Co-reporter:Shaun Smullen, Paul Evans
Tetrahedron 2017 Volume 73, Issue 37(Issue 37) pp:
Publication Date(Web):14 September 2017
DOI:10.1016/j.tet.2017.07.041
A new synthesis of both enantiomers of naturally occurring febrifugine (1) and its analogue halofuginone (3) are reported. This robust route relies on an enzymatic kinetic resolution (EKR)-cross metathesis (CM) sequence, performed on allylic alcohol 8. A diastereoselective Lewis acid-mediated cyclisation of resultant enone 6 affords piperidine 12. In relation to its enantiomeric excess, values of 87 and 88% e.e were obtained for both enantiomers of 12 and for (−)-12 this was increased to 98% e.e by conducting the resolution process twice. A bromination (featuring an in situ temporary alcohol protection), alkylation and amino-deprotection sequence finally afforded the target compounds (+)- and (−)-1, and (+)- and (−)-3. Studies demonstrate that, as their ammonium salts, the compounds are stereochemically stable. However, as their free-bases, particularly in chloroform, C-2 isomerisation occurs. This isomerisation has been taken advantage of synthetically to provide enantioenriched (+)-isofebrifugine (2) and both enantiomers of isohalofuginone (14).Download high-res image (248KB)Download full-size image
Co-reporter:Aisha Khalifa, Lorna Conway, Kimberly Geoghegan, Paul Evans
Tetrahedron Letters 2017 Volume 58, Issue 48(Issue 48) pp:
Publication Date(Web):29 November 2017
DOI:10.1016/j.tetlet.2017.10.053
•Synthesis of novel cyclic sulfonamides in high yield from simple precursors.•One-pot reductive intramolecular Heck reaction.•Tandem transformation using same palladium catalyst for two reactions.•Selectivity observed for nitro and alkene substitution patterns.A modified method is reported for the conversion of unsaturated sulfonamides into their cyclic saturated counterparts. This method utilises a single palladium catalyst for an intramolecular Heck reaction and subsequent transfer hydrogenation, which is achieved in one-pot following the addition of ammonium formate. Accordingly, a range of fourteen structural variations are reported and under optimal conditions the adducts were generated in typically good to excellent yields. Notably, discrimination of differentially substituted dienes can be accomplished in the case of compounds 28 and 29 and the process was only observed to fail with the more sterically hindered precursor 32.Download high-res image (148KB)Download full-size image
Co-reporter:Aaron Keeley, Shane McCauley, Paul Evans
Tetrahedron 2016 Volume 72(Issue 20) pp:2552-2559
Publication Date(Web):19 May 2016
DOI:10.1016/j.tet.2016.03.088
The combination of ring closing, or enyne metathesis with oxidation in order to prepare N-sulfonyl pyrroles is described. Reasonable to good yields were obtained for a variety of substituents and the procedure may also be conducted in one-pot. 2-Bromo N-sulfonyl adducts prepared in this manner were subjected to an intramolecular Heck-type cyclisation, forming cyclic sulfonamides.
Co-reporter:William Doherty and Paul Evans
The Journal of Organic Chemistry 2016 Volume 81(Issue 4) pp:1416-1424
Publication Date(Web):January 27, 2016
DOI:10.1021/acs.joc.5b02556
An operationally simple protocol for the synthesis of γ-hydroxy vinyl sulfones has been developed using a proline-based aldehyde aminooxylation, followed by a vinyl sulfone forming Horner–Wadsworth–Emmons olefination. The adducts, formed in high enantiopurity, were subsequently converted to γ-azido vinyl sulfones, and azide–alkyne click chemistry enabled the synthesis of vinyl sulfone-based triazoles as potential nonpeptidic cysteine protease inhibitors.
Co-reporter:Raed K. Zaidan, Shaun Smullen, Paul Evans
Tetrahedron Letters 2015 Volume 56(Issue 46) pp:6433-6435
Publication Date(Web):18 November 2015
DOI:10.1016/j.tetlet.2015.09.146
Both enantiomers of deoxyfebrifugine (4) and deoxyhalofuginone (5), analogues of the quinazolinone-containing biologically active compounds febrifugine (1) and halofuginone (3), have been prepared in a six-step reaction sequence featuring an organocatalyzed Mannich reaction as the key stereo-inducing step. The compounds were isolated as their dihydrobromide salts in 29–42% overall yield and in 74–80% enantiomeric excess.
Co-reporter:William Doherty, Jinju James, Paul Evans, Laura Martin, Nikoletta Adler, Derek Nolan and Andrew Knox
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 38) pp:7561-7571
Publication Date(Web):2014/08/12
DOI:10.1039/C4OB01412J
An improved, Weinreb amide-based, synthesis of anti-trypanosomal lysine-containing vinyl sulfones is described incorporating, as a feature, diversity at the ε-lysine amino group. Members of this family demonstrated moderate to good efficacy as anti-trypanosomal agents and a fluorescent dansyl (19) derivative was used to investigate subcellular localisation of the compound class.
Co-reporter:Noel P. McLaughlin, Paul Evans, Mark Pines
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 7) pp:1993-2004
Publication Date(Web):1 April 2014
DOI:10.1016/j.bmc.2014.02.040
The trans-2,3-disubstituted piperidine, quinazolinone-containing natural product febrifugine (also known as dichroine B) and its synthetic analogue, halofuginone, possess antimalarial activity. More recently studies have also shown that halofuginone acts as an agent capable of reducing fibrosis, an indication with clinical relevance for several disease states. This review summarizes historical isolation studies and the chemistry performed which culminated in the correct structural elucidation of naturally occurring febrifugine and its isomer isofebrifugine. It also includes the range of febrifugine analogues prepared for antimalarial evaluation, including halofuginone. Finally, a section detailing current opinion in the field of halofuginone’s human biology is included.
Co-reporter:Kimberly Geoghegan, Paul Evans
Tetrahedron Letters 2014 Volume 55(Issue 8) pp:1431-1433
Publication Date(Web):19 February 2014
DOI:10.1016/j.tetlet.2014.01.039
(+)-Perillyl alcohol (1) has been synthesised in four steps and 39% overall yield from commercially available limonene oxide (4). The sequence features, as its key step, a palladium(0)-mediated transformation of a secondary allylic acetate (6) into its primary isomer (7). An application of (+)-perillyl alcohol (1) in a formal synthesis of naturally occurring (−)-mesembrine (2) and (−)-mesembranol was demonstrated.
