence variants of transthyretin causes the human amyloid diseases senile
systemic amyloidosis or familial amyloid polyneuropathy, respectively. The late onset and variable penetrance of these diseases
has led to their designation as multigenic—implying that the expression levels and alleles of multiple gene products influence
the course of pathology. Here we show that the binding stoichiometry of three interacting molecules, retinol-binding protein,
vitamin A, and l-thyroxine, notably influenced transthyretin amyloidogenicity in vitro. At least 70 genes control retinol-binding protein, vitamin A, and l-thyroxine levels in plasma and have the potential to modulate the course of senile systemic amyloidosis or familial amyloid
polyneuropathy.
Co-reporter:Gayathri Ratnaswamy;Mary E. Huff;Severine Rion;Andrew I. Su
PNAS 2001 Volume 98 (Issue 5 ) pp:2334-2339
Publication Date(Web):2001-02-27
DOI:10.1073/pnas.041452598
Mutations at position 187 in secreted gelsolin enable aberrant
proteolysis at the 172–173 and 243–244 amide bonds, affording the
71-residue amyloidogenic peptide deposited in Familial Amyloidosis of
Finnish Type (FAF). Thermodynamic comparisons of two different domain 2
constructs were carried out to study possible effects of the mutations
on proteolytic susceptibility. In the construct we consider to be most
representative of domain 2 in the context of the full-length protein
(134–266), the D187N FAF variant is slightly destabilized relative to
wild type (WT) under the conditions of urea denaturation, but exhibits
a Tm identical to WT. The D187Y variant is
less stable to intermediate urea concentrations and exhibits a
Tm that is estimated to be ≈5°C lower
than WT (pH 7.4, Ca2+-free). Although the thermodynamic
data indicate that the FAF mutations may slightly destabilize domain 2,
these changes are probably not sufficient to shift the native to
denatured state equilibrium enough to enable the proteolysis leading to
FAF. Biophysical data indicate that these two FAF variants may have
different native state structures and possibly different pathways of
amyloidosis.
Co-reporter:Xin Jiang;Joel N. Buxbaum
PNAS 2001 Volume 98 (Issue 26 ) pp:14943-14948
Publication Date(Web):2001-12-18
DOI:10.1073/pnas.261419998
The transthyretin (TTR) amyloid diseases are of keen
interest, because there are >80 mutations that cause, and a few
mutations that suppress, disease. The V122I variant is the most common
amyloidogenic mutation worldwide, producing familial amyloidotic
cardiomyopathy primarily in individuals of African descent. The
substitution shifts the tetramer-folded monomer equilibrium toward
monomer (lowers tetramer stability) and lowers the kinetic barrier
associated with rate-limiting tetramer dissociation (pH 7; relative to
wild-type TTR) required for amyloid fibril formation. Fibril formation
is also accelerated because the folded monomer resulting from the
tetramer-folded monomer equilibrium rapidly undergoes partial
denaturation and self-assembles into amyloid (in vitro)
when subjected to a mild denaturation stress (e.g., pH 4.8).
Incorporation of the V122I mutation into a folded monomeric variant of
transthyretin reveals that this mutation does not destabilize the
tertiary structure or alter the rate of amyloidogenesis relative to the
wild-type monomer. The increase in the velocity of rate-limiting
tetramer dissociation coupled with the lowered tetramer stability
(increasing the mol fraction of folded monomer present at equilibrium)
may explain why V122I confers an apparent absolute anatomic risk for
cardiac amyloid deposition.
Co-reporter:
Nature Structural and Molecular Biology 2000 7(10) pp:824-826
Publication Date(Web):
DOI:10.1038/82815
Limitations of the three putative mechanisms of amyloid fibril assembly for explaining Sup35 prion formation have led to the introduction of a fourth model — nucleated conformational conversion — that invokes oligomeric intermediates for initiation and assembly.
Co-reporter:Derrick Sek Tong Ong, Jeffery W Kelly
Current Opinion in Cell Biology (April 2011) Volume 23(Issue 2) pp:231-238
Publication Date(Web):1 April 2011
DOI:10.1016/j.ceb.2010.11.002
Inheriting a mutant misfolding-prone protein that cannot be efficiently folded in a given cell type(s) results in a spectrum of human loss-of-function misfolding diseases. The inability of the biological protein maturation pathways to adapt to a specific misfolding-prone protein also contributes to pathology. Chemical and biological therapeutic strategies are presented that restore protein homeostasis, or proteostasis, either by enhancing the biological capacity of the proteostasis network or through small molecule stabilization of a specific misfolding-prone protein. Herein, we review the recent literature on therapeutic strategies to ameliorate protein misfolding diseases that function through either of these mechanisms, or a combination thereof, and provide our perspective on the promise of alleviating protein misfolding diseases by taking advantage of proteostasis adaptation.
Co-reporter:Xin Zhang, Jeffery W. Kelly
Journal of Molecular Biology (29 July 2014) Volume 426(Issue 15) pp:2736-2738
Publication Date(Web):29 July 2014
DOI:10.1016/j.jmb.2014.06.001
Co-reporter:Gareth J. Morgan, Jeffery W. Kelly
Journal of Molecular Biology (23 October 2016) Volume 428(Issue 21) pp:4280-4297
Publication Date(Web):23 October 2016
DOI:10.1016/j.jmb.2016.08.021
