Co-reporter:Daisuke Nishiyama, Ayako Ohara, Hiroaki Chiba, Hiroshi Kumagai, Shinya Oishi, Nobutaka Fujii, and Hiroaki Ohno
Organic Letters 2016 Volume 18(Issue 7) pp:1670-1673
Publication Date(Web):March 24, 2016
DOI:10.1021/acs.orglett.6b00536
A gold-catalyzed cyclization of 1-propargyl-1,2,3,4-tetrahydro-β-carboline led to formation the D-ring of strictamine. Functional group modifications of the resulting tetracyclic indolenine led to the formal total synthesis of (±)-strictamine.
Co-reporter:Taro Noguchi, Shinya Oishi, Kaori Honda, Yasumitsu Kondoh, Tamio Saito, Hiroaki Ohno, Hiroyuki Osada and Nobutaka Fujii
Chemical Communications 2016 vol. 52(Issue 49) pp:7653-7656
Publication Date(Web):11 May 2016
DOI:10.1039/C6CC03114E
We established a facile access to an unexplored mirror-image library of chiral natural product derivatives using D-protein technology. In this process, two chemical syntheses of mirror-image substances including a target protein and hit compound(s) allow the lead discovery from a virtual mirror-image library without the synthesis of numerous mirror-image compounds.
Co-reporter:Hiroaki Ohno, Daiki Minamiguchi, Shinya Nakamura, Keito Shu, Shiho Okazaki, Maho Honda, Ryosuke Misu, Hirotomo Moriwaki, Shinsuke Nakanishi, Shinya Oishi, Takayoshi Kinoshita, Isao Nakanishi, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 5) pp:1136-1141
Publication Date(Web):1 March 2016
DOI:10.1016/j.bmc.2016.01.043
Two classes of modified analogs of 4-(thiazol-5-yl)benzoic acid-type CK2 inhibitors were designed. The azabenzene analogs, pyridine- and pyridazine-carboxylic acid derivatives, showed potent protein kinase CK2 inhibitory activities [IC50 (CK2α) = 0.014–0.017 μM; IC50 (CK2α′) = 0.0046–0.010 μM]. Introduction of a 2-halo- or 2-methoxy-benzyloxy group at the 3-position of the benzoic acid moiety maintained the potent CK2 inhibitory activities [IC50 (CK2α) = 0.014–0.016 μM; IC50 (CK2α′) = 0.0088–0.014 μM] and led to antiproliferative activities [CC50 (A549) = 1.5–3.3 μM] three to six times higher than those of the parent compound.
Co-reporter:Saori Naoe, Yusuke Yoshida, Shinya Oishi, Nobutaka Fujii, and Hiroaki Ohno
The Journal of Organic Chemistry 2016 Volume 81(Issue 13) pp:5690-5698
Publication Date(Web):June 8, 2016
DOI:10.1021/acs.joc.6b00720
A total synthesis of (+)-conolidine has been achieved via the gold(I)-catalyzed cascade cyclization of a conjugated enyne. Remarkably, this strategy allowed for the simultaneous formation of the indole ring and the ethylidene-substituted piperidine moiety of (+)-conolidine under homogeneous gold catalysis in an enantioselective manner (88–91% ee).
Co-reporter:Masamitsu Taguchi, Yusuke Tokimizu, Shinya Oishi, Nobutaka Fujii, and Hiroaki Ohno
Organic Letters 2015 Volume 17(Issue 24) pp:6250-6253
Publication Date(Web):December 9, 2015
DOI:10.1021/acs.orglett.5b03254
Various N-propargylanilines bearing a conjugated diyne moiety at the 2-position were converted to tetracyclic fused carbazoles by treatment with a homogeneous gold(I) catalyst. This cascade reaction proceeds through indole formation with concomitant rearrangement of the N-propargyl group, intramolecular nucleophilic addition toward the resulting allene moiety, and subsequent hydroalkenylation. This transformation enables a one-pot synthesis of fused carbazoles from readily accessible substrates with 100% atom economy.
Co-reporter:Shinya Oishi; Tomoko Kuroyanagi; Tatsuhiko Kubo; Nicolas Montpas; Yasushi Yoshikawa; Ryosuke Misu; Yuka Kobayashi; Hiroaki Ohno; Nikolaus Heveker; Toshio Furuya
Journal of Medicinal Chemistry 2015 Volume 58(Issue 13) pp:5218-5225
Publication Date(Web):June 4, 2015
DOI:10.1021/acs.jmedchem.5b00216
The CXC chemokine receptor 7 (CXCR7)/ACKR3 is a chemokine receptor that recognizes stromal cell-derived factor 1 (SDF-1)/CXCL12 and interferon-inducible T-cell α chemoattractant (I-TAC)/CXCL11. Here, we report the development of novel CXCR7-selective ligands with a cyclic pentapeptide scaffold through an SAR study of CXC chemokine receptor 4 (CXCR4) selective antagonist FC131 [cyclo(-d-Tyr-l-Arg-l-Arg-l-Nal-Gly-), Nal = 3-(2-naphthyl)alanine]. Substitution of Gly with l-Pro switched the receptor preference of the peptides from CXCR4 to CXCR7. The SAR study led to the identification of a potent CXCR7 ligand, FC313 [cyclo(-d-Tyr-l-Arg-l-MeArg-l-Nal-l-Pro-)], which recruits β-arrestin to CXCR7. Investigations via receptor mutagenesis and molecular modeling experiments suggest a possible binding mode of the cyclic pentapeptide CXCR7 agonist.
Co-reporter:Shiho Okazaki, Shinya Oishi, Tsukasa Mizuhara, Kazuya Shimura, Hiroto Murayama, Hiroaki Ohno, Masao Matsuoka and Nobutaka Fujii
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 16) pp:4706-4713
Publication Date(Web):10 Mar 2015
DOI:10.1039/C5OB00301F
3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine (PD 404182) and 3,4-dihydro-2H-benzo[4,5]isothiazolo[2,3-a]pyrimidine are the heterocyclic antiretroviral agents against human immunodeficiency virus type 1 (HIV-1) infection. On the basis of similar structure–activity relationships of anti-HIV activities toward the early-stage of viral infection between these unique scaffolds, the transformations under the bioassay conditions were investigated. The distinctive S–N bond in the isothiazolopyrimidine scaffold was immediately cleaved under reductive conditions in the presence of GSH to generate a thiophenol derivative. A similar rapid conversion of PD 404182 into the same thiophenol derivative was observed, suggesting that pyrimidobenzothiazine and isothiazolopyrimidine scaffolds may work as prodrug forms of the common bioactive thiophenol derivatives.
Co-reporter:Ryosuke Misu, Koki Yamamoto, Ai Yamada, Taro Noguchi, Hiroaki Ohno, Takashi Yamamura, Hiroaki Okamura, Fuko Matsuda, Satoshi Ohkura, Shinya Oishi and Nobutaka Fujii
MedChemComm 2015 vol. 6(Issue 3) pp:469-476
Publication Date(Web):05 Jan 2015
DOI:10.1039/C4MD00514G
Neurokinin B (NKB) regulates the secretion of gonadotropin-releasing hormone (GnRH) in the hypothalamus via activation of the cognate neurokinin-3 receptor (NK3R). The stimulatory effect of NKB and the derivatives on gonadotropin secretion can potentially be used for development of novel regulatory and therapeutic agents for reproductive dysfunctions. Here, we report a comprehensive structure–activity relationship study on the NK3R-selective agonist peptide, senktide. Substitution of the N-terminal succinyl-Asp substructure in senktide with oxalyl-Glu, oxalyl-D-Glu or oxalyl-L-2-aminoadipic acid (Aad) increased receptor binding and NK3R activation. Among these modifications, the oxalyl-D-Glu substructure prevented neutral endopeptidase (NEP) 24.11-mediated degradation, thus providing a novel NK3R agonist peptide with favourable biological and stability properties.
Co-reporter:Ryota Nabika, Takashi L. Suyama, Andrew M. Hau, Ryosuke Misu, Hiroaki Ohno, Jane E. Ishmael, Kerry L. McPhail, Shinya Oishi, Nobutaka Fujii
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 2) pp:302-306
Publication Date(Web):15 January 2015
DOI:10.1016/j.bmcl.2014.11.044
Coibamide A is a highly potent antiproliferative cyclic depsipeptide, which was originally isolated from a Panamanian marine cyanobacterium. In this study, the synthesis of coibamide A has been investigated using Fmoc-based solid-phase peptide synthesis followed by the cleavage of the resulting linear peptide from the resin and its subsequent macrolactonization. The peptide sequence of the linear coibamide A precursor was constructed on a solid-support following the optimization of the coupling conditions, where numerous coupling agents were evaluated. The macrocyclization of the resulting linear peptide provided the [d-MeAla11]-epimer of coibamide A, which exhibited nanomolar cytotoxic activity towards a number of human cancer cell lines.
Co-reporter:Yusuke Tokimizu;Dr. Shinya Oishi;Dr. Nobutaka Fujii;Dr. Hiroaki Ohno
Angewandte Chemie International Edition 2015 Volume 54( Issue 27) pp:7862-7866
Publication Date(Web):
DOI:10.1002/anie.201502256
Abstract
Gold catalysis enables direct construction of tetracyclic fused indolines through the migration of a propargyl substituent from an aniline nitrogen atom to the C3-position of an indole from 2-alkynyl-N-propargylanilines. This reaction provides rapid access to fused three-dimensional indolines in a single operation with the formation of four bonds and three rings.
Co-reporter:Shiho Okazaki, Tsukasa Mizuhara, Kazuya Shimura, Hiroto Murayama, Hiroaki Ohno, Shinya Oishi, Masao Matsuoka, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2015 23(7) pp: 1447-1452
Publication Date(Web):
DOI:10.1016/j.bmc.2015.02.015
Co-reporter:Akira Iwata, Shinsuke Inuki, Shinya Oishi, Nobutaka Fujii, Hiroaki Ohno
Tetrahedron 2015 Volume 71(Issue 37) pp:6580-6585
Publication Date(Web):16 September 2015
DOI:10.1016/j.tet.2015.05.006
Herein, we report the palladium-catalyzed cyclization reactions of indoles bearing a propargyl chloride side chain at their 3-position. In the presence of an external nucleophile, such as a sulfonamide or malonate, indoles bearing a propargyl group at their 3-position gave fused tetracyclic spiroindolines preferentially. However, in the absence of an external nucleophile, the same substrates afforded spiroindoles. Our attempts to develop a catalytic asymmetric spirocyclization onto a propargylpalladium species are also presented in this paper.
Co-reporter:Yusuke Tokimizu;Dr. Shinya Oishi;Dr. Nobutaka Fujii;Dr. Hiroaki Ohno
Angewandte Chemie 2015 Volume 127( Issue 27) pp:7973-7977
Publication Date(Web):
DOI:10.1002/ange.201502256
Abstract
Gold catalysis enables direct construction of tetracyclic fused indolines through the migration of a propargyl substituent from an aniline nitrogen atom to the C3-position of an indole from 2-alkynyl-N-propargylanilines. This reaction provides rapid access to fused three-dimensional indolines in a single operation with the formation of four bonds and three rings.
Co-reporter:Yuka Matsuda;Saori Naoe;Dr. Shinya Oishi;Dr. Nobutaka Fujii;Dr. Hiroaki Ohno
Chemistry - A European Journal 2015 Volume 21( Issue 4) pp:1463-1467
Publication Date(Web):
DOI:10.1002/chem.201405903
Abstract
Indole synthesis by a gold(I)-catalyzed intermolecular formal [4+2] reaction between 1,3-diynes and pyrroles has been developed. This reaction involves the hydroarylation of 1,3-diynes with pyrroles followed by an intramolecular hydroarylation to give the 4,7-disubstituted indoles. This reaction can also be applied to the synthesis of carbazoles when indoles are used as the nucleophiles instead of pyrroles.
Co-reporter:Akira Iwata, Shinsuke Inuki, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
Chemical Communications 2014 vol. 50(Issue 3) pp:298-300
Publication Date(Web):28 Oct 2013
DOI:10.1039/C3CC46511J
Efficient palladium-catalysed cascade cyclisation to form spiroindoles is developed. Treatment of indoles bearing a propargyl chloride side chain at the 3-position with various external nucleophiles in the presence of a catalytic amount of Pd2(dba)3·CHCl3/dppb and Cs2CO3 in THF gives fused tetracyclic spiroindoles in moderate to good yields.
Co-reporter:Yusuke Tokimizu, Shinya Oishi, Nobutaka Fujii, and Hiroaki Ohno
Organic Letters 2014 Volume 16(Issue 11) pp:3138-3141
Publication Date(Web):May 29, 2014
DOI:10.1021/ol5012604
(Azido)ynamides were efficiently converted into indoloquinolines by the use of a gold catalyst. While ynamides bearing an allylsilane gave terminal alkenes, ynamides bearing a simple alkene gave cyclopropanes. This reaction proceeds through the formation of an α-amidino gold carbenoid.
Co-reporter:Ryosuke Misu ; Shinya Oishi ; Ai Yamada ; Takashi Yamamura ; Fuko Matsuda ; Koki Yamamoto ; Taro Noguchi ; Hiroaki Ohno ; Hiroaki Okamura ; Satoshi Ohkura
Journal of Medicinal Chemistry 2014 Volume 57(Issue 20) pp:8646-8651
Publication Date(Web):September 23, 2014
DOI:10.1021/jm500771w
Neurokinin B (NKB) regulates the release of gonadotropin-releasing hormone (GnRH) via activation of the neurokinin-3 receptor (NK3R). We evaluated the biological stability of NK3R selective agonists to develop novel NK3R agonists to regulate reproductive functions. On the basis of degradation profiles, several peptidomimetic derivatives were designed. The modification of senktide with (E)-alkene dipeptide isostere generated a novel potent NK3R agonist with high stability and prolonged bioactivity.
Co-reporter:Ryota Nabika, Shinya Oishi, Ryosuke Misu, Hiroaki Ohno, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 21) pp:6156-6162
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmc.2014.08.036
There are many natural peptides with multiple N-methylamino acids that exhibit potent attractive biological activities. N-methylation of a peptide bond(s) is also one of the standard approaches in medicinal chemistry of bioactive peptides, to improve the potency and physicochemical properties, especially membrane permeability. In this study, we investigated a facile synthesis process of N-methylated peptides via simultaneous N-methylation of several peptide bonds in the presence of peptide bonds that were not to be methylated. As a model study, we investigated the synthesis of the antiproliferative depsipeptide, IB-01212. We used a pseudoproline to protect the non-methylated peptide bond during a simultaneous N-methylation with MeI–Ag2O. Using further manipulations including a dimerization/cyclization process, IB-01212 and its derivatives were successfully synthesized. A preliminary structure–activity relationship study demonstrated that the symmetric structure contributed to the potent cytotoxic activity of IB-01212.
Co-reporter:Tomoki Takeuchi, Shinya Oishi, Masato Kaneda, Hiroaki Ohno, Shinya Nakamura, Isao Nakanishi, Masayoshi Yamane, Jun-ichi Sawada, Akira Asai, and Nobutaka Fujii
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 5) pp:566-571
Publication Date(Web):March 10, 2014
DOI:10.1021/ml500016j
Diaryl amine derivatives have been designed and synthesized as novel kinesin spindle protein (KSP) inhibitors based on planar carbazole-type KSP inhibitors with poor aqueous solubility. The new generation of inhibitors was found to show comparable inhibitory activity and high selectivity for KSP, and this was accompanied with improved solubility. Kinetic analysis and molecular modeling studies suggested that these inhibitors work in an ATP-competitive manner via binding to the secondary allosteric site formed by α4 and α6 helices of KSP. Comparative structural investigations on a series of compounds revealed that the higher solubility of diaryl amine-type inhibitors was attributed to fewer van der Waals interactions in the crystal packing and the hydrogen-bond acceptor nitrogen of the aniline moiety for favorable solvation.Keywords: Aqueous solubility; crystal packing; diphenylamine; kinesin spindle protein;
Co-reporter:Marcel Wieteck;Yusuke Tokimizu;Dr. Matthias Rudolph;Dr. Frank Rominger;Dr. Hiroaki Ohno;Dr. Nobutaka Fujii;Dr. A. Stephen K. Hashmi
Chemistry - A European Journal 2014 Volume 20( Issue 49) pp:16331-16336
Publication Date(Web):
DOI:10.1002/chem.201404987
Abstract
New and interesting polycyclic compounds have been synthesized from non-conjugated diyne systems by dual gold catalysis. A quaternary carbon center in the backbone and the accompanying Thorpe–Ingold effect enabled the unprecedented insertion of sp3 and sp2 CH bonds that for the first time were incorporated within the backbone of the diyne system and allowed the construction of complex polycyclic carbon scaffolds inaccessible by previous approaches in which the CH bonds for the insertion were situated at the other end of the alkyne.
Co-reporter:Masato Kaneda, Ryosuke Misu, Hiroaki Ohno, Akira Hirasawa, Nahoko Ieda, Yoshihisa Uenoyama, Hiroko Tsukamura, Kei-ichiro Maeda, Shinya Oishi, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2014 22(13) pp: 3325-3330
Publication Date(Web):
DOI:10.1016/j.bmc.2014.04.052
Co-reporter:Tomoki Takeuchi, Shinya Oishi, Masato Kaneda, Ryosuke Misu, Hiroaki Ohno, Jun-ichi Sawada, Akira Asai, Shinya Nakamura, Isao Nakanishi, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2014 22(12) pp: 3171-3179
Publication Date(Web):
DOI:10.1016/j.bmc.2014.04.008
Co-reporter:Yuji Yoshimitsu, Shinsuke Inuki, Shinya Oishi, Nobutaka Fujii, and Hiroaki Ohno
Organic Letters 2013 Volume 15(Issue 12) pp:3046-3049
Publication Date(Web):May 30, 2013
DOI:10.1021/ol401231y
Palladium-catalyzed medium-ring formation from a cyclic propargyl carbonate via a ring-opening and -closing cascade proceeded at the central carbon atom of the propargyl unit to provide a tetrahydro-2H-oxocine derivative bearing the core structure of laurencia oxacycle. The synthetic application of this reaction to a possible laurendecumallene B precursor is also presented.
Co-reporter:Zengye Hou, Shinya Oishi, Yamato Suzuki, Tatsuhide Kure, Isao Nakanishi, Akira Hirasawa, Gozoh Tsujimoto, Hiroaki Ohno and Nobutaka Fujii
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 20) pp:3288-3296
Publication Date(Web):11 Mar 2013
DOI:10.1039/C3OB40223A
Pyrazolo[4,3-b]indole derivatives have been designed as novel CK2 inhibitor compounds based on the binding mode analysis of a previously reported phenylpyrazole-type CK2 inhibitor. A series of pyrazolo[4,3-b]indoles and related dihydropyrazolo[4,3-b]indoles were efficiently prepared from simple starting materials using a gold-catalysed three-component annulation reaction as a key step. Several of the newly synthesized compounds displayed high levels of inhibitory activity, indicating that the pyrazolo[4,3-b]indole core represents a promising scaffold for the development of potent CK2 inhibitors.