Co-reporter:Roderick C. Jones;Helge Müller-Bunz, ;Donal F. O'Shea
Acta Crystallographica Section C 2014 Volume 70( Issue 2) pp:165-168
Publication Date(Web):
DOI:10.1107/S2053229614000084
The structural chemistry of the title compound, [Pd(C32H22N3)2], at 173 K is described. The compound is comprised of two deprotonated (3,5-diphenyl-1H-pyrrol-2-yl)(3,5-diphenylpyrrol-2-ylidene)amine ligands coordinated to a central PdII cation, which lies on an inversion centre and has distorted square-planar geometry. The Pd—N bond lengths range from 2.008 (4) to 2.014 (4) Å and the bite angle is 84.16 (14)°. The chelate plane makes a dihedral angle of 45.3 (2)° with respect to the central PdN4 plane, giving a stepped conformation to the molecule. The complex displays simple intramolecular C—H...N hydrogen bonds, while the unit cell consists of discrete monomeric Pd(C32H22N3)2 units which display intermolecular C—H...π interactions and limited intra- and intermolecular π–π stacking.
Co-reporter:Elizabeth Dunny ; William Doherty ; Paul Evans ; J. Paul G. Malthouse ; Derek Nolan ;Andrew J. S. Knox
Journal of Medicinal Chemistry 2013 Volume 56(Issue 17) pp:6638-6650
Publication Date(Web):August 16, 2013
DOI:10.1021/jm400294w
A series of vinyl sulfone-containing peptidomimetics were rationally designed, synthesized, and evaluated against Trypanosoma brucei brucei. These electrophilic compounds are likely to exert their antitrypanosomal activity via inhibition of trypanosomal cysteine proteases, TbCatB and rhodesain, through alkylation of a key cysteine residue within the protease active site. The series was designed to present complementary groups to naturally recognized peptide substrates while probing tolerance to a range of substitutions at the P1, P1′, and P2 positions. The most potent compound, 29 (EC50 = 70 nM, T. b. brucei whole cell assay), displayed minimal toxicity (>785 times selectivity) when assayed for cytotoxicity against the human promyelocytic leukemia (HL-60) cell line. Cells treated with compound 29, as with K777 (2), exhibited an increase in both the number of multinucleated cells and cells with swollen flagellar pockets. Computational analysis revealed a strong correlation between the hypothetical binding mode in TbCatB/rhodesain and trypanocidal activity in vitro.
Co-reporter:Paul Evans
Applied Organometallic Chemistry 2013 Volume 27( Issue 4) pp:261-262
Publication Date(Web):
DOI:10.1002/aoc.2923
Co-reporter:Kimberly Geoghegan, Shaun Smullen, and Paul Evans
The Journal of Organic Chemistry 2013 Volume 78(Issue 20) pp:10443-10451
Publication Date(Web):September 24, 2013
DOI:10.1021/jo401888f
The halonium ion mediated 1,2-Wagner–Meerwein-type rearrangement of a series of benzo-fused bi- and tricyclic sulfonamides is reported. During this rearrangement the carbon–carbon bond that migrates was selectively set in the intramolecular Mizoroki–Heck (IHR) synthesis of the starting materials. Consequently, this method constitutes a means to access the regioisomeric series of cyclic sulfonamides not observed during the Mizoroki–Heck reaction.
Co-reporter:Kimberly Geoghegan and Paul Evans
The Journal of Organic Chemistry 2013 Volume 78(Issue 7) pp:3410-3415
Publication Date(Web):March 15, 2013
DOI:10.1021/jo4000306
The synthesis of (+)-mesembrine (1) and (+)-mesembranol (2) has been achieved from the monoterpene (S)-(−)-perillyl alcohol. Key transformations include a diastereo- and regioselective Pd-mediated intramolecular Heck reaction, and a double reduction of the resultant cyclic sulfonamide, to afford the cis-3a-aryloctahydroindole skeleton.
Co-reporter:Aisling O’Byrne, Steven O’Reilly, Catherine Tighe, Paul Evans, Laura Ciuffini, M. Gabriella Santoro
Tetrahedron Letters 2012 Volume 53(Issue 44) pp:5936-5938
Publication Date(Web):31 October 2012
DOI:10.1016/j.tetlet.2012.08.101
Co-reporter:Kimberly Geoghegan;Dr. Paul Evans; Isabel Rozas; Ibon Alkorta
Chemistry - A European Journal 2012 Volume 18( Issue 42) pp:13379-13387
Publication Date(Web):
DOI:10.1002/chem.201201359
Abstract
Regioselectivity in the intramolecular Heck reaction of a series of N-sulfonyl-2,5-dihydro-3-substituted pyrroles was studied. These substrates are unbiased in terms of the formed ring size of the new heterocycle. Results indicate that high levels of regioselectivity are observed under a range of conditions, and that there is an underlying propensity for carbon–carbon bond formation at the most hindered end of the alkene. For two examples (3-Me and 3-tBu), DFT calculations were performed and indicate that in both cases, the modelled transition state for carbopalladation is energetically lower for the experimentally preferred isomer.
Co-reporter:Kimberly Geoghegan, Susan Kelleher, and Paul Evans
The Journal of Organic Chemistry 2011 Volume 76(Issue 7) pp:2187-2194
Publication Date(Web):March 7, 2011
DOI:10.1021/jo200023r
Herein is described an operationally simple process concerning the observation that, following either inter-, or intramolecular Heck olefination, stirring of the so formed substituted alkenyl product under an atmosphere of hydrogen efficiently effects alkene hydrogenation. Overall this two-operation, one-pot “reductive Heck” sequence is notable since direct reductive Heck processes, using additives such as formate salts, are restricted to a limited range of substrates. In total 25 examples are reported (yields ranging from 0 to 95%), which were selected in order to probe the scope and limitations of this method. Finally, the utility of this sequence was demonstrated in a short synthesis of the calcimimetic agent, cinacalcet.
Co-reporter:Johannes E. M. N. Klein, Kimberly Geoghegan, Nicolas Méral and Paul Evans
Chemical Communications 2010 vol. 46(Issue 6) pp:937-939
Publication Date(Web):14 Dec 2009
DOI:10.1039/B923175G
Described is an observation that the intramolecular Heck reaction of a trisubstituted alkene proceeds with high regioselectivity and leads to the preferential formation of a quaternary all-carbon-centre. This observation was subsequently applied in a short synthesis of (±)-mesembrane.
Co-reporter:Aisling O'Byrne, Cian Murray, Dearbhla Keegan, Carole Palacio, Paul Evans and Ben S. Morgan
