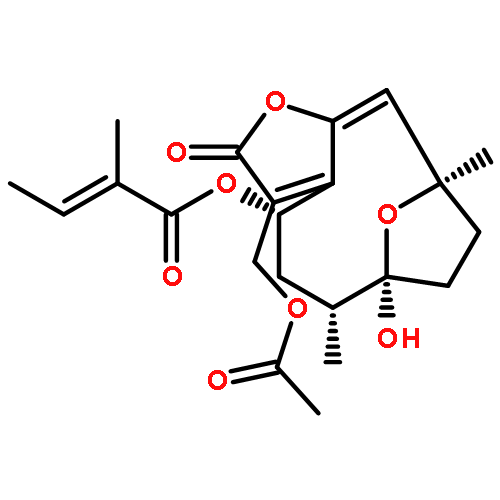
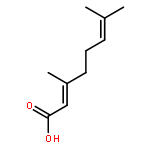
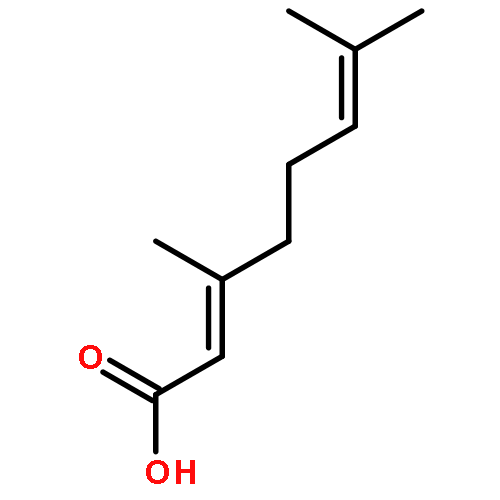
Co-reporter:Xufen Yu, Mingming Zhang, Thirunavukkarasu Annamalai, Priyanka Bansod, Gagandeep Narula, Yuk-Ching Tse-Dinh, Dianqing Sun
European Journal of Medicinal Chemistry 2017 Volume 125() pp:515-527
Publication Date(Web):5 January 2017
DOI:10.1016/j.ejmech.2016.09.053
•Fluoroquinophenoxazines were synthesized as bacterial topoisomerase IA inhibitors.•Some derivatives showed excellent inhibitory activity against topoisomerase IA.•CoMFA analysis was performed to investigate the 3D-QSAR of this chemical series.•The constructed CoMFA model produced reasonable and good statistics.New antibacterial agents with novel target and mechanism of action are urgently needed to combat problematic bacterial infections and mounting antibiotic resistances. Topoisomerase IA represents an attractive and underexplored antibacterial target, as such, there is a growing interest in developing selective and potent topoisomerase I inhibitors for antibacterial therapy. Based on our initial biological screening, fluoroquinophenoxazine 1 was discovered as a low micromolar inhibitor against E. coli topoisomerase IA. In the literature, fluoroquinophenoxazine analogs have been investigated as antibacterial and anticancer agents, however, their topoisomerase I inhibition was relatively underexplored and there is little structure-activity relationship (SAR) available. The good topoisomerase I inhibitory activity of 1 and the lack of SAR prompted us to design and synthesize a series of fluoroquinophenoxazine analogs to systematically evaluate the SAR and to probe the structural elements of the fluoroquinophenoxazine core toward topoisomerase I enzyme target recognition. In this study, a series of fluoroquinophenoxazine analogs was designed, synthesized, and evaluated as topoisomerase I inhibitors and antibacterial agents. Target-based assays revealed that the fluoroquinophenoxazine derivatives with 9-NH2 and/or 6-substituted amine functionalities generally exhibited good to excellent inhibitory activities against topoisomerase I with IC50s ranging from 0.24 to 3.9 μM. Notably, 11a bearing the 6-methylpiperazinyl and 9-amino motifs was identified as one of the most potent topoisomerase I inhibitors (IC50 = 0.48 μM), and showed broad spectrum antibacterial activity (MICs = 0.78–7.6 μM) against all the bacteria strains tested. Compound 11g with the 6-bipiperidinyl lipophilic side chain exhibited the most potent antituberculosis activity (MIC = 2.5 μM, SI = 9.8). In addition, CoMFA analysis was performed to investigate the 3D-QSAR of this class of fluoroquinophenoxazine derivatives. The constructed CoMFA model produced reasonable statistics (q2 = 0.688 and r2 = 0.806). The predictive power of the developed model was obtained using a test set of 7 compounds, giving a predictive correlation coefficient r2pred of 0.767. Collectively, these promising data demonstrated that fluoroquinophenoxazine derivatives have the potential to be developed as a new chemotype of potent topoisomerase IA inhibitors with antibacterial therapeutic potential.