Co-reporter:Tsukasa Mizuhara, Shinya Oishi, Hiroaki Ohno, Kazuya Shimura, Masao Matsuoka, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 7) pp:2079-2087
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmc.2013.01.016
To investigate the mechanism of action of the potent antiviral compound PD 404182, three novel photoaffinity probes equipped with a biotin or alkyne indicator were designed and synthesized based on previous structure–activity relationship studies. These probes retained the potent anti-HIV activity of the original pyrimidobenzothiazine derivatives. In photoaffinity labeling studies using HIV-1-infected H9 cells (H9IIIB), eight potential proteins were observed to bind PD 404182.
Co-reporter:Yuka Kobayashi, Shinya Oishi, Kazuya Kobayashi, Hiroaki Ohno, Hiroko Tsutsumi, Yoji Hata, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 14) pp:4296-4300
Publication Date(Web):15 July 2013
DOI:10.1016/j.bmc.2013.04.078
Deferriferrichrysin belongs to the siderophore peptide family which are Fe(III)-coordinating cyclic peptides. The common structure of this family is three consecutive hydroxamate moieties, such as Nδ-acetyl-Nδ-hydroxy-l-ornithine (Aho). We have designed two deferriferrichrysin derivatives where three Aho residues were arranged as: cyclo(-Aho-Gly-Aho-Gly-Aho-Gly-) and cyclo(-Aho-Ser-Aho-Ser-Aho-Ser-). Comparative evaluation of the physicochemical properties of their Fe(III) complexes revealed that naturally occurring deferriferrichrysin formed a more stable Fe(III) complex when compared with the two derivatives. This result shows that three consecutive Aho residues are indispensable for high affinity Fe(III) binding by deferriferrichrysin. Of note, the observed pH-dependent chromogenic response of the Fe(III) complexes of the derivatives suggests that these two derivatives should function as sensitive pH indicators in acidic environments.
Co-reporter:Ryosuke Misu, Shinya Oishi, Shohei Setsuda, Taro Noguchi, Masato Kaneda, Hiroaki Ohno, Barry Evans, Jean-Marc Navenot, Stephen C. Peiper, Nobutaka Fujii
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 9) pp:2628-2631
Publication Date(Web):1 May 2013
DOI:10.1016/j.bmcl.2013.02.098
Kisspeptins, endogenous peptide ligands for GPR54, play an important role in GnRH secretion. Since in vivo administration of kisspeptins induces increased plasma LH levels, GPR54 agonists hold promise as therapeutic agents for the treatment of hormonal secretion diseases. To facilitate the design of novel potent GPR54 ligands, residues in kisspeptins that involve in the interaction with GPR54 were investigated by kisspeptin-based photoaffinity probes. Herein, we report the design and synthesis of novel kisspeptin-based photoaffinity probes, and the application to crosslinking experiments for GPR54-expressing cells.
Co-reporter:Tsukasa Mizuhara, Takayuki Kato, Atsushi Hirai, Hideki Kurihara, Yasuhiro Shimada, Masahiko Taniguchi, Hideki Maeta, Hiroaki Togami, Kazuya Shimura, Masao Matsuoka, Shiho Okazaki, Tomoki Takeuchi, Hiroaki Ohno, Shinya Oishi, Nobutaka Fujii
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 16) pp:4557-4561
Publication Date(Web):15 August 2013
DOI:10.1016/j.bmcl.2013.06.026
The structure–activity relationship of phenylpyrazole derivative 1 was investigated for the development of novel anti-HIV agents. Initial efforts revealed that the diazenyl group can be replaced by an aminomethylene group. In addition, we synthesized various derivatives by the reductive amination of benzaldehydes with 5-aminopyrazoles and carried out parallel structural optimization on the benzyl group and the pyrazole ring. This optimization led to a six-fold more potent derivative 32j than the lead compound 1, and this derivative has a 3′,4′-dichloro-(1,1′-biphenyl)-3-yl group.
Co-reporter:Taro Noguchi, Shinya Oishi, Kaori Honda, Yasumitsu Kondoh, Tamio Saito, Tatsuhiko Kubo, Masato Kaneda, Hiroaki Ohno, Hiroyuki Osada, Nobutaka Fujii
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 13) pp:3802-3805
Publication Date(Web):1 July 2013
DOI:10.1016/j.bmcl.2013.04.094
MDM2 and MDMX are oncoproteins that negatively regulate the activity and stability of the tumor suppressor protein p53. The inhibitors of protein–protein interactions (PPIs) of MDM2–p53 and MDMX–p53 represent potential anticancer agents. In this study, a novel approach for identifying MDM2–p53 and MDMX–p53 PPI inhibitor candidates by affinity-based screening using a chemical array has been established. A number of compounds from an in-house compound library, which were immobilized onto a chemical array, were screened for interaction with fluorescence-labeled MDM2 and MDMX proteins. The subsequent fluorescent polarization assay identified several compounds that inhibited MDM2–p53 and MDMX–p53 interactions.
Co-reporter:Ryosuke Misu, Taro Noguchi, Hiroaki Ohno, Shinya Oishi, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2013 21(8) pp: 2413-2417
Publication Date(Web):
DOI:10.1016/j.bmc.2013.01.036
Co-reporter:Hiroaki Chiba;Yuki Sakai;Ayako Ohara;Dr. Shinya Oishi;Dr. Nobutaka Fujii;Dr. Hiroaki Ohno
Chemistry - A European Journal 2013 Volume 19( Issue 27) pp:8875-8883
Publication Date(Web):
DOI:10.1002/chem.201300687
Abstract
The total synthesis of the pentacyclic tetrahydroisoquinoline alkaloid quinocarcin, which possesses intriguing structural and biological features, has been achieved through a gold(I)-catalyzed regioselective hydroamination reaction. It is noteworthy that the regioselectivity of the intramolecular hydroamination of an unsymmetrical alkyne could be completely switched through substrate control. Other key features of this synthesis include the highly stereoselective synthesis of 2,5-cis-pyrrolidine through the intramolecular amination of the bromoallene and the Lewis acid mediated ring opening of dihydrobenzofuran.
Co-reporter:Yuji Yoshimitsu, Jun Miyagaki, Shinya Oishi, Nobutaka Fujii, Hiroaki Ohno
Tetrahedron 2013 69(21) pp: 4211-4220
Publication Date(Web):
DOI:10.1016/j.tet.2013.03.091
Co-reporter:Yamato Suzuki, Saori Naoe, Shinya Oishi, Nobutaka Fujii, and Hiroaki Ohno
Organic Letters 2012 Volume 14(Issue 1) pp:326-329
Publication Date(Web):December 16, 2011
DOI:10.1021/ol203072u
Polysubstituted dihydropyrazoles were directly obtained by a gold-catalyzed three-component annulation. This reaction consists of a Mannich-type coupling of alkynes with N,N′-disubstituted hydrazines and aldehydes/ketones followed by intramolecular hydroamination. Cascade cyclization using 1,2-dialkynylbenzene derivatives as the alkyne component was also performed producing fused tricyclic dihydropyrazoles in good yields.
Co-reporter:Kazuya Kobayashi ; Shinya Oishi ; Ryoko Hayashi ; Kenji Tomita ; Tatsuhiko Kubo ; Noriko Tanahara ; Hiroaki Ohno ; Yasushi Yoshikawa ; Toshio Furuya ; Masaru Hoshino
Journal of Medicinal Chemistry 2012 Volume 55(Issue 6) pp:2746-2757
Publication Date(Web):February 21, 2012
DOI:10.1021/jm2016914
A structure–activity relationship study on a highly potent CXC chemokine receptor type 4 (CXCR4) antagonist, FC131 [cyclo(-d-Tyr1-Arg2-Arg3-Nal4-Gly5-)], was carried out using a series of alkene isosteres of the d-Tyr1-l/d-Arg2 dipeptide to investigate the binding mode of FC131 and its derivatives with CXCR4. The structure–activity relationships of isostere-containing FC131 analogues were similar to those of the parent FC131 and its derivatives, suggesting that a trans-conformer of the d-Tyr1–Arg2 peptide bond is the dominant contributor to the bioactive conformations of FC131. Although NMR analysis demonstrated that the two conformations of the peptidomimetic containing the d-Tyr1-d-Arg2 isostere are possible, binding-mode prediction indicated that the orientations of the alkene motif within d-Tyr1-MeArg2 peptidomimetics depend on the chirality of Arg2 and the β-methyl group of the isostere unit, which makes the dominant contribution for binding to the receptor. The most potent FC122 [cyclo(-d-Tyr1-d-MeArg2-Arg3-Nal4-Gly5-)] bound with CXCR4 by a binding mode different from that of FC131.
Co-reporter:Ryo Masuda, Shinya Oishi, Noriko Tanahara, Hiroaki Ohno, Akira Hirasawa, Gozoh Tsujimoto, Yoshiaki Yano, Katsumi Matsuzaki, Jean-Marc Navenot, Stephen C. Peiper, and Nobutaka Fujii
Bioconjugate Chemistry 2012 Volume 23(Issue 6) pp:1259
Publication Date(Web):April 10, 2012
DOI:10.1021/bc300084h
CXC chemokine receptor 4 (CXCR4) is a G protein-coupled receptor implicated in cell entry of T-cell line-tropic HIV-1 strains. CXCR4 and its ligand stromal cell derived factor-1 (SDF-1)/CXCL12 play pivotal parts in many physiological processes and pathogenetic conditions (e.g., immune cell-homing and cancer metastasis). We previously developed the potent CXCR4 antagonist T140 from structure–activity relationship studies of the antimicrobial peptide polyphemusin II. T140 and its derivatives have been exploited in biological and biomedical studies for the SDF-1/CXCR4 axis. We investigated receptor localization upon ligand stimulation using fluorescent SDF-1 and T140 derivatives as well as a specific labeling technique for cellular-membrane CXCR4. Fluorescent T140 derivatives induced translocation of CXCR4 into the perinuclear region as observed by treatment with fluorescent SDF-1. T140 derivative-mediated internalization of CXCR4 was also monitored by the coiled-coil tag-probe system. These findings demonstrated that the CXCR4 antagonistic activity and anti-HIV activity of T140 derivatives were derived (at least in part) from antagonist-mediated receptor internalization.
Co-reporter:Tsukasa Mizuhara, Shinya Oishi, Hiroaki Ohno, Kazuya Shimura, Masao Matsuoka and Nobutaka Fujii
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 33) pp:6792-6802
Publication Date(Web):22 Jun 2012
DOI:10.1039/C2OB25904D
3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine (PD 404182) is a virucidal heterocyclic compound active against various viruses, including HCV, HIV, and simian immunodeficiency virus. Using facile synthetic approaches that we developed for the synthesis of pyrimido[1,2-c][1,3]benzothiazin-6-imines and related tricyclic derivatives, the parallel structural optimizations of the central 1,3-thiazin-2-imine core, the benzene part, and the cyclic amidine part of PD 404182 were investigated. Replacement of the 6-6-6 pyrimido[1,2-c][1,3]benzothiazin-6-imine framework with 5-6-6 or 6-6-5 derivatives led to a significant loss of anti-HIV activity, and introduction of a hydrophobic group at the 9- or 10-positions improved the potency. In addition, we demonstrated that the PD 404182 derivative exerts anti-HIV effects at an early stage of viral infection.
Co-reporter:Yamato Suzuki, Shinya Oishi, Yoshinori Takei, Misato Yasue, Ryosuke Misu, Saori Naoe, Zengye Hou, Tatsuhide Kure, Isao Nakanishi, Hiroaki Ohno, Akira Hirasawa, Gozoh Tsujimoto and Nobutaka Fujii
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 25) pp:4907-4915
Publication Date(Web):25 Apr 2012
DOI:10.1039/C2OB25298H
Two classes of fused nitrogen heterocycles were designed as CK2 inhibitor candidates on the basis of previous structure–activity relationship (SAR) studies. Various dipyrrolo[3,2-b:2′,3′-e]pyridine and benzo[g]indazole derivatives were prepared using transition-metal-catalysed cascade and/or multicomponent reactions. Biological evaluation of these candidates revealed that benzo[g]indazole is a promising scaffold for potent CK2 inhibitors. The inhibitory activities on cell proliferation of these potent CK2 inhibitors are also presented.
Co-reporter:Shinya Oishi and Nobutaka Fujii
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 30) pp:5720-5731
Publication Date(Web):19 Mar 2012
DOI:10.1039/C2OB25107H
The development of novel peptide and peptidomimetic ligands for the CXC chemokine receptor 4 (CXCR4) as therapeutic agents for HIV-1 infection, cancer, and immune system diseases has grown over the last decade. In this perspective article, the design of CXCR4 agonists and antagonists from endogenous stromal cell-derived factor-1 (SDF-1)/CXCL12 and horseshoe crab-derived antimicrobial peptides and their therapeutic and diagnostic applications are described.
Co-reporter:Tsukasa Mizuhara, Shinya Oishi, Hiroaki Ohno, Kazuya Shimura, Masao Matsuoka, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 21) pp:6434-6441
Publication Date(Web):1 November 2012
DOI:10.1016/j.bmc.2012.08.030
3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine (PD 404182) is an antiretroviral agent with submicromolar inhibitory activity against human immunodeficiency virus-1 (HIV-1) and HIV-2 infection. In the current study, the structure–activity relationships of accessory groups at the 3- and 9-positions of pyrimido[1,2-c][1,3]benzothiazin-6-imine were investigated for the development of more potent anti-HIV agents. Several different derivatives containing a 9-aryl group were designed and synthesized using Suzuki–Miyaura cross-coupling and Ullmann coupling reactions. Modification of the m-methoxyphenyl or benzo[d][1,3]dioxol-5-yl group resulted in improved anti-HIV activity. In addition, the 2,4-diazaspiro[5.5]undec-2-ene-fused benzo[e][1,3]thiazine derivatives were designed and tested for their anti-HIV activities. The most potent 9-(benzo[d][1,3]dioxol-5-yl) derivative was two–threefold more effective against several strains of HIV-1 and HIV-2 than the parent compound, PD 404182.
Co-reporter:Kazuya Kobayashi, Shinya Oishi, Yuka Kobayashi, Hiroaki Ohno, Hiroko Tsutsumi, Yoji Hata, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 8) pp:2651-2655
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmc.2012.02.033
Synthesis of Fmoc-protected Nδ-acetyl-Nδ-(tert-butoxy)-l-ornithine has revealed it to be a metal-chelating amino-acid precursor. This protected amino acid was compatible with the preparation of ferrichrome peptides by standard Fmoc-based solid-phase peptide synthesis. Evaluation of deferriferrichrysin for metal ion chelation revealed that zirconium(IV) and titanium(IV) formed complexes with deferriferrichrysin.
Co-reporter:Hiroaki Chiba, Yuki Sakai, Shinya Oishi, Nobutaka Fujii, Hiroaki Ohno
Tetrahedron Letters 2012 Volume 53(Issue 46) pp:6273-6276
Publication Date(Web):14 November 2012
DOI:10.1016/j.tetlet.2012.09.030
An unusual Lewis-acid-mediated ring-exchange reaction of dihydrobenzofurans is described. The fused tricyclic ring system is the key structural element for this reaction as it restricts C–N bond rotation and/or destabilizes the benzofuran ring. We achieved the formal total synthesis of (−)-quinocarcinamide using a combination of this reaction and the Au(I)-catalyzed 6-endo-dig hydroamination of an alkyne
Co-reporter:Zengye Hou, Yamato Suzuki, Shinya Oishi, Nobutaka Fujii, Hiroaki Ohno
Tetrahedron 2012 68(6) pp: 1695-1703
Publication Date(Web):
DOI:10.1016/j.tet.2011.12.059
Co-reporter:Saori Naoe, Yamato Suzuki, Kimio Hirano, Yusuke Inaba, Shinya Oishi, Nobutaka Fujii, and Hiroaki Ohno
The Journal of Organic Chemistry 2012 Volume 77(Issue 11) pp:4907-4916
Publication Date(Web):May 8, 2012
DOI:10.1021/jo300771f
The gold-catalyzed cascade intermolecular addition–intramolecular carbocyclization reaction of dialkynylbenzenes was developed. In this reaction, regioselective addition of an external nucleophile toward the terminal alkyne and subsequent 6-endo-dig cyclization proceeded to give the 1,3-disubstituted naphthalenes in good yields. The direct synthesis of disubstituted chrysenes via a gold-catalyzed addition and double cyclization cascade using a triyne-type substrate was also achieved.
Co-reporter:Hiroaki Chiba;Dr. Shinya Oishi;Dr. Nobutaka Fujii;Dr. Hiroaki Ohno
Angewandte Chemie 2012 Volume 124( Issue 36) pp:9303-9306
Publication Date(Web):
DOI:10.1002/ange.201205106
Co-reporter:Hiroaki Chiba;Dr. Shinya Oishi;Dr. Nobutaka Fujii;Dr. Hiroaki Ohno
Angewandte Chemie International Edition 2012 Volume 51( Issue 36) pp:9169-9172
Publication Date(Web):
DOI:10.1002/anie.201205106
Co-reporter:Tomoki Takeuchi ; Shinya Oishi ; Toshiaki Watanabe ; Hiroaki Ohno ; Jun-ichi Sawada ; Kenji Matsuno ; Akira Asai ; Naoya Asada ; Kazuo Kitaura
Journal of Medicinal Chemistry 2011 Volume 54(Issue 13) pp:4839-4846
Publication Date(Web):May 20, 2011
DOI:10.1021/jm200448n
The kinesin spindle protein (KSP) is a mitotic kinesin involved in the establishment of a functional bipolar mitotic spindle during cell division. It is considered to be an attractive target for cancer chemotherapy with reduced side effects. Based on natural product scaffold-derived fused indole-based inhibitors and known biphenyl-type KSP inhibitors, various carboline and carbazole derivatives were synthesized and biologically evaluated. β-Carboline and lactam-fused carbazole derivatives exhibited remarkably potent KSP inhibitory activity and mitotic arrest in prometaphase with formation of an irregular monopolar spindle. The planar tri- and tetracyclic analogs inhibited KSP ATPase in an ATP-competitive manner just like biphenyl-type inhibitors.
Co-reporter:Eriko Inokuchi, Ai Yamada, Kentaro Hozumi, Kenji Tomita, Shinya Oishi, Hiroaki Ohno, Motoyoshi Nomizu and Nobutaka Fujii
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 9) pp:3421-3427
Publication Date(Web):22 Feb 2011
DOI:10.1039/C0OB01193B