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 3) pp:539-545
Publication Date(Web):2009/11/30
DOI:10.1039/B916506A
The addition of 3,4-dimethoxybenzyl thiol 8, as a benzyl thiol surrogate, to racemic 4-hydroxycyclopent-2-enone 2 and 4-hydroxycyclohex-2-enone 15 gave the corresponding cis-adducts (±)-3-(3,4-dimethoxybenzylthio)-4-hydroxycyclopentanone 4b and (±)-3-(3,4-dimethoxybenzylthio)-4-hydroxycyclohexanone 16 with good diastereocontrol. In both cases, subsequent treatment with vinyl acetate, in the presence of a lipase enabled enantiomer resolution. Thus, (+)-16 and the acetate of its enantiomer, (–)-(1R,2S)-2-(3,4-dimethoxybenzylthio)-4-oxocyclohexyl acetate, (–)-17 were isolated in 98% enantiomeric excess. Based on the 1,4-dioxygenation pattern, (–)-17 can be used to prepare both enantiomers of 4-(tert-butyldimethylsilyloxy)cyclohex-2-enone 19. Firstly, saponification, with a sub-stoichiometric amount of NaOMe, followed by a one-pot silyl ether formation–sulfide elimination sequence gave (+)-19. Then using the same starting material a 6-step sequence, featuring a diastereoselective NaBH4 reduction and a Cope-type sulfoxide elimination, gave (–)-19.
Co-reporter:Elizabeth Dunny and Paul Evans
The Journal of Organic Chemistry 2010 Volume 75(Issue 15) pp:5334-5336
Publication Date(Web):July 1, 2010
DOI:10.1021/jo1007493
The naturally occurring PPAR-γ ligand 10-nitrooctadeca-9(E),12(Z)-dienoic acid (10-nitrolinoleic acid) (2a) was prepared as a single regio- and geometrical isomer in a practical eight-step, convergent sequence. The synthetic route featured a nitro aldol reaction between 9-oxononanoic acid methyl ester (3) and 1-nitronon-3(Z)-ene (4) in the key carbon−carbon bond forming step. The ability of 2a (and its methyl ester 9) to bind to PPAR-γ in a ligand-binding assay is reported.
Co-reporter:Noel P. McLaughlin, Eibhlín Butler, Paul Evans, Nigel P. Brunton, Anastasios Koidis, Dilip K. Rai
Tetrahedron 2010 66(51) pp: 9681-9687
Publication Date(Web):
DOI:10.1016/j.tet.2010.10.049
Co-reporter:Johannes E. M. N. Klein, Helge Müller-Bunz and Paul Evans
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 5) pp:986-995
Publication Date(Web):23 Jan 2009
DOI:10.1039/B819610A
The bromination of a series of cyclohexenyl substituted secondary amines 1a-i has been investigated using Br2, PHT and NBS. In the case of Br2 and NBS the secondary amines preferentially undergo N-bromination. In contrast, PHT cleanly affords the products of alkene dibromination. In the case of Br2 the N-bromo species then give the products of alkene dibromination, albeit less efficiently. On subsequent treatment with K2CO3 these dibromides form the corresponding hexahydroindoles 2a-h and octahydroquinoline 2i. The presence of an N-substituent bearing a stereogenic centre (1h and 1i) was studied and the products 2h and 2i were isolated with no diastereoselectivity. When NBS was used a novel cyclisation, forming bromo-substituted octahydroindoles 9a,b and d, was observed. In relation to this sequence it was shown that these products were not intermediates in the former Br2/PHT processes and that the reaction only proceeded in the presence of the succinimide by-product of N-bromination.
Co-reporter:Yvonne Kavanagh, Matthew O'Brien, Paul Evans
Tetrahedron 2009 65(39) pp: 8259-8268
Publication Date(Web):
DOI:10.1016/j.tet.2009.07.060
Co-reporter:Sarah A. Brusey, Emilie V. Banide, Steffen Dörrich, Paul O’Donohue, Yannick Ortin, Helge Müller-Bunz, Conor Long, Paul Evans and Michael J. McGlinchey
Organometallics 2009 Volume 28(Issue 21) pp:6308-6319
Publication Date(Web):September 30, 2009
DOI:10.1021/om900615m
The hexacarbonyldicobalt complexes of a range of 5-alkynyl-5H-dibenzo[a,d]cycloheptenes, 11a−e, readily undergo loss of a carbonyl ligand with concomitant formation of pentacarbonyldicobalt clusters, 12a−e, in which the vacant coordination site on cobalt is now occupied by the C(10)−C(11) double bond of the central seven-membered ring. These (η2-alkene)(μ-alkyne)pentacarbonyldicobalt complexes provide structural models of the first step of the proposed mechanism of the Pauson−Khand process for the formation of cyclopentenones via the coupling of an alkene, an alkyne, and a source of carbon monoxide. X-ray crystallographic data reveal that the distance between the alkene carbons and the nearest alkyne carbon is approximately 2.85 Å, slightly shorter than the theoretically predicted value of ∼2.95 Å. VT NMR data for the interconversion of 11b and 12b yielded activation energies for the forward and reverse processes, 29 and 15 kcal mol−1, respectively, and the enthalpy change for the endothermic process (14 kcal mol−1); these match very well the earlier predictions from DFT calculations. Although the ethynyl-hexacarbonyldicobalt complex, 11f, does not suffer facile elimination of CO, it does participate in an intermolecular PKR with norbornadiene. Likewise, attempts to extend the reach of the alkynyl moiety through incorporation of an additional methylene group yield only an intermolecular PKR product. It is suggested that the pentacarbonyl complexes, 12a−e, do not continue along the PKR pathway because of the unfavorable relative orientation of the cobalt-coordinated alkyne and alkene.
Co-reporter:Mazhar Iqbal, Patricia Duffy, Paul Evans, George Cloughley, Bernard Allan, Agustí Lledó, Xavier Verdaguer and Antoni Riera
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 24) pp:4649-4661
Publication Date(Web):04 Nov 2008
DOI:10.1039/B814619E
5-Alkylidenecyclopent-2-enones 15a–q may be prepared via a conjugate addition–Peterson olefination sequence, best achieved in one-pot, using exo-2-trimethylsilyl-3a,4,7,7a-tetrahydro-4,7-methanoinden-1-one 12, followed by a retro-Diels–Alder reaction. The geometry of the exocyclic alkene may be controlled according to the use of organometallic species in the conjugate addition step; organocuprate reagents are found to selectively lead to the formation of E-exocyclic alkene adducts, whereas Grignard reagents favour the formation of Z-alkenyl isomers. The use of enantiomerically enriched 12, accessed from an asymmetric Pauson–Khand reaction, affords the corresponding enantioenriched 5-alkylidenecyclopent-2-enones and this approach is exemplified by the short, stereoselective total syntheses of two cyclopentenone phytoprostanes 51 and 13,14-dehydrophytoprostane J165. The ability of this family of synthetic compounds to activate the peroxisome proliferator activated receptor-γ is reported.