•Aggregation of antibody LCs is associated with AL amyloidosis.•Full-length LCs are much less amyloidogenic than their variable domains.•LCs from AL amyloidosis patients are less kinetically stable than other LCs.•Amyloidogenesis may be initiated by endoproteolysis of kinetically unstable LCs.•Proteolytic fragments of LCs can form amyloid fibrils.Light chain (LC) amyloidosis (AL amyloidosis) appears to be caused by the misfolding, or misfolding and aggregation of an antibody LC or fragment thereof and is fatal if untreated. LCs are secreted from clonally expanded plasma cells, generally as disulfide-linked dimers, with each monomer comprising one constant and one variable domain. The energetic contribution of each domain and the role of endoproteolysis in AL amyloidosis remain unclear. To investigate why only some LCs form amyloid and cause organ toxicity, we measured the aggregation propensity and kinetic stability of LC dimers and their associated variable domains from AL amyloidosis patients and non-patients. All the variable domains studied readily form amyloid fibrils, whereas none of the full-length LC dimers, even those from AL amyloidosis patients, are amyloidogenic. Kinetic stability—that is, the free energy difference between the native state and the unfolding transition state—dictates the LC's unfolding rate. Full-length LC dimers derived from AL amyloidosis patients unfold more rapidly than other full-length LC dimers and can be readily cleaved into their component domains by proteases, whereas non-amyloidogenic LC dimers are more kinetically stable and resistant to endoproteolysis. Our data suggest that amyloidogenic LC dimers are kinetically unstable (unfold faster) and are thus susceptible to endoproteolysis that results in the release amyloidogenic LC fragments, whereas other LCs are not as amenable to unfolding and endoproteolysis and are therefore aggregation resistant. Pharmacologic kinetic stabilization of the full-length LC dimer could be a useful strategy to treat AL amyloidosis.Download high-res image (154KB)Download full-size image
Co-reporter:R. Luke Wiseman, Jeffery W. Kelly
Molecular Cell (11 December 2009) Volume 36(Issue 5) pp:730-731
Publication Date(Web):11 December 2009
DOI:10.1016/j.molcel.2009.11.023
In this issue of Molecular Cell, Foit et al. (2009) probe cellular protein folding using a split β-lactamase approach for evolving protein stability in the absence of any requirement for function.
Co-reporter:Steven M. Johnson, Stephen Connelly, Colleen Fearns, Evan T. Powers, Jeffery W. Kelly
Journal of Molecular Biology (10 August 2012) Volume 421(Issues 2–3) pp:185-203
Publication Date(Web):10 August 2012
DOI:10.1016/j.jmb.2011.12.060
Transthyretin (TTR) is one of the many proteins that are known to misfold and aggregate (i.e., undergo amyloidogenesis) in vivo. The process of TTR amyloidogenesis causes nervous system and/or heart pathology. While several of these maladies are associated with mutations that destabilize the native TTR quaternary and/or tertiary structure, wild-type TTR amyloidogenesis also leads to the degeneration of postmitotic tissue. Over the past 20 years, much has been learned about the factors that influence the propensity of TTR to aggregate. This biophysical information led to the development of a therapeutic strategy, termed “kinetic stabilization,” to prevent TTR amyloidogenesis. This strategy afforded the drug tafamidis which was recently approved by the European Medicines Agency for the treatment of TTR familial amyloid polyneuropathy, the most common familial TTR amyloid disease. Tafamidis is the first and currently the only medication approved to treat TTR familial amyloid polyneuropathy. Here we review the biophysical basis for the kinetic stabilization strategy and the structure-based drug design effort that led to this first-in-class pharmacologic agent.Download high-res image (335KB)Download full-size imageHighlights► Wild-type and mutant TTRs cause amyloid diseases in humans. ► Amyloid formation requires tetramer dissociation (rate limiting) and monomer misfolding. ► TTR tetramers can be kinetically stabilized by binding small molecules. ► Tafamidis, a kinetic stabilizer of TTR, is the first drug approved to treat TTR amyloidosis.
Co-reporter:Fernando L. Palhano ; Jiyong Lee ; Neil P. Grimster
Journal of the American Chemical Society () pp:
Publication Date(Web):April 23, 2013
DOI:10.1021/ja3115696
Protein misfolding and/or aggregation has been implicated as the cause of several human diseases, such as Alzheimer’s and Parkinson’s diseases and familial amyloid polyneuropathy. These maladies are referred to as amyloid diseases, named after the cross-β-sheet amyloid fibril aggregates or deposits common to these disorders. Epigallocatechin-3-gallate (EGCG), the principal polyphenol present in green tea, has been shown to be effective at preventing aggregation and is able to remodel amyloid fibrils comprising different amyloidogenic proteins, although the mechanistic underpinnings are unclear. Herein, we work toward an understanding of the molecular mechanism(s) by which EGCG remodels mature amyloid fibrils made up of Aβ1–40, IAPP8–24, or Sup35NM7–16. We show that EGCG amyloid remodeling activity in vitro is dependent on auto-oxidation of the EGCG. Oxidized and unoxidized EGCG binds to amyloid fibrils, preventing the binding of thioflavin T. This engagement of the hydrophobic binding sites in Aβ1–40, IAPP8–24, or Sup35NMAc7–16 Y→F amyloid fibrils seems to be sufficient to explain the majority of the amyloid remodeling observed by EGCG treatment, although how EGCG oxidation drives remodeling remains unclear. Oxidized EGCG molecules react with free amines within the amyloid fibril through the formation of Schiff bases, cross-linking the fibrils, which may prevent dissociation and toxicity, but these aberrant post-translational modifications do not appear to be the major driving force for amyloid remodeling by EGCG treatment. These insights into the molecular mechanism of action of EGCG provide boundary conditions for exploring amyloid remodeling in more detail.
.jpg)
![1H-Thieno[3,4-d]imidazole-4-pentanamide, hexahydro-2-oxo-N-2-propyn-1-yl-, (3aS,4S,6aR)-](/data/chemimg/103200/773888-45-2.png)
![1H-Thieno[3,4-d]imidazole-4-pentanamide, hexahydro-2-oxo-N-2-propyn-1-yl-, (3aS,4S,6aR)-](/data/chemimg/103200/773888-45-2_b.png)
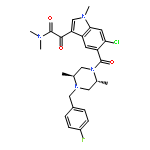
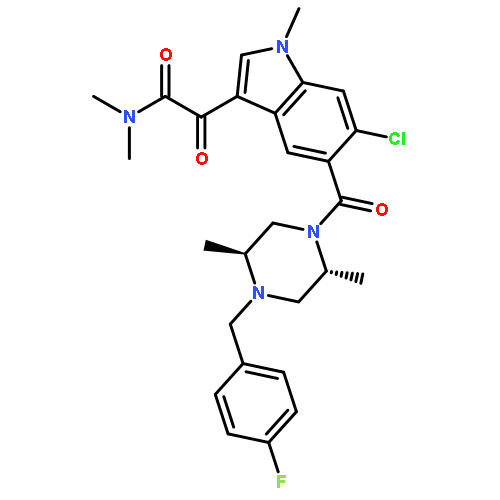
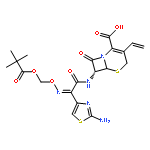
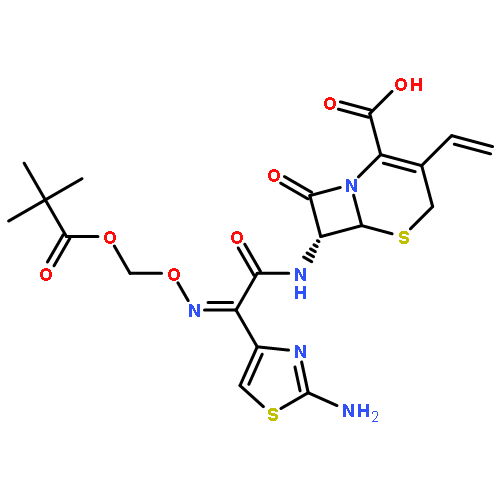