Amidine-type peptide bond isosteres were designed based on the substitution of the peptide bond carbonyl (CO) group with an imino (CNH) group. The positively-charged property of the isosteric part resembles a reduced amide-type peptidomimetic. The peptidyl amidine units were synthesized by the reduction of a key amidoxime (N-hydroxyamidine) precursor, which was prepared from nitrile oxide components as an aminoacyl or peptidyl equivalent. This nitrile oxide-mediated C–N bond formation was also used for peptide macrocyclization, in which the amidoxime group was converted to peptide bonds under mild acidic conditions. Syntheses of the cyclic RGD peptide and a peptidomimetic using both approaches, and their inhibitory activity against integrin-mediated cell attachment, are presented.
Co-reporter:Eriko Inokuchi, Shinya Oishi, Tatsuhiko Kubo, Hiroaki Ohno, Kazuya Shimura, Masao Matsuoka, and Nobutaka Fujii
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 6) pp:477
Publication Date(Web):March 28, 2011
DOI:10.1021/ml200047e
A series of FC131 [cyclo(-d-Tyr-Arg-Arg-Nal-Gly-)] analogues containing amidine type peptide bond isosteres were synthesized as selective CXC chemokine receptor type 4 (CXCR4) antagonists. An isosteric amidine substructure was constructed by a macrocyclization process using nitrile oxide-mediated C−N bond formation. All of the amidine-containing FC131 analogues exhibited potent SDF-1 binding inhibition to CXCR4. The Nal-Gly-substituted analogue was characterized as one of the most potent cyclic pentapeptide-based CXCR4 antagonists reported to date. The improved activity against human immunodeficiency virus (HIV) type-1 X4 strains suggested that addition of another basic amidine group to the peptide backbone effectively increases the selective binding of the peptides to CXCR4 receptor.Keywords: Amidine; chemokine; CXCR4 antagonist; FC131; nitrile oxide; peptidomimetics
Co-reporter:Shinya Oishi, Ryosuke Misu, Kenji Tomita, Shohei Setsuda, Ryo Masuda, Hiroaki Ohno, Yousuke Naniwa, Nahoko Ieda, Naoko Inoue, Satoshi Ohkura, Yoshihisa Uenoyama, Hiroko Tsukamura, Kei-ichiro Maeda, Akira Hirasawa, Gozoh Tsujimoto, and Nobutaka Fujii
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 1) pp:53
Publication Date(Web):October 25, 2010
DOI:10.1021/ml1002053
Kisspeptin is a member of the RFamide neuropeptide family that is implicated in gonadotropin secretion. Because kisspeptin-GPR54 signaling is implicated in the neuroendocrine regulation of reproduction, GPR54 ligands represent promising therapeutic agents against endocrine secretion disorders. In the present study, the selectivity profiles of GPR54 agonist peptides were investigated for several GPCRs, including RFamide receptors. Kisspeptin-10 exhibited potent binding and activation of neuropeptide FF receptors (NPFFR1 and NPFFR2). In contrast, short peptide agonists bound with much lower affinity to NPFFRs while showing relatively high selectivity toward GPR54. The possible localization of secondary kisspeptin targets was also demonstrated by variation in the levels of GnRH release from the median eminence and the type of GPR54 agonists used. Negligible affinity of the reported NPFFR ligands to GPR54 was observed and indicates the unidirectional cross-reactivity between both ligands.Keywords: GPR54; kisspeptin; Neuropeptide FF receptors; NPFFR1, NPFFR2
Co-reporter:Yuji Yoshimitsu, Shinya Oishi, Jun Miyagaki, Shinsuke Inuki, Hiroaki Ohno, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 18) pp:5402-5408
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmc.2011.07.061
Sphingosine kinases (SphKs) are oncogenic enzymes that regulate the critical balance between ceramide and sphingosine-1-phosphate. Much effort has been dedicated to develop inhibitors against these enzymes. Naturally occurring pachastrissamine (jaspine B) and all its stereoisomers were prepared and evaluated for their inhibitory effects against SphKs. All eight stereoisomers exhibited moderate to potent inhibitory activity against SphK1 and SphK2. Inhibitory effects were profiled against protein kinase C (PKC) isoforms by in vitro experiments. Atypical PKCs (PKCζ and PKCι) were inhibited by several pachastrissamine stereoisomers. The improved activity over N,N-dimethylsphingosine suggests that the cyclic scaffold in pachastrissamines facilitates potential favorable interactions with SphKs and PKCs.
Co-reporter:Ryo Masuda, Shinya Oishi, Hiroaki Ohno, Hiroyuki Kimura, Hideo Saji, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 10) pp:3216-3220
Publication Date(Web):15 May 2011
DOI:10.1016/j.bmc.2011.03.059
Diethylenetriaminepentaacetic acid (DTPA) is a useful chelating agent for radionuclides such as 68Ga, 99mTc and 111In, which are applicable to nuclear medicine imaging. In this study, we established a facile synthetic protocol for the production of mono-DTPA-conjugated peptide probes. A novel monoreactive DTPA precursor reagent was synthesized in two steps using the chemistry of the o-nitrobenzenesulfonyl (Ns) protecting group, and under mild conditions this DTPA precursor was incorporated onto an Nε-bromoacetylated Lys of a protected peptide resin. The site-specific DTPA conjugation was facilitated by using a highly acid-labile 4-methyltrityl (Mtt) protecting group for the target site of the bioactive peptide during the solid-phase synthesis. A combination of both techniques yielded peptides with disulfide bonds, such as octreotide and polyphemusin II-derived CXCR4 antagonists. DTPA–peptide conjugates were purified in a single step following cleavage from the resin and disulfide bond formation. This site-specific on-resin construction strategy was used for the design and synthesis of a novel In-DTPA-labeled CXCR4 antagonist, which exhibited highly potent inhibitory activity against SDF-1–CXCR4 binding.
Co-reporter:Kimio Hirano, Yusuke Inaba, Naoya Takahashi, Masanao Shimano, Shinya Oishi, Nobutaka Fujii, and Hiroaki Ohno
The Journal of Organic Chemistry 2011 Volume 76(Issue 5) pp:1212-1227
Publication Date(Web):January 21, 2011
DOI:10.1021/jo102507c
A direct, concise, and atom-economical synthetic method for the generation of fused indoles, using a gold-catalyzed cascade cyclization of diynes, has been developed. The reaction gave various fused indoles, such as aryl-annulated[a]carbazoles, dihydrobenzo[g]indoles, and azepino- or oxepinoindole derivatives in good to excellent yields, through an intramolecular cascade 5-endo-dig hydroamination followed by a 6- or 7-endo-dig cycloisomerization, without producing theoretical byproduct. Three of the resulting indoles exhibited potent antifungal activities against T. mentagrophytes and T. rubrum, demonstrating the practical application of the described cascade reaction for drug discovery.
Co-reporter:Shinsuke Inuki, Akira Iwata, Shinya Oishi, Nobutaka Fujii, and Hiroaki Ohno
The Journal of Organic Chemistry 2011 Volume 76(Issue 7) pp:2072-2083
Publication Date(Web):March 1, 2011
DOI:10.1021/jo102388e
Enantioselective total synthesis of the biologically important indole alkaloids (+)-lysergol, (+)-isolysergol, and (+)-lysergic acid is described. Key features of these total synthesis include (1) a facile synthesis of a chiral 1,3-amino alcohol via the Pd(0)- and In(I)-mediated reductive coupling reaction between l-serine-derived 2-ethynylaziridine and formaldehyde; (2) the Cr(II)/Ni(0)-mediated Nozaki−Hiyama−Kishi (NHK) reaction of an indole-3-acetaldehyde with iodoalkyne; and (3) Pd(0)-catalyzed domino cyclization of an allene bearing amino and bromoindolyl groups. This domino cyclization enabled direct construction of the C/D ring system of the ergot alkaloids skeleton, as well as the creation of the C5 stereogenic center with transfer of the allenic axial chirality to the central chirality.
Co-reporter:Akira Iwata, Shinsuke Inuki, Shinya Oishi, Nobutaka Fujii, and Hiroaki Ohno
The Journal of Organic Chemistry 2011 Volume 76(Issue 13) pp:5506-5512
Publication Date(Web):May 20, 2011
DOI:10.1021/jo2008324
Asymmetric formal synthesis of (+)-lysergic acid was achieved with a reductive ring-opening reaction of chiral 2-alkynyl-3-indolyloxirane with NaBH3CN as the key step. With Zn(OTf)2 as an additive, the ring-opening reaction proceeded regioselectively at the 3-position to give the corresponding propargyl alcohol, which was a precursor of the allenic amide for palladium-catalyzed domino cyclization to construct the ergot alkaloid core structure.
Co-reporter:Yusuke Tokimizu, Yusuke Ohta, Hiroaki Chiba, Shinya Oishi, Nobutaka Fujii, Hiroaki Ohno
Tetrahedron 2011 67(29) pp: 5168-5175
Publication Date(Web):
DOI:10.1016/j.tet.2011.05.051
Co-reporter:Yusuke Ohta, Yusuke Tokimizu, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
Organic Letters 2010 Volume 12(Issue 17) pp:3963-3965
Publication Date(Web):August 12, 2010
DOI:10.1021/ol1016756
A novel synthesis of 2-phenyl-4-[(triisopropylsilyl)methyl]quinazolines from monosubstituted arenes has been developed. Treatment of N-phenylbenzamidines with 5-nitro-1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one and K2CO3 in the presence of a catalytic amount of CuBr in benzene gives 2-phenyl-4-[(triisopropylsilyl)methyl]quinazolines in moderate to good yields.
Co-reporter:Shinya Oishi ; Toshiaki Watanabe ; Jun-ichi Sawada ; Akira Asai ; Hiroaki Ohno
Journal of Medicinal Chemistry 2010 Volume 53(Issue 13) pp:5054-5058
Publication Date(Web):June 3, 2010
DOI:10.1021/jm100476d
Mitotic kinesin spindle protein (KSP) is involved in the assembly of the bipolar spindle during cell division. On the basis of a common 2,3-fused indole substructure within the complex frameworks of terpendole E and other KSP inhibitors, the carbazoles with a bulky alkyl group were identified as a novel KSP inhibitory scaffold. Additionally, among several naturally occurring cell growth inhibitors with 2,3-fused indole structures, β-carboline alkaloids, harman and harmine, showed moderate inhibition of KSP.
Co-reporter:Shinya Oishi, Kentaro Watanabe, Saori Ito, Michinori Tanaka, Hiroki Nishikawa, Hiroaki Ohno, Kazuki Shimane, Kazuki Izumi, Yasuko Sakagami, Eiichi N. Kodama, Masao Matsuoka, Akira Asai and Nobutaka Fujii
MedChemComm 2010 vol. 1(Issue 4) pp:276-281
Publication Date(Web):20 Aug 2010
DOI:10.1039/C0MD00091D
Enfuvirtide is the first approved membrane fusion inhibitor against HIV-1. Although this drug is effective against multi-drug resistant strains, the emergence of enfuvirtide-resistant strains has been reported in patients who have received an enfuvirtide-containing regimen. Based on the high affinity of synthetic HIV-1 gp41 C-terminal heptad repeat (C-HR) peptides to the counterpart trimeric N-terminal heptad repeat (N-HR) coiled-coil structure, a novel screening approach has been established to facilitate the identification of potent fusion inhibitors against wild-type and enfuvirtide-resistant HIV-1. In this process, affinity selection using histidine-tagged N-HR peptides with the sequences derived from wild-type and resistant strains efficiently captured potent inhibitory peptides from a pool of highly water-soluble C-HR peptides with α-helix-inducible motifs. A highly potent peptide was found from a single amino acid substitution observed in an enfuvirtide-resistant variant as well as peptides with unprecedented modifications at the mutated site.
Co-reporter:Akinori Okano Dr.;Koji Tsukamoto Dr.;Shohei Kosaka Dr.;Hatsuo Maeda Dr.;Shinya Oishi Dr.;Tetsuaki Tanaka Dr. Dr.;Hiroaki Ohno Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 28) pp:8410-8418
Publication Date(Web):
DOI:10.1002/chem.201000653
Abstract
The palladium(0)-catalyzed direct construction of bicyclic heterocycles is described. Treatment of propargyl bromides that have nucleophilic functional groups connected by two or three carbon atoms with catalytic [Pd(PPh3)4] affords bis-cyclization products in good yields. The desired bicyclic heterocycles can be obtained selectively when using substrates with appropriate nucleophilic groups. We also describe the reaction of a 2-alkynylazetidine derivative with a catalytic amount of [Pd(PPh3)4] under base-free conditions, which affords the same fused heterocycles as the corresponding propargyl bromides.
Co-reporter:Yusuke Ohta, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
Organic Letters 2009 Volume 11(Issue 9) pp:1979-1982
Publication Date(Web):April 8, 2009
DOI:10.1021/ol900460m
Two direct synthetic methods of 1,2,3,4-tetrahydro-β-carboline derivatives have been developed. After initial indole formation by copper-catalyzed domino three-component coupling−cyclization using an appropriate ethynylaniline, aldehyde, and a secondary amine, treatment with t-BuOK/hexane or MsOH afforded the desired tetrahydro-β-carboline derivatives in moderate to good yields.
Co-reporter:Shinsuke Inuki, Yuji Yoshimitsu, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
Organic Letters 2009 Volume 11(Issue 19) pp:4478-4481
Publication Date(Web):September 9, 2009
DOI:10.1021/ol901904w
Palladium(0)-catalyzed cyclization of bromoallenes bearing hydroxyl and benzamide groups as internal nucleophiles stereoselectively provides functionalized tetrahydrofuran. With this bis-cyclization as the key step, a short total synthesis of pachastrissamine, a biologically active marine natural product, was achieved.
Co-reporter:Shinya Oishi, Hirotaka Kamitani, Yasuyo Kodera, Kentaro Watanabe, Kazuya Kobayashi, Tetsuo Narumi, Kenji Tomita, Hiroaki Ohno, Takeshi Naito, Eiichi Kodama, Masao Matsuoka and Nobutaka Fujii
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 14) pp:2872-2877
Publication Date(Web):04 Jun 2009
DOI:10.1039/B907983A
The α-helix structures of the anti-HIV fusion inhibitory peptides are stabilized by the amino acid sequence and by intrachain hydrogen bonds. The study of peptide analogues using (E)-alkene and (Z)-fluoroalkene dipeptide isosteres demonstrated the substantial, yet position-dependent, contribution of hydrogen bonds to the α-helix stability and anti-HIV bioactivity.
Co-reporter:Shinya Oishi, Yasuyo Kodera, Hiroki Nishikawa, Hirotaka Kamitani, Tsuyoshi Watabe, Hiroaki Ohno, Tadafumi Tochikura, Kazuki Shimane, Eiichi Kodama, Masao Matsuoka, Fuminori Mizukoshi, Hajime Tsujimoto, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 14) pp:4916-4920
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmc.2009.06.001
Feline immunodeficiency virus (FIV) is a pathogenic virus that causes an AIDS-like syndrome in the domestic cats. For viral entry and infection, fusion between the virus and the cell membrane is the critical process and this process is mediated by an envelope glycoprotein gp40. We have identified fusion inhibitory peptides from the heptad repeat-2 (HR2) of gp40. Remodeling of the original sequences using α-helix-inducible motifs revealed the interactive residues of gp40. Comparative analysis of HR2 peptides derived from four FIV strains demonstrated that the interactive surface of the Shizuoka strain-derived HR2 peptides provides the highest affinity of all the FIV strains examined.Feline immunodeficiency virus (FIV) is a pathogenic virus that causes an AIDS-like syndrome in the domestic cats. For viral entry and infection, fusion between the virus and the cell membrane is the critical process and this process is mediated by an envelope glycoprotein gp40. We have identified fusion inhibitory peptides from the heptad repeat-2 (HR2) of gp40. Remodeling of the original sequences using α-helix-inducible motifs revealed the interactive residues of gp40. Comparative analysis of HR2 peptides derived from four FIV strains demonstrated that the interactive surface of the Shizuoka strain-derived HR2 peptides provides the highest affinity of all the FIV strains examined.
Co-reporter:Kazumi Kajiwara, Kentaro Watanabe, Rei Tokiwa, Tomoko Kurose, Hiroaki Ohno, Hiroko Tsutsumi, Yoji Hata, Kazuki Izumi, Eiichi Kodama, Masao Matsuoka, Shinya Oishi, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 23) pp:7964-7970
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmc.2009.10.017
The bioorganic synthesis of an end-capped anti-HIV peptide from a recombinant protein was investigated. Cyanogen bromide-mediated cleavage of two Met-Gln sites across the target anti-HIV sequence generated an HIV-1 fusion inhibitor (SC35EK) analog bearing an N-terminal pyroglutamate (pGlu) residue and a C-terminal homoserine lactone (Hsl) residue. The end-capped peptide, pGlu-SC35EK-Hsl, had similar bioactivity and biophysical properties to the parent peptide, and an improved resistance to peptidase-mediated degradation was observed compared with the non-end-capped peptide obtained using standard recombinant technology.An anti-HIV peptide with N- and C-terminal end-capping groups was synthesized by cyanogen bromide-mediated cleavage of a recombinant protein.
Co-reporter:Michinori Tanaka, Kazumi Kajiwara, Rei Tokiwa, Kentaro Watanabe, Hiroaki Ohno, Hiroko Tsutsumi, Yoji Hata, Kazuki Izumi, Eiichi Kodama, Masao Matsuoka, Shinya Oishi, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 21) pp:7487-7492
Publication Date(Web):1 November 2009
DOI:10.1016/j.bmc.2009.09.015
Bioorganic synthesis of N- and C-terminal end-capped peptides by two simultaneous S-cyanocysteine-mediated cleavages of recombinant proteins is described. This approach is demonstrated in the preparation of anti-HIV fusion inhibitory peptides.Anti-HIV fusion inhibitory peptides with N- and C-terminal end-capping groups was synthesized by two simultaneous S-cyanocysteine-mediated cleavages of recombinant proteins.
Co-reporter:Yamato Suzuki, Yusuke Ohta, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
The Journal of Organic Chemistry 2009 Volume 74(Issue 11) pp:4246-4251
Publication Date(Web):May 8, 2009
DOI:10.1021/jo900681p