Co-reporter:Emilie V. Banide;Helge Müller-Bunz Dr.;Anthony R. Manning Dr. Dr.;Michael J. McGlinchey Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 16) pp:
Publication Date(Web):13 MAR 2007
DOI:10.1002/anie.200605171
Caught in the act: The [(dibenzosuberenol)Co2(CO)6] cluster 1 (see scheme) loses a carbonyl ligand to yield 2, the first structurally characterized alkyne–dicobaltpentacarbonyl–alkene complex. This finding provides experimental support for the first step of the proposed mechanism of the Pauson–Khand reaction and yields crucial structural data relevant to computational studies of the process.
Co-reporter:Emilie V. Banide;Helge Müller-Bunz Dr.;Anthony R. Manning Dr. Dr.;Michael J. McGlinchey Dr.
Angewandte Chemie 2007 Volume 119(Issue 16) pp:
Publication Date(Web):13 MAR 2007
DOI:10.1002/ange.200605171
Ertappt! Der Dibenzosuberenol-Co2(CO)6-Cluster 1 (siehe Schema) geht unter Verlust eines Carbonylliganden in 2 über. Dieser erste strukturanalytisch charakterisierte Alkin-Dicobaltpentacarbonyl-Alken-Komplex untermauert den ersten Schritt in einem Mechanismusvorschlag für die Pauson-Khand-Reaktion und liefert wichtige Strukturdaten für computerchemische Studien.
Co-reporter:Johannes E. M. N. Klein, Kimberly Geoghegan, Nicolas Méral and Paul Evans
Chemical Communications 2010 - vol. 46(Issue 6) pp:NaN939-939
Publication Date(Web):2009/12/14
DOI:10.1039/B923175G
Described is an observation that the intramolecular Heck reaction of a trisubstituted alkene proceeds with high regioselectivity and leads to the preferential formation of a quaternary all-carbon-centre. This observation was subsequently applied in a short synthesis of (±)-mesembrane.
Co-reporter:William Doherty, Jinju James, Paul Evans, Laura Martin, Nikoletta Adler, Derek Nolan and Andrew Knox
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 38) pp:NaN7571-7571
Publication Date(Web):2014/08/12
DOI:10.1039/C4OB01412J
An improved, Weinreb amide-based, synthesis of anti-trypanosomal lysine-containing vinyl sulfones is described incorporating, as a feature, diversity at the ε-lysine amino group. Members of this family demonstrated moderate to good efficacy as anti-trypanosomal agents and a fluorescent dansyl (19) derivative was used to investigate subcellular localisation of the compound class.
Co-reporter:Aisling O'Byrne, Cian Murray, Dearbhla Keegan, Carole Palacio, Paul Evans and Ben S. Morgan
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 3) pp:NaN545-545
Publication Date(Web):2009/11/30
DOI:10.1039/B916506A
The addition of 3,4-dimethoxybenzyl thiol 8, as a benzyl thiol surrogate, to racemic 4-hydroxycyclopent-2-enone 2 and 4-hydroxycyclohex-2-enone 15 gave the corresponding cis-adducts (±)-3-(3,4-dimethoxybenzylthio)-4-hydroxycyclopentanone 4b and (±)-3-(3,4-dimethoxybenzylthio)-4-hydroxycyclohexanone 16 with good diastereocontrol. In both cases, subsequent treatment with vinyl acetate, in the presence of a lipase enabled enantiomer resolution. Thus, (+)-16 and the acetate of its enantiomer, (–)-(1R,2S)-2-(3,4-dimethoxybenzylthio)-4-oxocyclohexyl acetate, (–)-17 were isolated in 98% enantiomeric excess. Based on the 1,4-dioxygenation pattern, (–)-17 can be used to prepare both enantiomers of 4-(tert-butyldimethylsilyloxy)cyclohex-2-enone 19. Firstly, saponification, with a sub-stoichiometric amount of NaOMe, followed by a one-pot silyl ether formation–sulfide elimination sequence gave (+)-19. Then using the same starting material a 6-step sequence, featuring a diastereoselective NaBH4 reduction and a Cope-type sulfoxide elimination, gave (–)-19.
Co-reporter:Johannes E. M. N. Klein, Helge Müller-Bunz and Paul Evans
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 5) pp:NaN995-995
Publication Date(Web):2009/01/23
DOI:10.1039/B819610A
The bromination of a series of cyclohexenyl substituted secondary amines 1a-i has been investigated using Br2, PHT and NBS. In the case of Br2 and NBS the secondary amines preferentially undergo N-bromination. In contrast, PHT cleanly affords the products of alkene dibromination. In the case of Br2 the N-bromo species then give the products of alkene dibromination, albeit less efficiently. On subsequent treatment with K2CO3 these dibromides form the corresponding hexahydroindoles 2a-h and octahydroquinoline 2i. The presence of an N-substituent bearing a stereogenic centre (1h and 1i) was studied and the products 2h and 2i were isolated with no diastereoselectivity. When NBS was used a novel cyclisation, forming bromo-substituted octahydroindoles 9a,b and d, was observed. In relation to this sequence it was shown that these products were not intermediates in the former Br2/PHT processes and that the reaction only proceeded in the presence of the succinimide by-product of N-bromination.
Co-reporter:Mazhar Iqbal, Patricia Duffy, Paul Evans, George Cloughley, Bernard Allan, Agustí Lledó, Xavier Verdaguer and Antoni Riera
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 24) pp:NaN4661-4661
Publication Date(Web):2008/11/04
DOI:10.1039/B814619E
5-Alkylidenecyclopent-2-enones 15a–q may be prepared via a conjugate addition–Peterson olefination sequence, best achieved in one-pot, using exo-2-trimethylsilyl-3a,4,7,7a-tetrahydro-4,7-methanoinden-1-one 12, followed by a retro-Diels–Alder reaction. The geometry of the exocyclic alkene may be controlled according to the use of organometallic species in the conjugate addition step; organocuprate reagents are found to selectively lead to the formation of E-exocyclic alkene adducts, whereas Grignard reagents favour the formation of Z-alkenyl isomers. The use of enantiomerically enriched 12, accessed from an asymmetric Pauson–Khand reaction, affords the corresponding enantioenriched 5-alkylidenecyclopent-2-enones and this approach is exemplified by the short, stereoselective total syntheses of two cyclopentenone phytoprostanes 51 and 13,14-dehydrophytoprostane J165. The ability of this family of synthetic compounds to activate the peroxisome proliferator activated receptor-γ is reported.