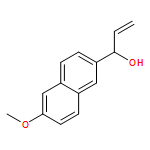
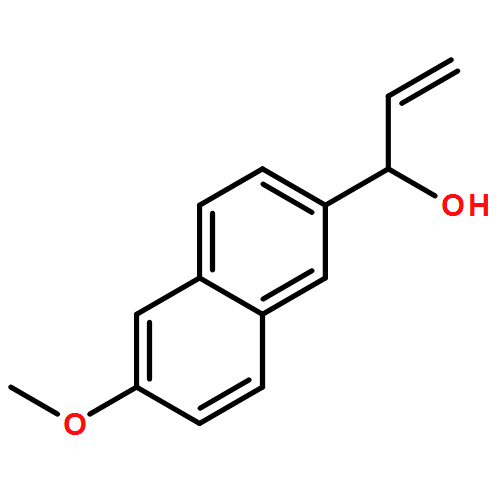




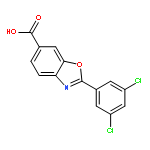
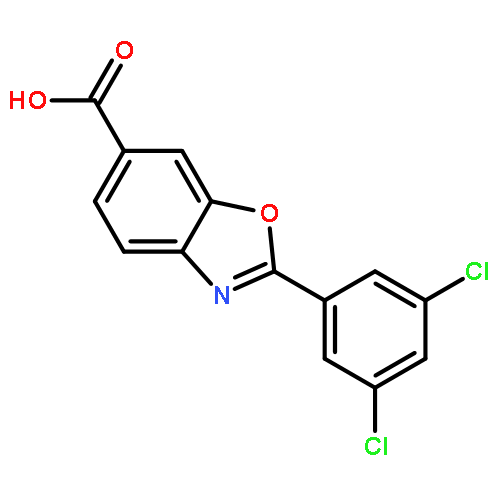