Efficient methods for the synthesis of pyrrole-fused indole derivatives via domino copper-catalyzed multicomponent coupling and bis-cyclization have been developed. The mono- or bis-aminomethylated pyrroloindoles and dipyrrolopyridines were selectively obtained in moderate to excellent yields by a controlled Mannich-type reaction−cyclization of 4,6-diethynyl-1,3-phenylenediamine or its pyridine congener with paraformaldehyde and a secondary amine. The high-yielding bis-cyclization of terminal alkynes without using Mannich-type reactions is also presented.
Co-reporter:Kazuya Kobayashi, Tetsuo Narumi, Shinya Oishi, Hiroaki Ohno and Nobutaka Fujii
The Journal of Organic Chemistry 2009 Volume 74(Issue 12) pp:4626-4629
Publication Date(Web):May 15, 2009
DOI:10.1021/jo9005602
A novel synthetic approach to Xaa-Yaa-type (Z)-trifluoromethylalkene dipeptide isostere (CF3-ADI) has been developed. Starting from readily available l-phenylalanine and l-alanine, several CF3-ADIs were obtained through nucleophilic trifluoromethylation of γ-keto esters and SN2′ alkylation of trifluoromethylated mesylates. The influence of a trifluoromethyl group on the diastereoselectivity of the SN2′ reaction is also discussed.
Co-reporter:Toshiaki Watanabe, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
The Journal of Organic Chemistry 2009 Volume 74(Issue 13) pp:4720-4726
Publication Date(Web):June 2, 2009
DOI:10.1021/jo9003376
An efficient catalytic system has been developed for the synthesis of carbazoles by one-pot N-arylation and oxidative biaryl coupling. A significant substituent effect of the diarylamine intermediate on oxidative coupling was observed. Mechanistic studies of oxidative coupling, including trapping of reaction intermediates and kinetic isotope effect experiments, are also presented.
Co-reporter:Yusuke Ohta, Yushi Kubota, Tsuyoshi Watabe, Hiroaki Chiba, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
The Journal of Organic Chemistry 2009 Volume 74(Issue 16) pp:6299-6302
Publication Date(Web):July 2, 2009
DOI:10.1021/jo901090u
A novel copper-catalyzed synthesis of 3-(aminomethyl)isoquinoline-fused polycyclic compounds, through four-component coupling, cyclization, and oxidation, has been developed. A Mannich-type reaction of 2-ethynylbenzaldehyde with paraformaldehyde and a secondary amine followed by treatment with a diamine component gave tricyclic isoquinolines through cascade cyclization and oxidation. Construction of fused isoquinolines of various ring sizes is also presented.
Co-reporter:Akinori Okano, Tsuyoshi Mizutani, Shinya Oishi, Tetsuaki Tanaka, Hiroaki Ohno and Nobutaka Fujii
Chemical Communications 2008 (Issue 30) pp:3534-3536
Publication Date(Web):20 Jun 2008
DOI:10.1039/B805845H
Treatment of allenic bromoalkenes bearing a nucleophilic moiety with a catalytic amount of palladium(0) in the presence of TBAF or Cs2CO3 in MeCNaffords bicyclic heterocycles in good to high yields, through zipper-mode cascade cyclisation.
Co-reporter:Yusuke Ohta, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
Chemical Communications 2008 (Issue 7) pp:835-837
Publication Date(Web):04 Jan 2008
DOI:10.1039/B718201E
Copper(I)-catalysed domino four-component coupling–cyclisation using 2-ethynylbenzaldehydes, paraformaldehyde, secondary amine, and t-BuNH2 in DMF leads to direct and efficient formation of 3-(aminomethyl)isoquinolines in good to high yields.
Co-reporter:Kenji Tomita ; Shinya Oishi ; Hiroaki Ohno ; Stephen C. Peiper
Journal of Medicinal Chemistry 2008 Volume 51(Issue 23) pp:7645-7649
Publication Date(Web):November 14, 2008
DOI:10.1021/jm800930w
Kisspeptin−GPR54 signaling is involved in the suppression of cancer metastasis and regulation of hormonal secretion. Recently, matrix metalloproteinase mediated deactivation of kisspeptins through hydrolysis of the Gly-Leu peptide bond has been reported. In the present report, GPR54 agonistic peptides having several nonhydrolyzable Gly-Leu dipeptide isosteres were designed and synthesized. (E)-Alkene- and hydroxyethylene-type isostere-containing analogues maintained the original activity with higher stability in murine serum and resistance to MMP-9-mediated cleavage.
Co-reporter:Shinya Oishi ; Saori Ito ; Hiroki Nishikawa ; Kentaro Watanabe ; Michinori Tanaka ; Hiroaki Ohno ; Kazuki Izumi ; Yasuko Sakagami ; Eiichi Kodama ; Masao Matsuoka
Journal of Medicinal Chemistry 2008 Volume 51(Issue 3) pp:388-391
Publication Date(Web):January 16, 2008
DOI:10.1021/jm701109d
Reported herein are the design, biological activities, and biophysical properties of a novel HIV-1 membrane fusion inhibitor. α-Helix-inducible X-EE-XX-KK motifs were applied to design an enfuvirtide analogue 2 that exhibited highly potent anti-HIV activity against wild-type HIV-1, enfuvirtide-resistant HIV-1 strains, and an HIV-2 strain in vitro. Indispensable residues for bioactivity of enfuvirtide, including the residues interacting with the N-terminal heptad repeat and the C-terminal hydrophobic residues, were identified.
Co-reporter:Hiroki Nishikawa, Shinya Oishi, Mizuno Fujita, Kentaro Watanabe, Rei Tokiwa, Hiroaki Ohno, Eiichi Kodama, Kazuki Izumi, Keiko Kajiwara, Takeshi Naitoh, Masao Matsuoka, Akira Otaka, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 20) pp:9184-9187
Publication Date(Web):15 October 2008
DOI:10.1016/j.bmc.2008.09.018
Emergence of multi-drug resistant HIV-1 is a serious problem for AIDS treatment. Recently, the virus-cell membrane fusion process has been identified as a promising target for the development of novel drugs against these resistant variants. In this study, we identified a 29-residue peptide fusion inhibitor, SC29EK, which shows activity comparable to the previously reported inhibitor SC35EK. Some residues in SC29EK not required for interaction with virus gp41 heptad repeat 1 (HR1) were replaced with a non-proteinogenic amino acid, 2-aminoisobutyric acid (Aib), to stabilize the α-helix structure and to provide resistance to peptidases.Minimal sequence of potential HIV-1 fusion inhibitors was identified using glycoprotein gp41-derived peptides containing α-helix-inducible EK motifs. One of the N-terminal motifs was replaced with an α-helix-inducible motif containing 2-aminoisobutyric acids.
Co-reporter:Satoshi Ueda, Manabu Kato, Shinsuke Inuki, Hiroaki Ohno, Barry Evans, Zi-xuan Wang, Stephen C. Peiper, Kazuki Izumi, Eiichi Kodama, Masao Matsuoka, Hideko Nagasawa, Shinya Oishi, Nobutaka Fujii
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 14) pp:4124-4129
Publication Date(Web):15 July 2008
DOI:10.1016/j.bmcl.2008.05.092
The design and synthesis of novel non-peptide CXCR4 antagonists is described. The peptide backbone of highly potent cyclic peptide-based CXCR4 antagonists was entirely replaced by an indole framework, which was expected to reproduce the disposition of the key pharmacophores consistent with those of potential bioactive conformations of the original peptides. A structure–activity relationship study on a series of modified indoles identified novel small-molecule antagonists having three pharmacophore functional groups through the appropriate linkers.
Co-reporter:Toshiaki Watanabe, Satoshi Ueda, Shinsuke Inuki, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
Chemical Communications 2007 (Issue 43) pp:4516-4518
Publication Date(Web):05 Sep 2007
DOI:10.1039/B707899D
One-pot N-arylation and oxidative coupling can be promoted by a common palladium catalyst in the presence of appropriate additives: palladium-catalyzed N-arylation of anilines with aryl triflates under the standard conditions followed by addition of acetic acid under oxygen or air atmosphere afforded various types of functionalized carbazoles in good to excellent yields.
Co-reporter:Jérôme Cluzeau, Shinya Oishi, Hiroaki Ohno, Zixuan Wang, Barry Evans, Stephen C. Peiper and Nobutaka Fujii
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 12) pp:1915-1923
Publication Date(Web):14 May 2007
DOI:10.1039/B702649H
The four diastereomers of 2,5-bis[(3-guanidino)propyl]-1-[3-(4-hydroxyphenyl)propionyl]-7-(2-naphthylacetyl)-1,4,7-triazacycloundec-9-en-3-one (54–57) and of 2,5-bis[(3-guanidino)propyl]-1-(4-hydroxyphenylacetyl)-7-(2-naphthylacetyl)-1,4,7-triazacycloundec-9-en-3-one (58–61) were synthesized by a divergent methodology from L- and D-glutamic acids. The 11-membered ring core was made by ring closing metathesis of linear bis(allylamines), and the guanidyl functions were introduced by a simultaneous double Mitsunobu reaction using bis(Boc)guanidine. These compounds were designed to mimic cyclic pentapeptide FC131 (c[Gly-D-Tyr-Arg-Arg-Nal]).
Co-reporter:Hiroaki Ohno Dr.;Yusuke Ohta;Shinya Oishi Dr. Dr.
Angewandte Chemie 2007 Volume 119(Issue 13) pp:
Publication Date(Web):15 FEB 2007
DOI:10.1002/ange.200604342
Ring an Ring: Obige Kupfer(I)-katalysierte Dreikomponentenreaktion führt zu 2-(Aminomethyl)indol-Derivaten, wobei zwei C-N- und eine C-C-Bindung gebildet werden und theoretisch nur Wasser als Beiprodukt entsteht. Dieser Weg zum Indolsystem eignet sich in Kombination mit einer palladiumkatalysierten C-H-Funktionalisierung auch für die Synthese polycyclischer Indole (siehe Schema).
Co-reporter:Hiroaki Ohno Dr.;Yusuke Ohta;Shinya Oishi Dr. Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 13) pp:
Publication Date(Web):15 FEB 2007
DOI:10.1002/anie.200604342
Tying up loose ends: A copper(I)-catalyzed three-component reaction has been developed that leads to the formation of 2-(aminomethyl)indole derivatives. Water is the sole theoretical by-product in this reaction in which two CN bonds and one CC bond are formed. This route to the indole nucleus in combination with a palladium-catalyzed functionalization of a CH bond is also useful for the synthesis of polycyclic indoles (see scheme).
Co-reporter:Hiroaki Ohno Dr.;Yusuke Ohta;Shinya Oishi Dr. Dr.
Angewandte Chemie 2007 Volume 119(Issue 18) pp:
Publication Date(Web):23 APR 2007
DOI:10.1002/ange.200790078
Co-reporter:Hiroaki Ohno Dr.;Yusuke Ohta;Shinya Oishi Dr. Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 18) pp:
Publication Date(Web):23 APR 2007
DOI:10.1002/anie.200790078
Co-reporter:Tetsuo Narumi, Ayumu Niida, Kenji Tomita, Shinya Oishi, Akira Otaka, Hiroaki Ohno and Nobutaka Fujii
Chemical Communications 2006 (Issue 45) pp:4720-4722
Publication Date(Web):28 Sep 2006
DOI:10.1039/B608596B
By a novel one-pot reaction sequence involving consecutive organocopper-mediated reduction, transmetalation and asymmetric alkylation, a highly diastereoselective synthesis of functionalized (Z)-fluoroalkene dipeptide isosteres was achieved in good to excellent yields.
Co-reporter:Hirokazu Tamamura, Hiroshi Tsutsumi, Hiroyuki Masuno, Satoko Mizokami, Kenichi Hiramatsu, Zixuan Wang, John O. Trent, Hideki Nakashima, Naoki Yamamoto, Stephen C. Peiper and Nobutaka Fujii
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 12) pp:2354-2357
Publication Date(Web):2006/05/12
DOI:10.1039/B603818B
A linear type of several low molecular weight CXCR4 antagonists were developed based on T140 analogs, which were previously found to be strong CXCR4 antagonists that block X4-HIV-1 entry and have inhibitory activities against cancer metastasis/progression and rheumatoid arthritis.
Co-reporter:Ayumu Niida, Zixuan Wang, Kenji Tomita, Shinya Oishi, Hirokazu Tamamura, Akira Otaka, Jean-Marc Navenot, James R. Broach, Stephen C. Peiper, Nobutaka Fujii
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 1) pp:134-137
Publication Date(Web):1 January 2006
DOI:10.1016/j.bmcl.2005.09.054
Metastin has been identified as a metastasis suppressor gene product that mediates its function through a G protein coupled receptor, GPR54. To refine insight into the critical pharmacophore for the activation of GPR54, we have conducted alanine and d-amino acid scanning on a biologically active metastin fragment (45–54). Based on these data and structures of peptides previously reported to activate GPR54, a series of shortened metastin (45–54) derivatives were synthesized and tested for the ability to induce GPR54 signaling. These biological experiments were performed in yeast containing human GPR54 that was coupled to the pheromone response pathway and a pheromone responsive lacZ reporter gene. Compounds 32, 33, and 39, which possess an N-terminal basic group and a C-terminal RW-amide motif, were strong agonists, similar to the level of metastin. This may provide an approach to reverse the pro-metastatic effect of metastin deletion in multiple malignant tumors.
Co-reporter:Hirokazu Tamamura, Ai Esaka, Teppei Ogawa, Takanobu Araki, Satoshi Ueda, Zixuan Wang, John O. Trent, Hiroshi Tsutsumi, Hiroyuki Masuno, Hideki Nakashima, Naoki Yamamoto, Stephen C. Peiper, Akira Otaka and Nobutaka Fujii
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 24) pp:4392-4394
Publication Date(Web):15 Nov 2005
DOI:10.1039/B513145F
Structure–activity relationship studies on CXCR4 antagonists, which were previously found by using cyclic pentapeptide libraries, were performed to optimize side-chain functional groups, involving conformationally constrained analogues. In addition, a new lead of cyclic pentapeptides with the introduction of a novel pharmacophore was developed.
Co-reporter:Satoshi Ueda;Mizuno Fujita;Hirokazu Tamamura Dr. ;Akira Otaka
ChemBioChem 2005 Volume 6(Issue 11) pp:
Publication Date(Web):5 OCT 2005
DOI:10.1002/cbic.200500272
Newly developed phenacyl-type protection (Mapoc) for amines provides a facile and efficient synthetic protocol for peptides/proteins, in which sequential native chemical ligation (NCL) in the same reaction vessel can be conducted in a purification-free manner. A synthetic strategy of sequential NCL followed by oxidation with DMSO for disulfide-bond formation was successfully applied to the one-pot preparation of the human-brain natriuretic peptide (hBNP).
Co-reporter:Hirokazu Tamamura, Terukazu Kato, Akira Otaka and Nobutaka Fujii
Organic & Biomolecular Chemistry 2003 vol. 1(Issue 14) pp:2468-2473
Publication Date(Web):10 Jun 2003
DOI:10.1039/B304842J
Several β-secretase inhibitors were designed based on hydroxyethylamine dipeptide isostere (HDI) structures and were synthesized by a methodology using the aza-Payne rearragement and O,N-acyl transfer reactions to study their structure–activity relationships. Among these pseudopeptides, effective compounds were developed as the first β-secretase inhibitors containing the HDI transition state mimic with potent enzyme inhibitory activity (IC50 < 100 nM).
Co-reporter:Nobutaka Fujii ;Shinya Oishi;Kenichi Hiramatsu;Takanobu Araki;Satoshi Ueda;Hirokazu Tamamura Dr.;Akira Otaka Dr.;Shuichi Kusano Dr.;Shigemi Terakubo;Hideki Nakashima ;James A. Broach Dr.;John O. Trent Dr.;Zi-xuan Wang Dr.;Stephen C. Peiper
Angewandte Chemie 2003 Volume 115(Issue 28) pp:
Publication Date(Web):16 JUL 2003
DOI:10.1002/ange.200351024
Peptide auf das Wesentliche beschränkt: Das Pentapeptid cyclo(-L-Nal 1-Gly 2-D-Tyr 3-L-Arg 4-L-Arg 5-) wurde durch Kombination einer konformationsbasierten mit einer sequenzbasierten Peptid-Bibliothek als möglicher CXCR4-Antagonist ermittelt (Nal=L-3-(2-Naphthyl)alanin; Bild: Projektion der fünf energetisch günstigsten Strukturen). Mit einem IC50-Wert von 4 nM ist es vergleichbar effektiv wie die 14-Aminosäuren-Stammverbindung T140.
Co-reporter:Nobutaka Fujii ;Shinya Oishi;Kenichi Hiramatsu;Takanobu Araki;Satoshi Ueda;Hirokazu Tamamura Dr.;Akira Otaka Dr.;Shuichi Kusano Dr.;Shigemi Terakubo;Hideki Nakashima ;James A. Broach Dr.;John O. Trent Dr.;Zi-xuan Wang Dr.;Stephen C. Peiper
Angewandte Chemie International Edition 2003 Volume 42(Issue 28) pp:
Publication Date(Web):16 JUL 2003
DOI:10.1002/anie.200351024
Efficient downsizing of peptides: By combination of two orthogonal “conformation-based” and “sequence-based” libraries, the cyclic pentapeptide cyclo(-L-Nal 1-Gly 2-D-Tyr 3-L-Arg 4-L-Arg 5-) (Nal=L-3-(2-naphthyl)alanine; see overlay of the five lowest energy structures), which exhibited strong CXCR4 antagonism (IC50=4 nM) comparable to that of a 14-residue lead compound, T140, was discovered.
Co-reporter:Hirokazu Tamamura, Akane Omagari, Kenichi Hiramatsu, Shinya Oishi, Hiromu Habashita, Taisei Kanamoto, Kazuyo Gotoh, Naoki Yamamoto, Hideki Nakashima, Akira Otaka, Nobutaka Fujii
Bioorganic & Medicinal Chemistry 2002 Volume 10(Issue 5) pp:1417-1426
Publication Date(Web):May 2002
DOI:10.1016/S0968-0896(01)00419-9
We have previously found that a 14-amino acid residue-peptide, T140, inhibits infection of target cells by T cell line-tropic HIV-1 (X4-HIV-1) through its specific binding to a chemokine receptor, CXCR4. Here, the importance of an l-3-(2-naphthyl)alanine (Nal) residue at position 3 in T140 for high anti-HIV activity and inhibitory activity against Ca2+ mobilization induced by stromal cell-derived factor (SDF)-1α-stimulation through CXCR4 has initially been shown by the synthesis and biological evaluation of several analogues, where Nal3 is substituted by diverse aromatic amino acids. Next, the order of the N-terminal 3 residues (Arg1-Arg2-Nal3) has been proved to be important from the structure–activity relationship (SAR) study shuffling these residues. Based on these results, we have found 10-residue peptides possessing modest anti-HIV activity by systematic antiviral evaluation of a series of synthetic, shortened analogues of T140.Graphic
Co-reporter:Hirokazu Tamamura, Kenichi Hiramatsu, Kazuhide Miyamoto, Akane Omagari, Shinya Oishi, Hideki Nakashima, Naoki Yamamoto, Yoshihiro Kuroda, Terumichi Nakagawa, Akira Otaka, Nobutaka Fujii
Bioorganic & Medicinal Chemistry Letters 2002 Volume 12(Issue 6) pp:923-928
Publication Date(Web):25 March 2002
DOI:10.1016/S0960-894X(02)00041-0