![Carbamic acid, N-[(5R,6E)-5-azido-7-(phenylsulfonyl)-6-hepten-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3723200/1863875-10-8.png)
![Carbamic acid, N-[(5R,6E)-5-azido-7-(phenylsulfonyl)-6-hepten-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3723200/1863875-10-8_b.png)
![Carbamic acid, N-[(1S)-1-[1-[(1S)-5-[[(1,1-dimethylethoxy)carbonyl]amino]-1-[(1E)-2-(phenylsulfonyl)ethenyl]pentyl]-1H-1,2,3-triazol-4-yl]-2-phenylethyl]-, phenylmethyl ester](/data/chemimg/3721800/1863875-22-2.png)
![Carbamic acid, N-[(1S)-1-[1-[(1S)-5-[[(1,1-dimethylethoxy)carbonyl]amino]-1-[(1E)-2-(phenylsulfonyl)ethenyl]pentyl]-1H-1,2,3-triazol-4-yl]-2-phenylethyl]-, phenylmethyl ester](/data/chemimg/3721800/1863875-22-2_b.png)


![Cyclopentanone, 3-hydroxy-4-[(phenylmethyl)thio]-, (3S,4R)-](http://img.cochemist.com/ccimg/885700/885612-84-0.png)
![Cyclopentanone, 3-hydroxy-4-[(phenylmethyl)thio]-, (3S,4R)-](http://img.cochemist.com/ccimg/885700/885612-84-0_b.png)
![Cyclopentanone, 3-hydroxy-4-[(phenylmethyl)thio]-, (3R,4S)-rel-](http://img.cochemist.com/ccimg/885700/885612-80-6.png)
![Cyclopentanone, 3-hydroxy-4-[(phenylmethyl)thio]-, (3R,4S)-rel-](http://img.cochemist.com/ccimg/885700/885612-80-6_b.png)