Co-reporter:Allan M. Prior, Xufen Yu, Eun-Jung Park, Tamara P. Kondratyuk, Yan Lin, John M. Pezzuto, Dianqing Sun
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 24(Issue 24) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.bmcl.2017.11.010
In our ongoing effort of discovering anticancer and chemopreventive agents, a series of 2-arylindole derivatives were synthesized and evaluated toward aromatase and quinone reductase 1 (QR1). Biological evaluation revealed that several compounds (e.g., 2d, IC50 = 1.61 μM; 21, IC50 = 3.05 μM; and 27, IC50 = 3.34 μM) showed aromatase inhibitory activity with half maximal inhibitory concentration (IC50) values in the low micromolar concentrations. With regard to the QR1 induction activity, 11 exhibited the highest QR1 induction ratio (IR) with a low concentration to double activity (CD) value (IR = 8.34, CD = 2.75 μM), while 7 showed the most potent CD value of 1.12 μM. A dual acting compound 24 showed aromatase inhibition (IC50 = 9.00 μM) as well as QR1 induction (CD = 5.76 μM) activities. Computational docking studies using CDOCKER (Discovery Studio 3.5) provided insight in regard to the potential binding modes of 2-arylindoles within the aromatase active site. Predominantly, the 2-arylindoles preferred binding with the 2-aryl group toward a small hydrophobic pocket within the active site. The C-5 electron withdrawing group on indole was predicted to have an important role and formed a hydrogen bond with Ser478 (OH). Alternatively, meta-pyridyl analogs may orient with the pyridyl 3′-nitrogen coordinating with the heme group.Download high-res image (64KB)Download full-size image
Co-reporter:Lissa S. Tsutsumi, Ghee T. Tan, Dianqing Sun
Tetrahedron Letters 2017 Volume 58, Issue 27(Issue 27) pp:
Publication Date(Web):5 July 2017
DOI:10.1016/j.tetlet.2017.05.084
•Solid-phase synthesis of wollamides A, B and desotamide B has been developed.•Optimization studies toward synthesis of cyclohexapeptides were performed.•Cyclization efficiency including terminal residues and coupling reagents was studied.•The 1H NMR discrepancies between the free base and TFA salt forms of wollamides were noted.Solid-phase synthesis of antibacterial cyclohexapeptides including wollamides A, B and desotamide B has been developed. Briefly, the protected linear hexapeptides were assembled on 2-chlorotrityl chloride resin using standard Fmoc chemistry and diisopropylcarbodiimide/hydroxybenzotriazole coupling reagents, cleaved off-resin with hexafluoroisopropanol/dichloromethane to keep side-chain protecting groups intact, and cyclized in solution. Final global removal of all protecting groups using a cocktail of trifluoroacetic acid/triisopropylsilane/dichloromethane afforded the desired cyclic hexapeptides, which were characterized by 1H, 13C NMR, and HRMS. Subsequent investigation of macrocyclization parameters such as terminal residues, coupling reagents, and cyclization concentration revealed the optimized conditions for the synthesis of this class of cyclic hexapeptides.Download high-res image (128KB)Download full-size image
Co-reporter:Hao Lin, David F. Bruhn, Marcus M. Maddox, Aman P. Singh, Richard E. Lee, Dianqing Sun
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 16) pp:4070-4076
Publication Date(Web):15 August 2016
DOI:10.1016/j.bmcl.2016.06.075
Bacterial infections, caused by Mycobacterium tuberculosis and other problematic bacterial pathogens, continue to pose a significant threat to global public health. As such, new chemotype antibacterial agents are desperately needed to fuel and strengthen the antibacterial drug discovery and development pipeline. As part of our antibacterial research program to develop natural product-inspired new antibacterial agents, here we report synthesis, antibacterial evaluation, and structure–activity relationship studies of an extended chemical library of macrocyclic diarylheptanoids with diverse amine, amide, urea, and sulfonamide functionalities. Results of this study have produced macrocyclic geranylamine and 4-fluorophenethylamine substituted derivatives, exhibiting moderate to good activity against M. tuberculosis and selected Gram-positive bacterial pathogens.