A 14-residue peptide, T140, strongly inhibits the T-cell line-tropic HIV-1 (X4-HIV-1) infection, since this peptide functions as a specific antagonist against a chemokine receptor, CXCR4. T140 takes an antiparallel β-sheet structure with a type II′ β-turn. In the present paper, we have designed and synthesized several T140 analogues, in which an (E)-alkene dipeptide isostere was inserted into the type II′ β-turn moiety, as a bridging study to develop nonpeptidic CXCR4 inhibitors. It has been proven that the turn region of T140 can be replaced by the above surrogate with the maintenance of strong anti-HIV activity.Graphic
Co-reporter:Akira Otaka Dr.;Miki Nakamura;Daisuke Nameki;Eiichi Kodama Dr.;Susumu Uchiyama Dr.;Syota Nakamura;Hiroaki Nakano;Hirokazu Tamamura Dr.;Yuji Kobayashi Dr.;Masao Matsuoka Dr. Dr.
Angewandte Chemie 2002 Volume 114(Issue 16) pp:
Publication Date(Web):21 AUG 2002
DOI:10.1002/1521-3757(20020816)114:16<3061::AID-ANGE3061>3.0.CO;2-P
Substitution an der äußeren Oberfläche des Sechs-Helix-Peptidbündels verbesserte die Löslichkeit und erhöhte die Anti-HIV-1-Aktivität von SC-Peptiden. Die Reste E und K an den Positionen b, c, f und g (siehe Schema) stabilisieren die für die Inhibierung essenzielle α-Helixkonformation; die Reste Z an den Positionen a, d und e wechselwirken mit dem inneren Strang.
Co-reporter:Akira Otaka Dr.;Miki Nakamura;Daisuke Nameki;Eiichi Kodama Dr.;Susumu Uchiyama Dr.;Syota Nakamura;Hiroaki Nakano;Hirokazu Tamamura Dr.;Yuji Kobayashi Dr.;Masao Matsuoka Dr. Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 16) pp:
Publication Date(Web):21 AUG 2002
DOI:10.1002/1521-3773(20020816)41:16<2937::AID-ANIE2937>3.0.CO;2-J
Substitution in the outer surface of the six-helix peptide bundle improved the solubility and enhanced the anti-HIV-1 activity of SC peptides. The E and K residues at positions b, c, f, and g (see scheme) stabilize the α-helix conformation critical to inhibition; the Z residues at positions a, d, and e interact with the inner strand.
Co-reporter:Hirokazu Tamamura, Makiko Sugioka, Yoshihiko Odagaki, Akane Omagari, Yukiko Kan, Shinya Oishi, Hideki Nakashima, Naoki Yamamoto, Stephen C. Peiper, Nobuyuki Hamanaka, Akira Otaka, Nobutaka Fujii
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 17) pp:2409
Publication Date(Web):3 September 2001
DOI:10.1016/S0960-894X(01)00434-6
Co-reporter:Hirokazu Tamamura, Makiko Sugioka, Yoshihiko Odagaki, Akane Omagari, Yukiko Kan, Shinya Oishi, Hideki Nakashima, Naoki Yamamoto, Stephen C Peiper, Nobuyuki Hamanaka, Akira Otaka, Nobutaka Fujii
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 3) pp:359-362
Publication Date(Web):2 February 2001
DOI:10.1016/S0960-894X(00)00664-8
We report the solution structure of T140, a truncated polyphemusin peptide analogue that efficiently inhibits infection of target cells by T-cell line-tropic strains of HIV-1 through its specific binding to a chemokine receptor, CXCR4. Nuclear magnetic resonance analysis and molecular dynamic calculations revealed that T140 has a rigidly structured conformation constituted by an antiparallel β-sheet and a type II′ β-turn. A protuberance is formed on one side of the β-sheet by the side-chain functional groups of the three amino acid residues (l-3-(2-naphthyl)alanine3, Tyr5 and Arg14), each of which is indispensable for strong anti-HIV activity. These findings provide a rationale to dissect the structural basis for the ability of this compound to block the interaction between CXCR4 and envelope glycoproteins from T-tropic strains of HIV-1.Graphic
Co-reporter:Hirokazu Tamamura, Akane Omagari, Shinya Oishi, Taisei Kanamoto, Naoki Yamamoto, Stephen C Peiper, Hideki Nakashima, Akira Otaka, Nobutaka Fujii
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 23) pp:2633-2637
Publication Date(Web):4 December 2000
DOI:10.1016/S0960-894X(00)00535-7
A polyphemusin peptide analogue, T22 ([Tyr5,12, Lys7]-polyphemusin II), and its shortened potent analogues, T134 (des-[Cys8,13, Tyr9,12]-[d-Lys10, Pro11, l-citrulline16]-T22 without C-terminal amide) and T140 {[l-3-(2-naphthyl)alanine3]-T134}, strongly inhibit the T-cell line-tropic (T-tropic) HIV-1 infection through their specific binding to a chemokine receptor, CXCR4. T22 is an extremely basic peptide possessing five Arg and three Lys residues in the molecule. In our previous study, we found that there is an apparent correlation in the T22-related peptides between the number of total positive charges and anti-HIV activity or cytotoxicity. Here, we have conducted the conventional Ala-scanning study in order to define the anti-HIV activity pharmacophore of T140 (the strongest analogue among our compounds) and identified four indispensable amino acid residues (Arg2, Nal3, Tyr5, and Arg14). Based on this result, a series of l-citrulline (Cit)-substituted analogues of T140 with decreased net positive charges have been synthesized and evaluated in terms of anti-HIV activity and cytotoxicity. As a result, novel effective inhibitors, TC14003 and TC14005, possessing higher selectivity indexes (SIs, 50% cytotoxic concentration/50% effective concentration) than that of T140 have been developed.
Co-reporter:Hyunsuk Shim, Shinya Oishi, Nobutaka Fujii
Seminars in Cancer Biology (April 2009) Volume 19(Issue 2) pp:123-134
Publication Date(Web):1 April 2009
DOI:10.1016/j.semcancer.2008.11.004
Chemokines (chemotactic cytokines) are a family of proteins associated with the trafficking and activation of leukocytes and other cell types in immune surveillance and inflammatory response. Besides their roles in the immune system, they play pleiotropic roles in tumor initiation, promotion, and progression. Chemokines can be classified into four subfamilies of chemokines, CXC, CC, C, or CX3C, based on their number and spacing of conserved cysteine residues near the N-terminus. This CXC subfamily can be further subclassified into two groups, depending on the presence or absence of a tripeptide motif glutamic acid–leucine–arginine (ELR) in the N-terminal domain. ELR−CXCL12, which binds to CXCR4 has been frequently implicated in various cancers. Over the past several years, studies have increasingly shown that the CXCR4/CXCL12 axis plays critical roles in tumor progression, such as invasion, angiogenesis, survival, homing to metastatic sites. This review focuses on involvement of CXCR4/CXCL12 interaction in neuroectodermal cancers and their therapeutic potentials. As an attractive therapeutic target of CXCR4/CXCL12 axis for cancer chemotherapy, development history and application of CXCR4 antagonists are described.
Co-reporter:Tsuyoshi Watabe, Yukihiro Terakawa, Kentaro Watanabe, Hiroaki Ohno, ... Nobutaka Fujii
Journal of Molecular Biology (25 September 2009) Volume 392(Issue 3) pp:657-665
Publication Date(Web):25 September 2009
DOI:10.1016/j.jmb.2009.07.027
The S138A substitution of fusion inhibitory peptides derived from the C-terminal heptad repeat (C-HR) of the human immunodeficiency virus type 1 (HIV-1) gp41 leads to enhanced binding affinity to the N-terminal heptad repeat (N-HR). As such, these peptides exhibit highly potent anti-HIV-1 activity. X-ray crystallographic analysis was performed to understand the effect of the substitution on binding affinity. The comparison of the native and S138A crystal structures indicated that the increase in the hydrophobicity of the S138A substitution may aid the stabilization of the N-HR/C-HR complex through additional hydrophobic contacts. Free-energy calculations suggest that the difference between the desolvation free energies of the C-HR-derived peptides with and without the S138A mutation dominates the observed difference in anti-HIV-1 activity.
Co-reporter:Shinya Oishi, Hirotaka Kamitani, Yasuyo Kodera, Kentaro Watanabe, Kazuya Kobayashi, Tetsuo Narumi, Kenji Tomita, Hiroaki Ohno, Takeshi Naito, Eiichi Kodama, Masao Matsuoka and Nobutaka Fujii
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 14) pp:NaN2877-2877
Publication Date(Web):2009/06/04
DOI:10.1039/B907983A
The α-helix structures of the anti-HIV fusion inhibitory peptides are stabilized by the amino acid sequence and by intrachain hydrogen bonds. The study of peptide analogues using (E)-alkene and (Z)-fluoroalkene dipeptide isosteres demonstrated the substantial, yet position-dependent, contribution of hydrogen bonds to the α-helix stability and anti-HIV bioactivity.
Co-reporter:Akinori Okano, Tsuyoshi Mizutani, Shinya Oishi, Tetsuaki Tanaka, Hiroaki Ohno and Nobutaka Fujii
Chemical Communications 2008(Issue 30) pp:NaN3536-3536
Publication Date(Web):2008/06/20
DOI:10.1039/B805845H
Treatment of allenic bromoalkenes bearing a nucleophilic moiety with a catalytic amount of palladium(0) in the presence of TBAF or Cs2CO3 in MeCNaffords bicyclic heterocycles in good to high yields, through zipper-mode cascade cyclisation.
Co-reporter:Eriko Inokuchi, Ai Yamada, Kentaro Hozumi, Kenji Tomita, Shinya Oishi, Hiroaki Ohno, Motoyoshi Nomizu and Nobutaka Fujii
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 9) pp:NaN3427-3427
Publication Date(Web):2011/02/22
DOI:10.1039/C0OB01193B
Amidine-type peptide bond isosteres were designed based on the substitution of the peptide bond carbonyl (CO) group with an imino (CNH) group. The positively-charged property of the isosteric part resembles a reduced amide-type peptidomimetic. The peptidyl amidine units were synthesized by the reduction of a key amidoxime (N-hydroxyamidine) precursor, which was prepared from nitrile oxide components as an aminoacyl or peptidyl equivalent. This nitrile oxide-mediated C–N bond formation was also used for peptide macrocyclization, in which the amidoxime group was converted to peptide bonds under mild acidic conditions. Syntheses of the cyclic RGD peptide and a peptidomimetic using both approaches, and their inhibitory activity against integrin-mediated cell attachment, are presented.
Co-reporter:Akira Iwata, Shinsuke Inuki, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
Chemical Communications 2014 - vol. 50(Issue 3) pp:NaN300-300
Publication Date(Web):2013/10/28
DOI:10.1039/C3CC46511J
Efficient palladium-catalysed cascade cyclisation to form spiroindoles is developed. Treatment of indoles bearing a propargyl chloride side chain at the 3-position with various external nucleophiles in the presence of a catalytic amount of Pd2(dba)3·CHCl3/dppb and Cs2CO3 in THF gives fused tetracyclic spiroindoles in moderate to good yields.
Co-reporter:Yamato Suzuki, Shinya Oishi, Yoshinori Takei, Misato Yasue, Ryosuke Misu, Saori Naoe, Zengye Hou, Tatsuhide Kure, Isao Nakanishi, Hiroaki Ohno, Akira Hirasawa, Gozoh Tsujimoto and Nobutaka Fujii
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 25) pp:NaN4915-4915
Publication Date(Web):2012/04/25
DOI:10.1039/C2OB25298H
Two classes of fused nitrogen heterocycles were designed as CK2 inhibitor candidates on the basis of previous structure–activity relationship (SAR) studies. Various dipyrrolo[3,2-b:2′,3′-e]pyridine and benzo[g]indazole derivatives were prepared using transition-metal-catalysed cascade and/or multicomponent reactions. Biological evaluation of these candidates revealed that benzo[g]indazole is a promising scaffold for potent CK2 inhibitors. The inhibitory activities on cell proliferation of these potent CK2 inhibitors are also presented.
Co-reporter:Zengye Hou, Shinya Oishi, Yamato Suzuki, Tatsuhide Kure, Isao Nakanishi, Akira Hirasawa, Gozoh Tsujimoto, Hiroaki Ohno and Nobutaka Fujii
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 20) pp:NaN3296-3296
Publication Date(Web):2013/03/11
DOI:10.1039/C3OB40223A
Pyrazolo[4,3-b]indole derivatives have been designed as novel CK2 inhibitor compounds based on the binding mode analysis of a previously reported phenylpyrazole-type CK2 inhibitor. A series of pyrazolo[4,3-b]indoles and related dihydropyrazolo[4,3-b]indoles were efficiently prepared from simple starting materials using a gold-catalysed three-component annulation reaction as a key step. Several of the newly synthesized compounds displayed high levels of inhibitory activity, indicating that the pyrazolo[4,3-b]indole core represents a promising scaffold for the development of potent CK2 inhibitors.
Co-reporter:Jérôme Cluzeau, Shinya Oishi, Hiroaki Ohno, Zixuan Wang, Barry Evans, Stephen C. Peiper and Nobutaka Fujii
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 12) pp:NaN1923-1923
Publication Date(Web):2007/05/14
DOI:10.1039/B702649H
The four diastereomers of 2,5-bis[(3-guanidino)propyl]-1-[3-(4-hydroxyphenyl)propionyl]-7-(2-naphthylacetyl)-1,4,7-triazacycloundec-9-en-3-one (54–57) and of 2,5-bis[(3-guanidino)propyl]-1-(4-hydroxyphenylacetyl)-7-(2-naphthylacetyl)-1,4,7-triazacycloundec-9-en-3-one (58–61) were synthesized by a divergent methodology from L- and D-glutamic acids. The 11-membered ring core was made by ring closing metathesis of linear bis(allylamines), and the guanidyl functions were introduced by a simultaneous double Mitsunobu reaction using bis(Boc)guanidine. These compounds were designed to mimic cyclic pentapeptide FC131 (c[Gly-D-Tyr-Arg-Arg-Nal]).
Co-reporter:Yusuke Ohta, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
Chemical Communications 2008(Issue 7) pp:NaN837-837
Publication Date(Web):2008/01/04
DOI:10.1039/B718201E
Copper(I)-catalysed domino four-component coupling–cyclisation using 2-ethynylbenzaldehydes, paraformaldehyde, secondary amine, and t-BuNH2 in DMF leads to direct and efficient formation of 3-(aminomethyl)isoquinolines in good to high yields.
Co-reporter:Toshiaki Watanabe, Satoshi Ueda, Shinsuke Inuki, Shinya Oishi, Nobutaka Fujii and Hiroaki Ohno
Chemical Communications 2007(Issue 43) pp:NaN4518-4518
Publication Date(Web):2007/09/05
DOI:10.1039/B707899D
One-pot N-arylation and oxidative coupling can be promoted by a common palladium catalyst in the presence of appropriate additives: palladium-catalyzed N-arylation of anilines with aryl triflates under the standard conditions followed by addition of acetic acid under oxygen or air atmosphere afforded various types of functionalized carbazoles in good to excellent yields.
Co-reporter:Shiho Okazaki, Shinya Oishi, Tsukasa Mizuhara, Kazuya Shimura, Hiroto Murayama, Hiroaki Ohno, Masao Matsuoka and Nobutaka Fujii
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 16) pp:NaN4713-4713
Publication Date(Web):2015/03/10
DOI:10.1039/C5OB00301F
3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine (PD 404182) and 3,4-dihydro-2H-benzo[4,5]isothiazolo[2,3-a]pyrimidine are the heterocyclic antiretroviral agents against human immunodeficiency virus type 1 (HIV-1) infection. On the basis of similar structure–activity relationships of anti-HIV activities toward the early-stage of viral infection between these unique scaffolds, the transformations under the bioassay conditions were investigated. The distinctive S–N bond in the isothiazolopyrimidine scaffold was immediately cleaved under reductive conditions in the presence of GSH to generate a thiophenol derivative. A similar rapid conversion of PD 404182 into the same thiophenol derivative was observed, suggesting that pyrimidobenzothiazine and isothiazolopyrimidine scaffolds may work as prodrug forms of the common bioactive thiophenol derivatives.
Co-reporter:Shinya Oishi and Nobutaka Fujii
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 30) pp:NaN5731-5731
Publication Date(Web):2012/03/19
DOI:10.1039/C2OB25107H
The development of novel peptide and peptidomimetic ligands for the CXC chemokine receptor 4 (CXCR4) as therapeutic agents for HIV-1 infection, cancer, and immune system diseases has grown over the last decade. In this perspective article, the design of CXCR4 agonists and antagonists from endogenous stromal cell-derived factor-1 (SDF-1)/CXCL12 and horseshoe crab-derived antimicrobial peptides and their therapeutic and diagnostic applications are described.
Co-reporter:Tsukasa Mizuhara, Shinya Oishi, Hiroaki Ohno, Kazuya Shimura, Masao Matsuoka and Nobutaka Fujii
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 33) pp:NaN6802-6802
Publication Date(Web):2012/06/22
DOI:10.1039/C2OB25904D
3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine (PD 404182) is a virucidal heterocyclic compound active against various viruses, including HCV, HIV, and simian immunodeficiency virus. Using facile synthetic approaches that we developed for the synthesis of pyrimido[1,2-c][1,3]benzothiazin-6-imines and related tricyclic derivatives, the parallel structural optimizations of the central 1,3-thiazin-2-imine core, the benzene part, and the cyclic amidine part of PD 404182 were investigated. Replacement of the 6-6-6 pyrimido[1,2-c][1,3]benzothiazin-6-imine framework with 5-6-6 or 6-6-5 derivatives led to a significant loss of anti-HIV activity, and introduction of a hydrophobic group at the 9- or 10-positions improved the potency. In addition, we demonstrated that the PD 404182 derivative exerts anti-HIV effects at an early stage of viral infection.
Co-reporter:Taro Noguchi, Shinya Oishi, Kaori Honda, Yasumitsu Kondoh, Tamio Saito, Hiroaki Ohno, Hiroyuki Osada and Nobutaka Fujii
Chemical Communications 2016 - vol. 52(Issue 49) pp:NaN7656-7656
Publication Date(Web):2016/05/11
DOI:10.1039/C6CC03114E
We established a facile access to an unexplored mirror-image library of chiral natural product derivatives using D-protein technology. In this process, two chemical syntheses of mirror-image substances including a target protein and hit compound(s) allow the lead discovery from a virtual mirror-image library without the synthesis of numerous mirror-image compounds.