![5H-Dibenzo[a,d]cyclohepten-5-ol, 5-ethynyl-](http://img.cochemist.com/ccimg/87400/87383-80-0.png)
![5H-Dibenzo[a,d]cyclohepten-5-ol, 5-ethynyl-](http://img.cochemist.com/ccimg/87400/87383-80-0_b.png)


![7H-Pyrido[1,2-b][1,2]benzisothiazole, 8,9-dihydro-, 5,5-dioxide](http://img.cochemist.com/ccimg/830400/830319-78-3.png)
![7H-Pyrido[1,2-b][1,2]benzisothiazole, 8,9-dihydro-, 5,5-dioxide](http://img.cochemist.com/ccimg/830400/830319-78-3_b.png)
![2-Pyrrolidinol, 1-[(2-bromophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/830400/830319-68-1.png)
![2-Pyrrolidinol, 1-[(2-bromophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/830400/830319-68-1_b.png)
![Carbamic acid, N-[(5S,6E)-5-azido-7-(phenylsulfonyl)-6-hepten-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3723100/1863875-08-4.png)
![Carbamic acid, N-[(5S,6E)-5-azido-7-(phenylsulfonyl)-6-hepten-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3723100/1863875-08-4_b.png)


![Pyrrolo[1,2-b][1,2]benzisothiazole, 1,2,3,9b-tetrahydro-, 5,5-dioxide](http://img.cochemist.com/ccimg/830400/830319-72-7.png)
![Pyrrolo[1,2-b][1,2]benzisothiazole, 1,2,3,9b-tetrahydro-, 5,5-dioxide](http://img.cochemist.com/ccimg/830400/830319-72-7_b.png)
![Cyclohexanone, 4-hydroxy-3-[(phenylmethyl)thio]-, (3R,4S)-](http://img.cochemist.com/ccimg/794600/794526-34-4.png)
![Cyclohexanone, 4-hydroxy-3-[(phenylmethyl)thio]-, (3R,4S)-](http://img.cochemist.com/ccimg/794600/794526-34-4_b.png)








![2-Cyclohexen-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (1R,4S)-rel-](http://img.cochemist.com/ccimg/758000/757997-98-1.png)
![2-Cyclohexen-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (1R,4S)-rel-](http://img.cochemist.com/ccimg/758000/757997-98-1_b.png)
![2-Cyclopenten-1-one, 4-[(phenylmethyl)thio]-, (4R)-](http://img.cochemist.com/ccimg/794600/794526-38-8.png)
![2-Cyclopenten-1-one, 4-[(phenylmethyl)thio]-, (4R)-](http://img.cochemist.com/ccimg/794600/794526-38-8_b.png)
![Cyclohexanone, 4-(acetyloxy)-3-[(phenylmethyl)thio]-, (3S,4R)-](http://img.cochemist.com/ccimg/794600/794526-36-6.png)
![Cyclohexanone, 4-(acetyloxy)-3-[(phenylmethyl)thio]-, (3S,4R)-](http://img.cochemist.com/ccimg/794600/794526-36-6_b.png)


![CARBAMIC ACID, [(4E)-5-(PHENYLSULFONYL)-4-PENTENYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/850700/850606-87-0.png)
![CARBAMIC ACID, [(4E)-5-(PHENYLSULFONYL)-4-PENTENYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/850700/850606-87-0_b.png)
![1H-Pyrrole, 2,5-dihydro-1-[(2-nitrophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/219600/219583-73-0.png)
![1H-Pyrrole, 2,5-dihydro-1-[(2-nitrophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/219600/219583-73-0_b.png)












![2-Cyclohexen-1-one, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (4R)-](http://img.cochemist.com/ccimg/164600/164577-42-8.png)
![2-Cyclohexen-1-one, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (4R)-](http://img.cochemist.com/ccimg/164600/164577-42-8_b.png)
![1H-Pyrrole, 1-[(4-methylphenyl)sulfonyl]-3-phenyl-](http://img.cochemist.com/ccimg/153700/153633-35-3.png)
![1H-Pyrrole, 1-[(4-methylphenyl)sulfonyl]-3-phenyl-](http://img.cochemist.com/ccimg/153700/153633-35-3_b.png)
![PYRROLO[1,2-B][1,2]BENZISOTHIAZOLE, 2,3-DIHYDRO-, 5,5-DIOXIDE](http://img.cochemist.com/ccimg/830400/830319-71-6.png)
![PYRROLO[1,2-B][1,2]BENZISOTHIAZOLE, 2,3-DIHYDRO-, 5,5-DIOXIDE](http://img.cochemist.com/ccimg/830400/830319-71-6_b.png)
![PYRIDINE, 1-[(2-BROMOPHENYL)SULFONYL]-1,2,3,4-TETRAHYDRO-](http://img.cochemist.com/ccimg/830400/830319-77-2.png)
![PYRIDINE, 1-[(2-BROMOPHENYL)SULFONYL]-1,2,3,4-TETRAHYDRO-](http://img.cochemist.com/ccimg/830400/830319-77-2_b.png)
![2-PIPERIDINONE, 1-[(2-BROMOPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/830400/830319-73-8.png)
![2-PIPERIDINONE, 1-[(2-BROMOPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/830400/830319-73-8_b.png)