Co-reporter:Li Feng ; Marcus M. Maddox ; Md. Zahidul Alam ; Lissa S. Tsutsumi ; Gagandeep Narula ; David F. Bruhn ; Xiaoqian Wu ; Shayna Sandhaus ; Robin B. Lee ; Charles J. Simmons ; Yuk-Ching Tse-Dinh ; Julian G. Hurdle ; Richard E. Lee
Journal of Medicinal Chemistry 2014 Volume 57(Issue 20) pp:8398-8420
Publication Date(Web):September 19, 2014
DOI:10.1021/jm500853v
On the basis of recently reported abyssinone II and olympicin A, a series of chemically modified flavonoid phytochemicals were synthesized and evaluated against Mycobacterium tuberculosis and a panel of Gram-positive and -negative bacterial pathogens. Some of the synthesized compounds exhibited good antibacterial activities against Gram-positive pathogens including methicillin resistant Staphylococcus aureus with minimum inhibitory concentration as low as 0.39 μg/mL. SAR analysis revealed that the 2-hydrophobic substituent and the 4-hydrogen bond donor/acceptor of the 4-chromanone scaffold together with the hydroxy groups at 5- and 7-positions enhanced antibacterial activities; the 2′,4′-dihydroxylated A ring and the lipophilic substituted B ring of chalcone derivatives were pharmacophoric elements for antibacterial activities. Mode of action studies performed on selected compounds revealed that they dissipated the bacterial membrane potential, resulting in the inhibition of macromolecular biosynthesis; further studies showed that selected compounds inhibited DNA topoisomerase IV, suggesting complex mechanisms of actions for compounds in this series.
Co-reporter:Hao Lin, Thirunavukkarasu Annamalai, Priyanka Bansod, Yuk-Ching Tse-Dinh and Dianqing Sun
MedChemComm 2013 vol. 4(Issue 12) pp:1613-1618
Publication Date(Web):15 Oct 2013
DOI:10.1039/C3MD00238A
Naturally occurring anziaic acid has very recently been reported as a topoisomerase I inhibitor with antibacterial activity. Herein total synthesis of anziaic acid and its structural analogues is described and the preliminary structure–activity relationship (SAR) has been developed based on topoisomerase inhibition and whole cell antibacterial activity.
Co-reporter:Li Shen, Marcus M Maddox, Sudip Adhikari, David F Bruhn, Manish Kumar, Robin E Lee, Julian G Hurdle, Richard E Lee and Dianqing Sun
The Journal of Antibiotics 2013 66(6) pp:319-325
Publication Date(Web):April 3, 2013
DOI:10.1038/ja.2013.21
The natural product engelhardione is an underexplored chemotype for developing novel treatments for bacterial infections; we therefore explored this natural product scaffold for chemical diversification and structure–activity relationship studies. Macrocyclic engelhardione and structural regioisomers were synthesized using a series of aldol condensations and selective hydrogenations to generate the 1,7-diarylheptan-3-one derivatives, followed by microwave-assisted intramolecular Ullmann coupling to afford a series of macrocyclic diaryl ether analogs. An extended macrocyclic chemical library was then produced by oxime formation, reductive amination and O-alkylation. Antibacterial evaluation revealed that the reductive amination derivatives 7b and 7d showed moderate activities (minimum inhibitory concentrations: 12.5–25 μg ml−1) against Mycobacterium tuberculosis and Gram-positive pathogens, as well as anti-Gram-negative activity against an efflux impaired Escherichia coli strain. These results provide validated leads for further optimization and development.
Co-reporter:Xufen Yu, Eun-Jung Park, Tamara P. Kondratyuk, John M. Pezzuto and Dianqing Sun
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 44) pp:8835-8847
Publication Date(Web):13 Sep 2012
DOI:10.1039/C2OB26456K
Development of small molecule drug-like inhibitors blocking both nitric oxide synthase and NFκB could offer a synergistic therapeutic approach in the prevention and treatment of inflammation and cancer. During the course of evaluating the biological potential of a commercial compound library, 2-phenylindole (1) displayed inhibitory activity against nitrite production and NFκB with IC50 values of 38.1 ± 1.8 and 25.4 ± 2.1 μM, respectively. Based on this lead, synthesis and systematic optimization have been undertaken in an effort to find novel and more potent nitric oxide synthase and NFκB inhibitors with antiinflammatory and cancer preventive potential. First, chemical derivatizations of 1 and 2-phenylindole-3-carboxaldehyde (4) were performed to generate a panel of N-alkylated indoles and 3-oxime derivatives 2–7. Second, a series of diversified 2-arylindole derivatives (10) were synthesized from an array of substituted 2-iodoanilines (8) and terminal alkynes (9) by applying a one-pot palladium catalyzed Sonogashira-type alkynylation and base-assisted cycloaddition. Subsequent biological evaluations revealed 3-carboxaldehyde oxime and cyano substituted 2-phenylindoles 5 and 7 exhibited the strongest nitrite inhibitory activities (IC50 = 4.4 ± 0.5 and 4.8 ± 0.4 μM, respectively); as well as NFκB inhibition (IC50 = 6.9 ± 0.8 and 8.5 ± 2.0 μM, respectively). In addition, the 6′-MeO-naphthalen-2′-yl indole derivative 10at displayed excellent inhibitory activity against NFκB with an IC50 value of 0.6 ± 0.2 μM.