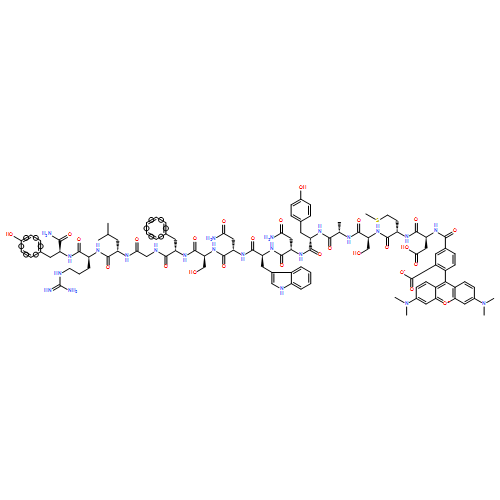


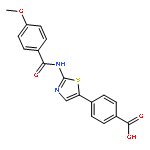
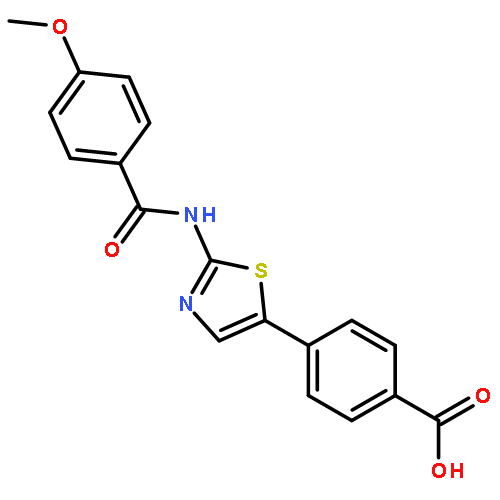
![7-(trifluoromethyl)-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/1232200/1232100-30-9.png)
![7-(trifluoromethyl)-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/1232200/1232100-30-9_b.png)
![(2E)-3-PHENYL-1-[3-(TRIFLUOROMETHYL)PHENYL]-2-PROPEN-1-ONE](/data/chemimg/348700/1150112-11-0.png)
![(2E)-3-PHENYL-1-[3-(TRIFLUOROMETHYL)PHENYL]-2-PROPEN-1-ONE](/data/chemimg/348700/1150112-11-0_b.png)


![5-((3-Chlorophenyl)amino)benzo[c][2,6]naphthyridine-8-carboxylic acid](http://img.cochemist.com/ccimg/1009900/1009820-21-6.png)
![5-((3-Chlorophenyl)amino)benzo[c][2,6]naphthyridine-8-carboxylic acid](http://img.cochemist.com/ccimg/1009900/1009820-21-6_b.png)
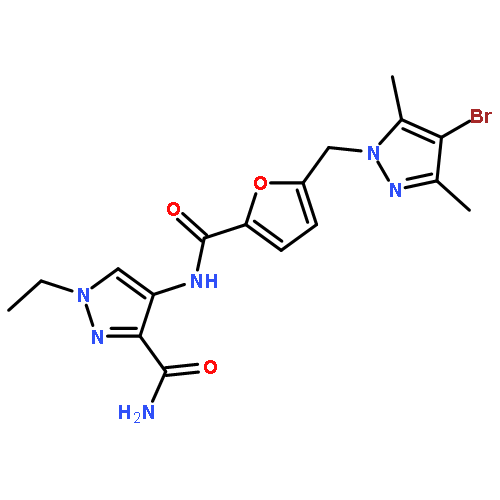


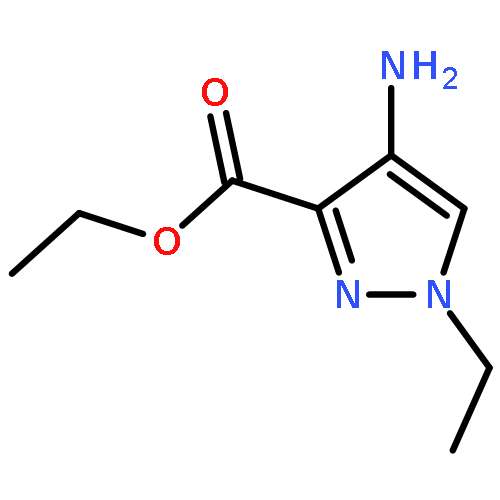




![Iodonium, bis[3-(trifluoromethyl)phenyl]-, tetrafluoroborate(1-) (1:1)](/data/chemimg/120700/888323-36-2.png)
![Iodonium, bis[3-(trifluoromethyl)phenyl]-, tetrafluoroborate(1-) (1:1)](/data/chemimg/120700/888323-36-2_b.png)