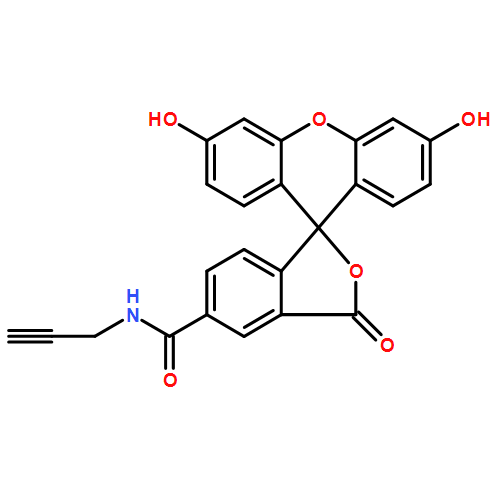






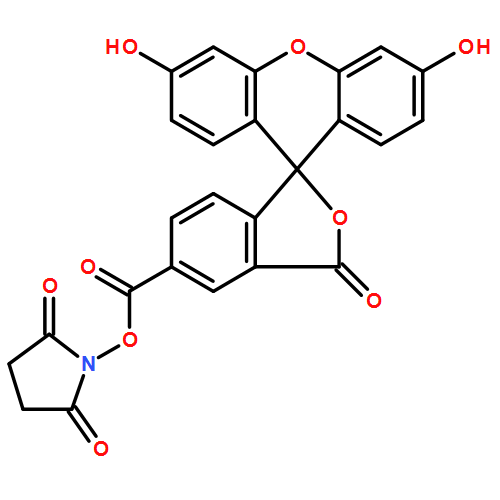


![Ethanol, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/964400/86520-52-7.png)
![Ethanol, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/964400/86520-52-7_b.png)
![[(2R,3R,4S,5S,6R)-3,4,5-triacetyloxy-6-azidooxan-2-yl]methyl acetate](http://img.cochemist.com/ccimg/65900/65864-60-0.png)
![[(2R,3R,4S,5S,6R)-3,4,5-triacetyloxy-6-azidooxan-2-yl]methyl acetate](http://img.cochemist.com/ccimg/65900/65864-60-0_b.png)
















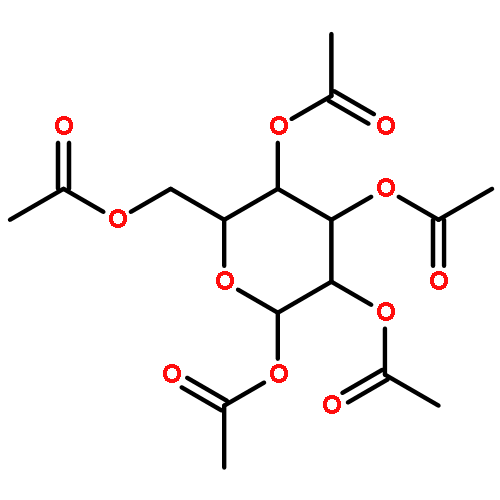
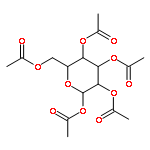


![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8.png)
![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8_b.png)