Co-reporter:Li Shen, Charles J. Simmons, Dianqing Sun
Tetrahedron Letters 2012 Volume 53(Issue 32) pp:4173-4178
Publication Date(Web):8 August 2012
DOI:10.1016/j.tetlet.2012.05.142
Microwave-assisted synthesis of macrocyclic diaryl ethers via intramolecular and/or bimolecular Ullmann coupling is described. Using the optimized conditions, a panel of macrocycles, with different substitution patterns, ring sizes, and linkers, has been successfully synthesized using microwave irradiation. To the best of our knowledge, this work represents the first examples of the microwave-assisted synthesis of macrocyclic diaryl ethers via intramolecular and/or bimolecular Ullmann coupling.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter: Dianqing Sun; Julian G. Hurdle;Robin Lee; Richard Lee; Mark Cushman; John M. Pezzuto
ChemMedChem 2012 Volume 7( Issue 9) pp:1541-1545
Publication Date(Web):
DOI:10.1002/cmdc.201200253
Co-reporter:Li Shen, Eun-Jung Park, Tamara P. Kondratyuk, Daniela Guendisch, Laura Marler, John M. Pezzuto, Anthony D. Wright, Dianqing Sun
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 21) pp:6182-6195
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmc.2011.09.020
Callophycin A was originally isolated from the red algae Callophycusoppositifolius and shown to mediate anticancer and cytotoxic effects. In our collaborative effort to identify potential chemopreventive and anticancer agents with enhanced potency and selectivity, we employed a tetrahydro-β-carboline-based template inspired by callophycin A for production of a chemical library. Utilizing a parallel synthetic approach, 50 various functionalized tetrahydro-β-carboline derivatives were prepared and assessed for activities related to cancer chemoprevention and cancer treatment: induction of quinone reductase 1 (QR1) and inhibition of aromatase, nitric oxide (NO) production, tumor necrosis factor (TNF)-α-induced NFκB activity, and MCF7 breast cancer cell proliferation. Biological results showed that the n-pentyl urea S-isomer 6a was the strongest inducer of QR1 with an induction ratio (IR) value of 4.9 at 50 μM [the concentration to double the activity (CD) = 3.8 μM] and its corresponding R-isomer 6f had an IR value of 4.3 (CD = 0.2 μM). The isobutyl carbamate derivative 3d with R stereochemistry demonstrated the most potent inhibitory activity of NFκB, with the half maximal inhibitory concentration (IC50) value of 4.8 μM, and also showed over 60% inhibition at 50 μM of NO production (IC50 = 2.8 μM). The R-isomer urea derivative 6j, having an appended adamantyl group, exhibited the most potent MCF7 cell proliferation inhibitory activity (IC50 = 14.7 μM). The S-isomer 12a of callophycin A showed the most potent activity in aromatase inhibition (IC50 = 10.5 μM).
Co-reporter:Li Shen, Dianqing Sun
Tetrahedron Letters 2011 Volume 52(Issue 35) pp:4570-4574
Publication Date(Web):31 August 2011
DOI:10.1016/j.tetlet.2011.06.112
The total synthesis of the macrocyclic natural product engelhardione is reported. This effort led to the structural revision of the published structure of engelhardione to that of pterocarine. The revision reflects the change of the substitution pattern of one phenyl ether ring from the meta to the para position. To confirm, pterocarine (2) and its close regioisomer 3 were subsequently synthesized for comparison. Moreover, to the best of our knowledge, our synthesis of 1 represents the first example of a 14-membered macrocyclic diarylheptanoid with a meta–meta substitution pattern at the diphenyl ether moiety.