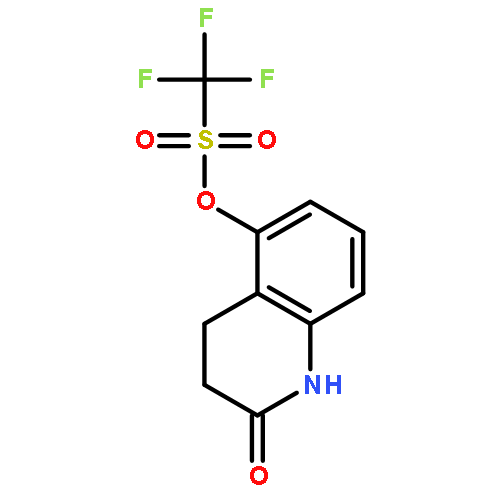
![Urea, [7-(trifluoromethyl)-9H-carbazol-2-yl]-](http://img.cochemist.com/ccimg/872700/872604-33-6.png)
![Urea, [7-(trifluoromethyl)-9H-carbazol-2-yl]-](http://img.cochemist.com/ccimg/872700/872604-33-6_b.png)
![Urea, [7-(trifluoromethyl)-9H-carbazol-3-yl]-](http://img.cochemist.com/ccimg/872700/872604-30-3.png)
![Urea, [7-(trifluoromethyl)-9H-carbazol-3-yl]-](http://img.cochemist.com/ccimg/872700/872604-30-3_b.png)

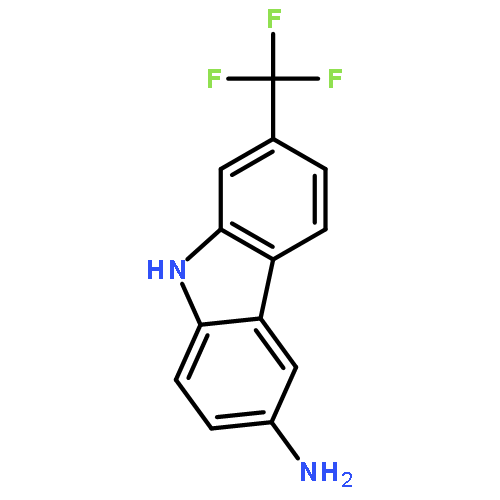
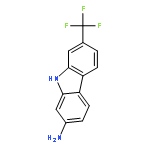

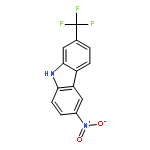
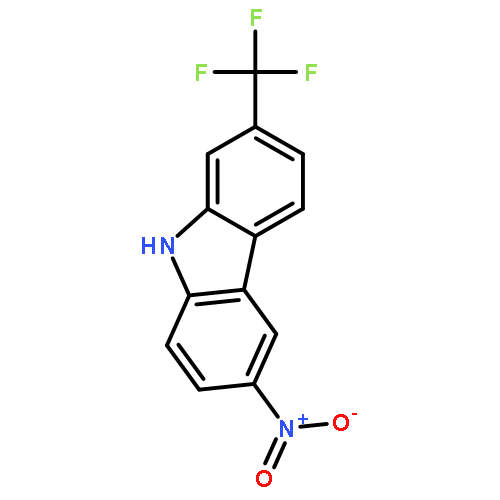
![6-(4-(2-(Piperidin-1-yl)ethoxy)phenyl)-3-(pyridin-4-yl)pyrazolo[1,5-a]pyrimidine](http://img.cochemist.com/ccimg/866500/866405-64-3.png)
![6-(4-(2-(Piperidin-1-yl)ethoxy)phenyl)-3-(pyridin-4-yl)pyrazolo[1,5-a]pyrimidine](http://img.cochemist.com/ccimg/866500/866405-64-3_b.png)
![Chloro[2-dicyclohexyl(2',6'-dimethoxybiphenyl)phosphine] gold(I)](http://img.cochemist.com/ccimg/854100/854045-95-7.png)
![Chloro[2-dicyclohexyl(2',6'-dimethoxybiphenyl)phosphine] gold(I)](http://img.cochemist.com/ccimg/854100/854045-95-7_b.png)
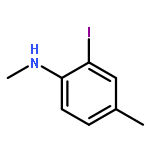



![CYCLO[3-(2-NAPHTHALENYL)-L-ALANYL-D-ALANYL-D-TYROSYL-L-ARGINYL-L-ARGINYL]](http://img.cochemist.com/ccimg/722600/722546-66-9.png)
![CYCLO[3-(2-NAPHTHALENYL)-L-ALANYL-D-ALANYL-D-TYROSYL-L-ARGINYL-L-ARGINYL]](http://img.cochemist.com/ccimg/722600/722546-66-9_b.png)
![Cyclo[3-(2-naphthalenyl)-L-alanyl-L-alanyl-D-tyrosyl-L-arginyl-L-arginyl]](http://img.cochemist.com/ccimg/722600/722546-63-6.png)
![Cyclo[3-(2-naphthalenyl)-L-alanyl-L-alanyl-D-tyrosyl-L-arginyl-L-arginyl]](http://img.cochemist.com/ccimg/722600/722546-63-6_b.png)
![Ethanone, 1-[2,4-bis(phenylmethoxy)phenyl]-2-bromo-](http://img.cochemist.com/ccimg/719300/719288-40-1.png)
![Ethanone, 1-[2,4-bis(phenylmethoxy)phenyl]-2-bromo-](http://img.cochemist.com/ccimg/719300/719288-40-1_b.png)
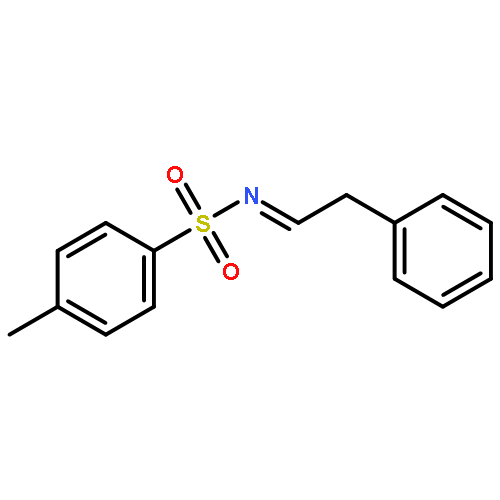
![Ethanone, 2-[[(4-methoxyphenyl)methyl]thio]-1-phenyl-](http://img.cochemist.com/ccimg/675600/675576-48-4.png)
![Ethanone, 2-[[(4-methoxyphenyl)methyl]thio]-1-phenyl-](http://img.cochemist.com/ccimg/675600/675576-48-4_b.png)

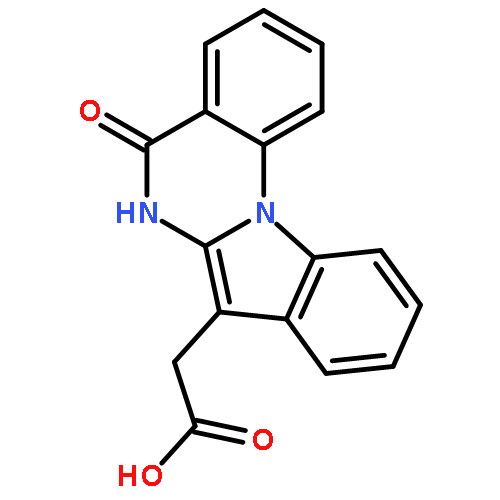
![chlorogold-tris[4-(trifluoromethyl)phenyl]phosphane (1:1)](http://img.cochemist.com/ccimg/385900/385815-83-8.png)
![chlorogold-tris[4-(trifluoromethyl)phenyl]phosphane (1:1)](http://img.cochemist.com/ccimg/385900/385815-83-8_b.png)
![SU 9516;3-[1-(3H-IMIDAZOL-4-YL)-METH-(Z)-YLIDENE]-5-METHOXY-1,3-DIHYDRO-INDOL-2-ONE](http://img.cochemist.com/ccimg/377100/377090-84-1.png)
![SU 9516;3-[1-(3H-IMIDAZOL-4-YL)-METH-(Z)-YLIDENE]-5-METHOXY-1,3-DIHYDRO-INDOL-2-ONE](http://img.cochemist.com/ccimg/377100/377090-84-1_b.png)
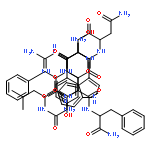
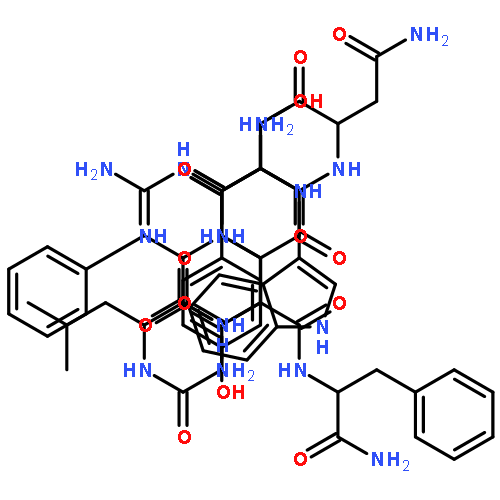


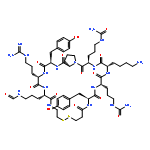
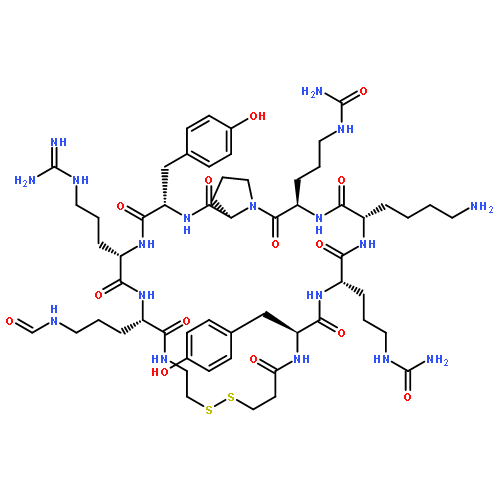




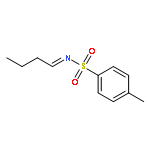
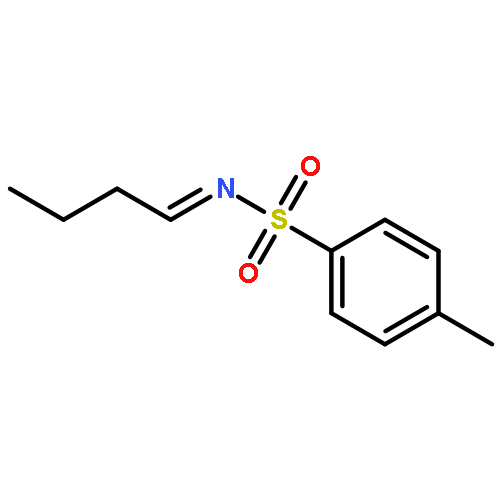
![Pyrimidine, 1,4,5,6-tetrahydro-2-[2-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/326500/326480-86-8.png)
![Pyrimidine, 1,4,5,6-tetrahydro-2-[2-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/326500/326480-86-8_b.png)

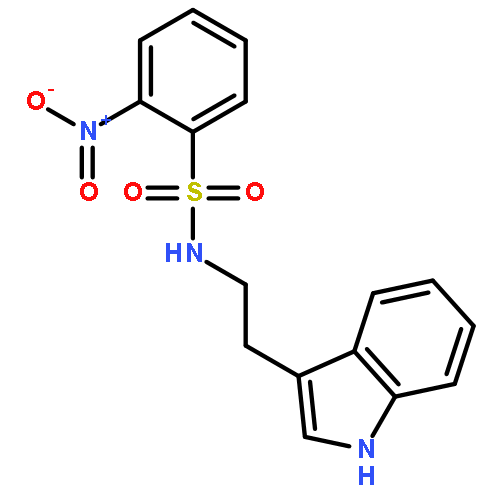
![2-Furancarboxylic acid,5-[(4-bromo-3,5-dimethyl-1H-pyrazol-1-yl)methyl]-](http://img.cochemist.com/ccimg/307000/306935-28-4.png)
![2-Furancarboxylic acid,5-[(4-bromo-3,5-dimethyl-1H-pyrazol-1-yl)methyl]-](http://img.cochemist.com/ccimg/307000/306935-28-4_b.png)

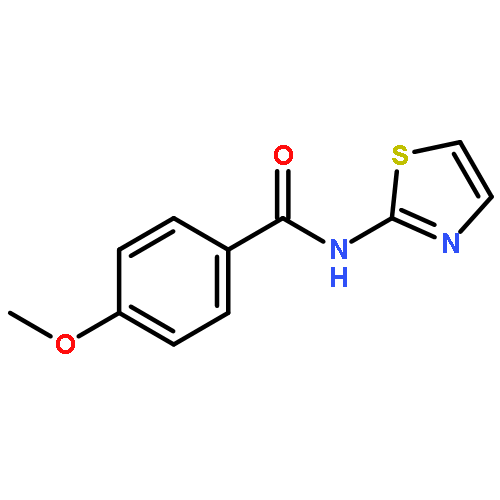
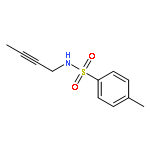
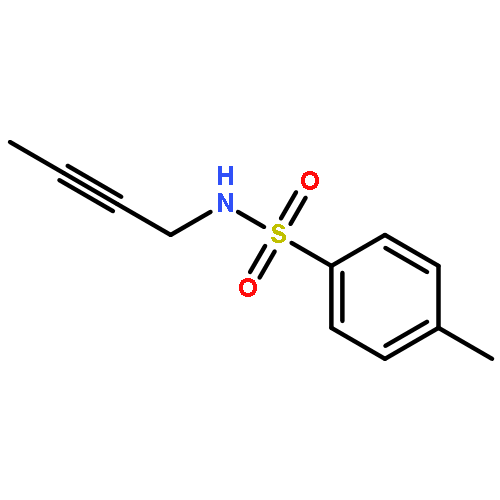


![Silane, [(3-bromo-2-thienyl)ethynyl]trimethyl-](http://img.cochemist.com/ccimg/198500/198489-77-9.png)
![Silane, [(3-bromo-2-thienyl)ethynyl]trimethyl-](http://img.cochemist.com/ccimg/198500/198489-77-9_b.png)


![4-QUINOLINECARBOXAMIDE, 3-AMINO-2-PHENYL-N-[(1S)-1-PHENYLPROPYL]-](http://img.cochemist.com/ccimg/174700/174636-16-9.png)
![4-QUINOLINECARBOXAMIDE, 3-AMINO-2-PHENYL-N-[(1S)-1-PHENYLPROPYL]-](http://img.cochemist.com/ccimg/174700/174636-16-9_b.png)






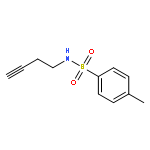
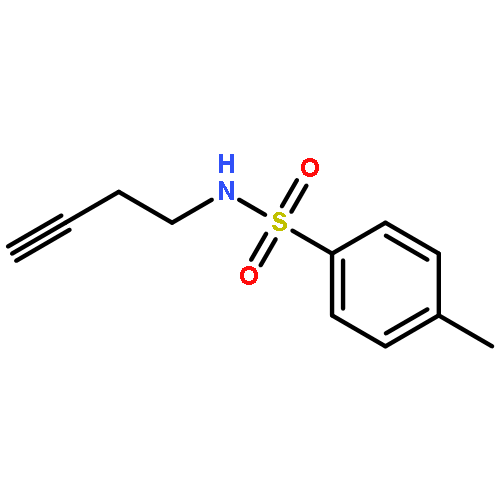










![Benzene, 1-[(2-ethynylphenyl)ethynyl]-2-(phenylethynyl)-](http://img.cochemist.com/ccimg/143200/143192-63-6.png)
![Benzene, 1-[(2-ethynylphenyl)ethynyl]-2-(phenylethynyl)-](http://img.cochemist.com/ccimg/143200/143192-63-6_b.png)
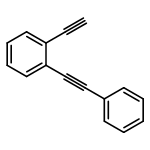
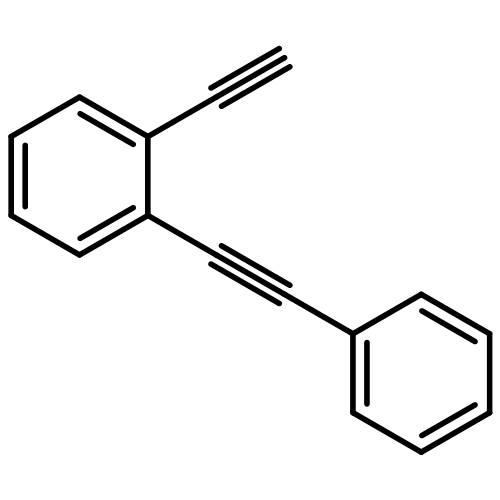
![Indolo[3,2-d][1]benzazepin-6(5H)-one,9-bromo-7,12-dihydro-](http://img.cochemist.com/ccimg/142300/142273-20-9.png)
![Indolo[3,2-d][1]benzazepin-6(5H)-one,9-bromo-7,12-dihydro-](http://img.cochemist.com/ccimg/142300/142273-20-9_b.png)