![Phosphonic acid, [(2-naphthalenylthio)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/183500/183444-04-4.png)
![Phosphonic acid, [(2-naphthalenylthio)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/183500/183444-04-4_b.png)
![L-ALANINAMIDE, N-[(PHENYLMETHOXY)CARBONYL]-L-ALANYL-N-(2-PHENYLETHYL)-](http://img.cochemist.com/ccimg/84900/84899-49-0.png)
![L-ALANINAMIDE, N-[(PHENYLMETHOXY)CARBONYL]-L-ALANYL-N-(2-PHENYLETHYL)-](http://img.cochemist.com/ccimg/84900/84899-49-0_b.png)
![2-FURANMETHANAMINE, N-[2-(1-CYCLOHEXEN-1-YL)ETHYL]-](http://img.cochemist.com/ccimg/628800/628706-86-5.png)
![2-FURANMETHANAMINE, N-[2-(1-CYCLOHEXEN-1-YL)ETHYL]-](http://img.cochemist.com/ccimg/628800/628706-86-5_b.png)




![2-PYRROLIDINEMETHANOL, 1-[(4-METHYLPHENYL)SULFONYL]-5-PHENYL-, (2S,5S)-](http://img.cochemist.com/ccimg/830400/830319-93-2.png)
![2-PYRROLIDINEMETHANOL, 1-[(4-METHYLPHENYL)SULFONYL]-5-PHENYL-, (2S,5S)-](http://img.cochemist.com/ccimg/830400/830319-93-2_b.png)






![BENZENE, 4-[(1E)-2-BROMOETHENYL]-1,2-DIMETHOXY-](http://img.cochemist.com/ccimg/69800/69731-27-7.png)
![BENZENE, 4-[(1E)-2-BROMOETHENYL]-1,2-DIMETHOXY-](http://img.cochemist.com/ccimg/69800/69731-27-7_b.png)
![2-PYRROLIDINONE, 1-[(2-BROMOPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/830400/830319-67-0.png)
![2-PYRROLIDINONE, 1-[(2-BROMOPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/830400/830319-67-0_b.png)


![5-(phenylethynyl)-5h-dibenzo[a,d][7]annulen-5-ol](http://img.cochemist.com/ccimg/941800/941716-31-0.png)
![5-(phenylethynyl)-5h-dibenzo[a,d][7]annulen-5-ol](http://img.cochemist.com/ccimg/941800/941716-31-0_b.png)
![1H-PYRROLE, 1-[(2-BROMOPHENYL)SULFONYL]-2,3-DIHYDRO-](http://img.cochemist.com/ccimg/830400/830319-69-2.png)
![1H-PYRROLE, 1-[(2-BROMOPHENYL)SULFONYL]-2,3-DIHYDRO-](http://img.cochemist.com/ccimg/830400/830319-69-2_b.png)


















![Pyrrolidine, 1-[(4-methylphenyl)sulfonyl]-3-phenyl-](http://img.cochemist.com/ccimg/157000/156908-33-7.png)
![Pyrrolidine, 1-[(4-methylphenyl)sulfonyl]-3-phenyl-](http://img.cochemist.com/ccimg/157000/156908-33-7_b.png)
![1H-Pyrrole, 1-[(2-bromophenyl)sulfonyl]-2,5-dihydro-](http://img.cochemist.com/ccimg/139100/139021-59-3.png)
![1H-Pyrrole, 1-[(2-bromophenyl)sulfonyl]-2,5-dihydro-](http://img.cochemist.com/ccimg/139100/139021-59-3_b.png)


![Piperidine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/176700/176650-38-7.png)
![Piperidine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/176700/176650-38-7_b.png)










![2-Cyclohexen-1-one, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (4S)-](http://img.cochemist.com/ccimg/119500/119478-74-9.png)
![2-Cyclohexen-1-one, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (4S)-](http://img.cochemist.com/ccimg/119500/119478-74-9_b.png)














![Pyrrolidine, 3-cyclohexyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/157000/156908-46-2.png)
![Pyrrolidine, 3-cyclohexyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/157000/156908-46-2_b.png)
![4-[TERT-BUTYL(DIMETHYL)SILYL]OXYCYCLOHEX-2-EN-1-ONE](/data/chemimg/394400/119414-47-0.png)
![4-[TERT-BUTYL(DIMETHYL)SILYL]OXYCYCLOHEX-2-EN-1-ONE](/data/chemimg/394400/119414-47-0_b.png)