Co-reporter:Xufen Yu, Eun-Jung Park, Tamara P. Kondratyuk, John M. Pezzuto and Dianqing Sun
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 44) pp:NaN8847-8847
Publication Date(Web):2012/09/13
DOI:10.1039/C2OB26456K
Development of small molecule drug-like inhibitors blocking both nitric oxide synthase and NFκB could offer a synergistic therapeutic approach in the prevention and treatment of inflammation and cancer. During the course of evaluating the biological potential of a commercial compound library, 2-phenylindole (1) displayed inhibitory activity against nitrite production and NFκB with IC50 values of 38.1 ± 1.8 and 25.4 ± 2.1 μM, respectively. Based on this lead, synthesis and systematic optimization have been undertaken in an effort to find novel and more potent nitric oxide synthase and NFκB inhibitors with antiinflammatory and cancer preventive potential. First, chemical derivatizations of 1 and 2-phenylindole-3-carboxaldehyde (4) were performed to generate a panel of N-alkylated indoles and 3-oxime derivatives 2–7. Second, a series of diversified 2-arylindole derivatives (10) were synthesized from an array of substituted 2-iodoanilines (8) and terminal alkynes (9) by applying a one-pot palladium catalyzed Sonogashira-type alkynylation and base-assisted cycloaddition. Subsequent biological evaluations revealed 3-carboxaldehyde oxime and cyano substituted 2-phenylindoles 5 and 7 exhibited the strongest nitrite inhibitory activities (IC50 = 4.4 ± 0.5 and 4.8 ± 0.4 μM, respectively); as well as NFκB inhibition (IC50 = 6.9 ± 0.8 and 8.5 ± 2.0 μM, respectively). In addition, the 6′-MeO-naphthalen-2′-yl indole derivative 10at displayed excellent inhibitory activity against NFκB with an IC50 value of 0.6 ± 0.2 μM.

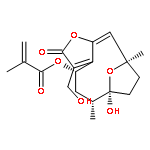
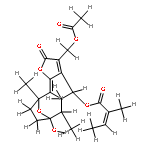
![(4S,6R,7S,10R,E)-3-(acetoxymethyl)-7-methoxy-6,10-dimethyl-2-oxo-2,4,5,6,7,8,9,10-octahydro-7,10-epoxycyclodeca[b]furan-4-yl methacrylate](http://img.cochemist.com/ccimg/883900/883872-71-7.png)
![(4S,6R,7S,10R,E)-3-(acetoxymethyl)-7-methoxy-6,10-dimethyl-2-oxo-2,4,5,6,7,8,9,10-octahydro-7,10-epoxycyclodeca[b]furan-4-yl methacrylate](http://img.cochemist.com/ccimg/883900/883872-71-7_b.png)
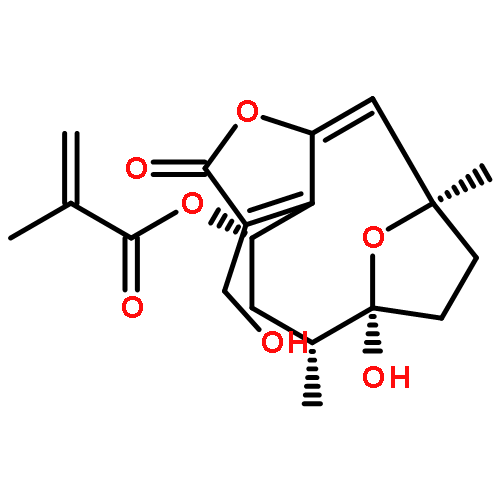
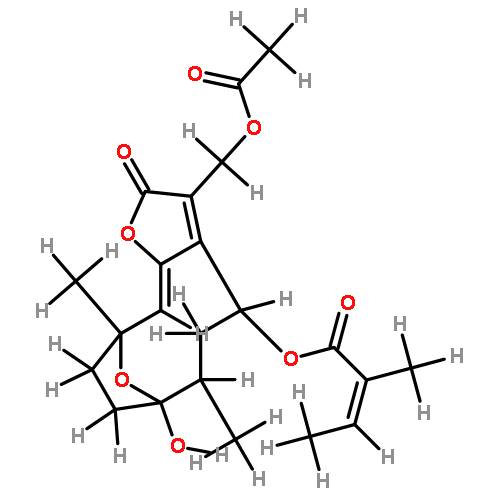
![(4S,6R,7S,10R,E)-3-(acetoxymethyl)-7-hydroxy-6,10-dimethyl-2-oxo-2,4,5,6,7,8,9,10-octahydro-7,10-epoxycyclodeca[b]furan-4-yl methacrylate](http://img.cochemist.com/ccimg/67700/67667-71-4.png)
![(4S,6R,7S,10R,E)-3-(acetoxymethyl)-7-hydroxy-6,10-dimethyl-2-oxo-2,4,5,6,7,8,9,10-octahydro-7,10-epoxycyclodeca[b]furan-4-yl methacrylate](http://img.cochemist.com/ccimg/67700/67667-71-4_b.png)