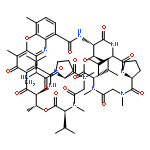









![2,5-Ethano-2H-azocino[4,3-b]indol-6(3H)-one,4-ethylidene-1,4,5,7-tetrahydro-, (2R,4E,5S)-](http://img.cochemist.com/ccimg/100500/100414-81-1.png)
![2,5-Ethano-2H-azocino[4,3-b]indol-6(3H)-one,4-ethylidene-1,4,5,7-tetrahydro-, (2R,4E,5S)-](http://img.cochemist.com/ccimg/100500/100414-81-1_b.png)


![Ethanol, 2-[(2-bromophenyl)amino]-](http://img.cochemist.com/ccimg/87800/87762-20-7.png)
![Ethanol, 2-[(2-bromophenyl)amino]-](http://img.cochemist.com/ccimg/87800/87762-20-7_b.png)
![3,6-Imino-1H-2-oxa-11c-azanaphth[1,2,3-cd]azulene-5-carboxylic acid, 2a,3,4,5,6,6a,7,11b-octahydro-11-methoxy-12-methyl-,](/data/chemimg/1982000/84573-33-1.png)
![3,6-Imino-1H-2-oxa-11c-azanaphth[1,2,3-cd]azulene-5-carboxylic acid, 2a,3,4,5,6,6a,7,11b-octahydro-11-methoxy-12-methyl-,](/data/chemimg/1982000/84573-33-1_b.png)


![Magnesium, bromo[3-(trimethylsilyl)-2-propynyl]-](http://img.cochemist.com/ccimg/78100/78012-45-0.png)
![Magnesium, bromo[3-(trimethylsilyl)-2-propynyl]-](http://img.cochemist.com/ccimg/78100/78012-45-0_b.png)


![2H-1-Benzopyran-6-ol, 3,4-dihydro-2,5,7,8-tetramethyl-2-[(4S,8S)-4,8,12-trimethyltridecyl]-, (2S)-](http://img.cochemist.com/ccimg/77200/77171-97-2.png)
![2H-1-Benzopyran-6-ol, 3,4-dihydro-2,5,7,8-tetramethyl-2-[(4S,8S)-4,8,12-trimethyltridecyl]-, (2S)-](http://img.cochemist.com/ccimg/77200/77171-97-2_b.png)




![2H,6H-PYRIMIDO[1,2-C][1,3]BENZOXAZINE-6-THIONE, 3,4-DIHYDRO-](http://img.cochemist.com/ccimg/65800/65739-85-7.png)
![2H,6H-PYRIMIDO[1,2-C][1,3]BENZOXAZINE-6-THIONE, 3,4-DIHYDRO-](http://img.cochemist.com/ccimg/65800/65739-85-7_b.png)
![2H,6H-PYRIMIDO[1,2-C][1,3]BENZOXAZIN-6-ONE, 3,4-DIHYDRO-](http://img.cochemist.com/ccimg/65800/65739-84-6.png)
![2H,6H-PYRIMIDO[1,2-C][1,3]BENZOXAZIN-6-ONE, 3,4-DIHYDRO-](http://img.cochemist.com/ccimg/65800/65739-84-6_b.png)















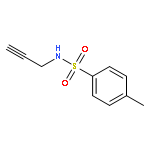
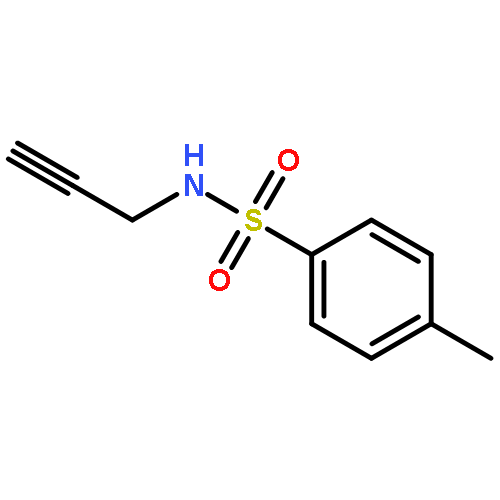
![7-Bromo-4,5-dihydro-1H-benzo[b]azepin-2(3H)-one](http://img.cochemist.com/ccimg/53900/53841-99-9.png)
![7-Bromo-4,5-dihydro-1H-benzo[b]azepin-2(3H)-one](http://img.cochemist.com/ccimg/53900/53841-99-9_b.png)

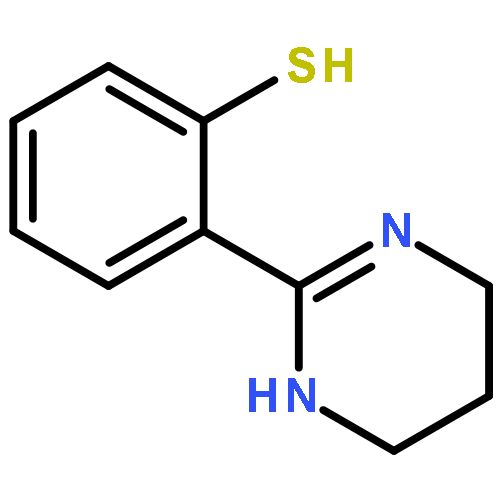

![6H-PYRIMIDO[1,2-C]QUINAZOLIN-6-ONE, 2,3,4,7-TETRAHYDRO-](http://img.cochemist.com/ccimg/51400/51300-33-5.png)
![6H-PYRIMIDO[1,2-C]QUINAZOLIN-6-ONE, 2,3,4,7-TETRAHYDRO-](http://img.cochemist.com/ccimg/51400/51300-33-5_b.png)


![2H-Pyrimido[2,1-b]benzothiazole, 3,4-dihydro-](http://img.cochemist.com/ccimg/42200/42142-60-9.png)
![2H-Pyrimido[2,1-b]benzothiazole, 3,4-dihydro-](http://img.cochemist.com/ccimg/42200/42142-60-9_b.png)




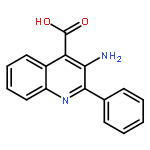
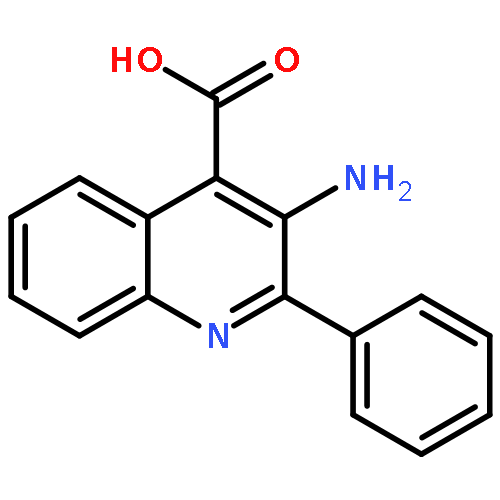
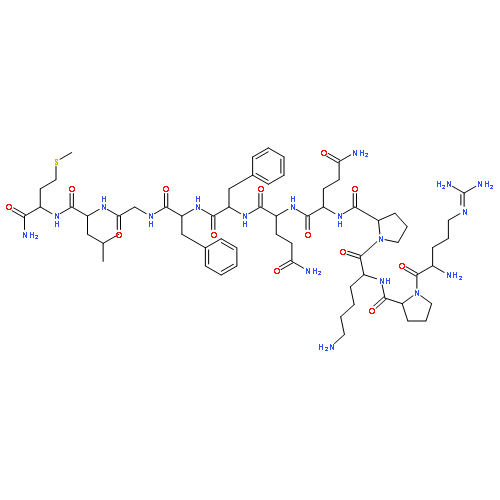
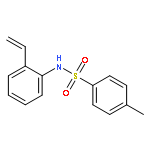
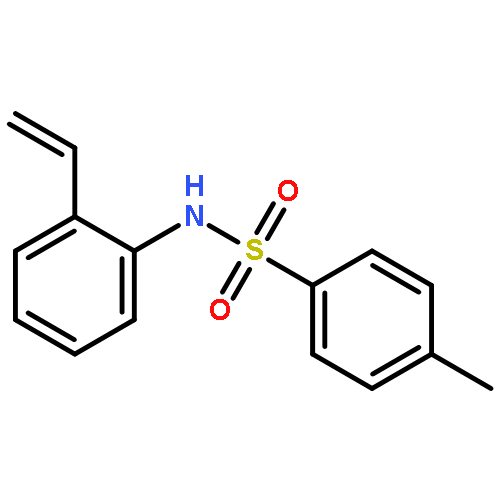


![L-Leucine, N-[(phenylmethoxy)carbonyl]-L-phenylalanyl-, 1,1-dimethylethyl ester](/data/chemimg/1180800/29842-94-2.png)
![L-Leucine, N-[(phenylmethoxy)carbonyl]-L-phenylalanyl-, 1,1-dimethylethyl ester](/data/chemimg/1180800/29842-94-2_b.png)







![Benzene, [(3-butynyloxy)methyl]-](http://img.cochemist.com/ccimg/22300/22273-77-4.png)
![Benzene, [(3-butynyloxy)methyl]-](http://img.cochemist.com/ccimg/22300/22273-77-4_b.png)
![1H-Indole, 1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/22100/22051-21-4.png)
![1H-Indole, 1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/22100/22051-21-4_b.png)





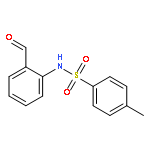
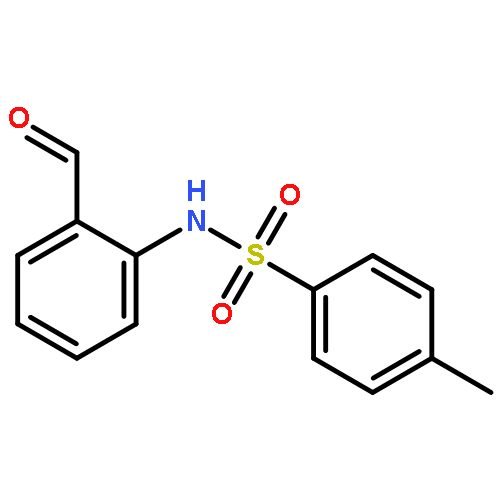






![4,9-dihydro-3h-pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/4900/4894-26-2.png)
![4,9-dihydro-3h-pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/4900/4894-26-2_b.png)





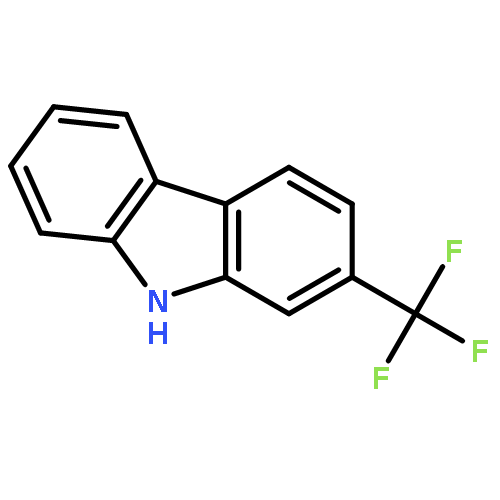







![((6aR,9S)-7-methyl-4,6,6a,7,8,9-hexahydroindolo[4,3-fg]quinolin-9-yl)methanol](http://img.cochemist.com/ccimg/500/478-93-3.png)
![((6aR,9S)-7-methyl-4,6,6a,7,8,9-hexahydroindolo[4,3-fg]quinolin-9-yl)methanol](http://img.cochemist.com/ccimg/500/478-93-3_b.png)

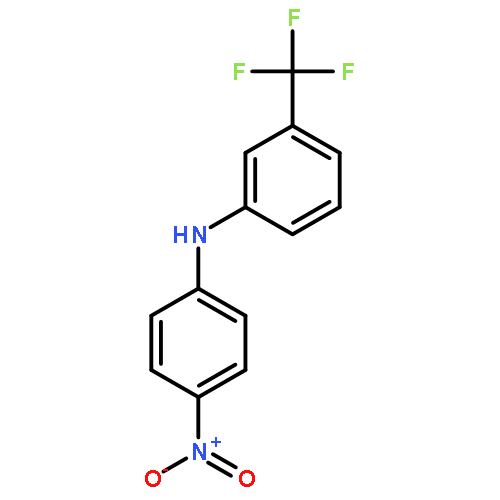
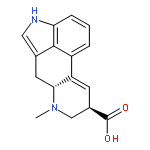
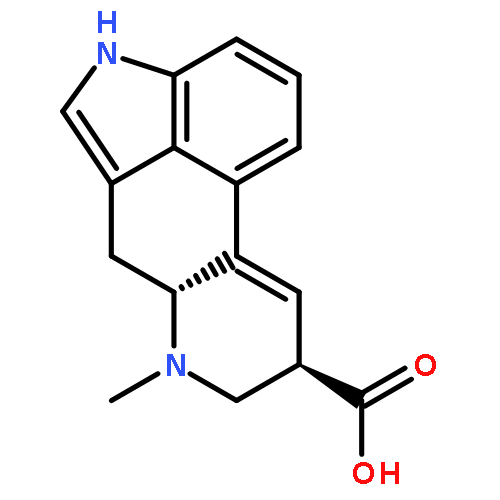

![4H-Dibenzo[de,g]quinoline-10,11-diol,5,6,6a,7-tetrahydro-6-methyl-, (6aR)-](http://img.cochemist.com/ccimg/100/58-00-4.png)
![4H-Dibenzo[de,g]quinoline-10,11-diol,5,6,6a,7-tetrahydro-6-methyl-, (6aR)-](http://img.cochemist.com/ccimg/100/58-00-4_b.png)
![Benzenamine, 4-methoxy-2-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/874800/874796-35-7.png)
![Benzenamine, 4-methoxy-2-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/874800/874796-35-7_b.png)
![(Acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate](http://img.cochemist.com/ccimg/866700/866641-66-9.png)
![(Acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate](http://img.cochemist.com/ccimg/866700/866641-66-9_b.png)
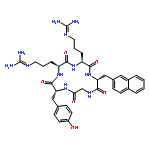
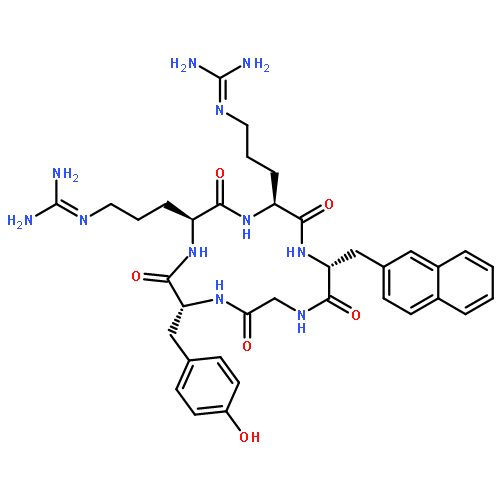
dimethyl-](http://img.cochemist.com/ccimg/110800/110796-98-0.png)
dimethyl-](http://img.cochemist.com/ccimg/110800/110796-98-0_b.png)
![[1,2,4]Triazolo[1,5-c]quinazolin-5-amine,9-chloro-2-(2-furanyl)-](http://img.cochemist.com/ccimg/104700/104615-18-1.png)
![[1,2,4]Triazolo[1,5-c]quinazolin-5-amine,9-chloro-2-(2-furanyl)-](http://img.cochemist.com/ccimg/104700/104615-18-1_b.png)
![3,4-DIHYDRO-2H-PYRIMIDO[1,2-C][1,3]BENZOTHIAZIN-6-IMINE](http://img.cochemist.com/ccimg/72600/72596-74-8.png)
![3,4-DIHYDRO-2H-PYRIMIDO[1,2-C][1,3]BENZOTHIAZIN-6-IMINE](http://img.cochemist.com/ccimg/72600/72596-74-8_b.png)