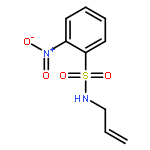
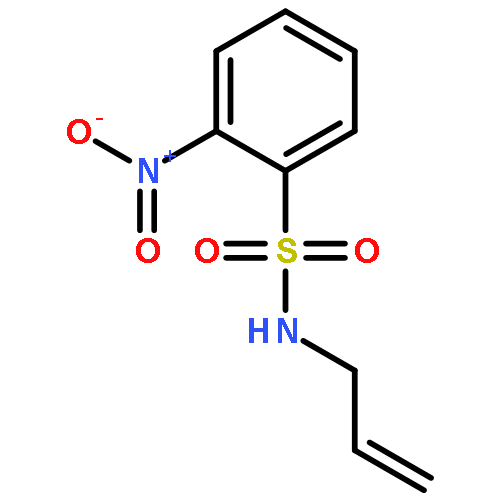
![1-[(2-nitrophenyl)sulfonyl]-1H-pyrrole](http://img.cochemist.com/ccimg/51200/51144-95-7.png)
![1-[(2-nitrophenyl)sulfonyl]-1H-pyrrole](http://img.cochemist.com/ccimg/51200/51144-95-7_b.png)






![Pyrrolidine, 3-(3,4-dimethoxyphenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/830400/830319-65-8.png)
![Pyrrolidine, 3-(3,4-dimethoxyphenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/830400/830319-65-8_b.png)
![Pyrrolidine, 3-(4-methoxyphenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/830400/830319-66-9.png)
![Pyrrolidine, 3-(4-methoxyphenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/830400/830319-66-9_b.png)
![2-Pyrrolidinemethanol, 1-[(4-methylphenyl)sulfonyl]-5-phenyl-, (2S,5R)-](http://img.cochemist.com/ccimg/830400/830319-92-1.png)
![2-Pyrrolidinemethanol, 1-[(4-methylphenyl)sulfonyl]-5-phenyl-, (2S,5R)-](http://img.cochemist.com/ccimg/830400/830319-92-1_b.png)
![2-Pyrrolidinone, 5-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-, (5S)-](http://img.cochemist.com/ccimg/133000/132974-83-5.png)
![2-Pyrrolidinone, 5-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-, (5S)-](http://img.cochemist.com/ccimg/133000/132974-83-5_b.png)
![5-[(E)-2-BROMOETHENYL]-1,3-BENZODIOXOLE](http://img.cochemist.com/ccimg/77200/77150-95-9.png)
![5-[(E)-2-BROMOETHENYL]-1,3-BENZODIOXOLE](http://img.cochemist.com/ccimg/77200/77150-95-9_b.png)
![1H-Pyrrole, 1-[(2-bromo-4,5-dimethoxyphenyl)sulfonyl]-2,5-dihydro-](http://img.cochemist.com/ccimg/830400/830319-62-5.png)
![1H-Pyrrole, 1-[(2-bromo-4,5-dimethoxyphenyl)sulfonyl]-2,5-dihydro-](http://img.cochemist.com/ccimg/830400/830319-62-5_b.png)


![2-Piperidinol, 1-[(2-bromophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/830400/830319-75-0.png)
![2-Piperidinol, 1-[(2-bromophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/830400/830319-75-0_b.png)
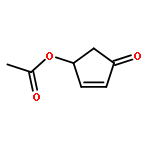
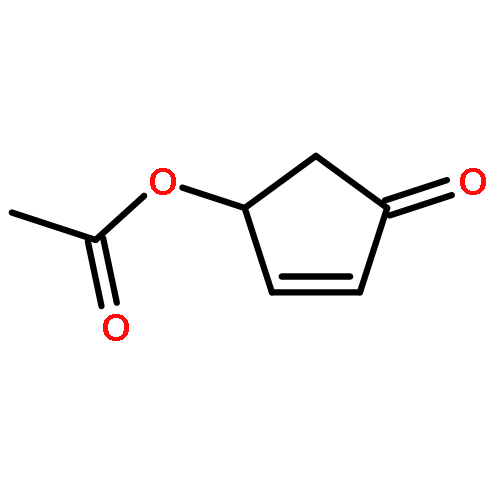




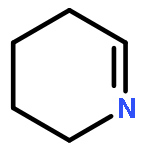
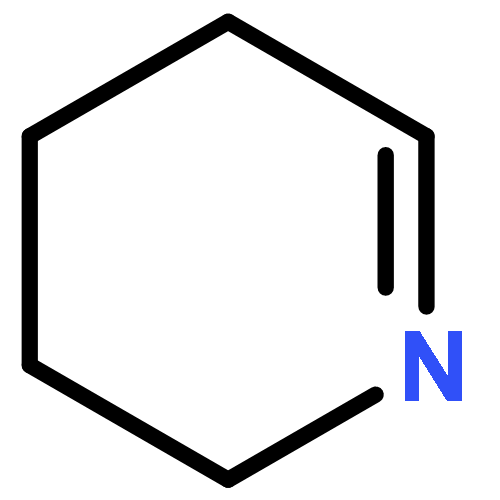




![7-Oxabicyclo[4.1.0]heptane,1-methyl-4-(1-methylethenyl)-, (1S,4R,6R)-](http://img.cochemist.com/ccimg/7000/6909-30-4.png)
![7-Oxabicyclo[4.1.0]heptane,1-methyl-4-(1-methylethenyl)-, (1S,4R,6R)-](http://img.cochemist.com/ccimg/7000/6909-30-4_b.png)


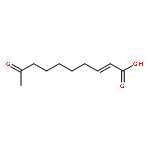


![Pyrrolidine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24600/24517-59-7.png)
![Pyrrolidine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24600/24517-59-7_b.png)








![L-Alanine,N-[(phenylmethoxy)carbonyl]-D-phenylalanyl- (9CI)](http://img.cochemist.com/ccimg/16100/16088-00-9.png)
![L-Alanine,N-[(phenylmethoxy)carbonyl]-D-phenylalanyl- (9CI)](http://img.cochemist.com/ccimg/16100/16088-00-9_b.png)

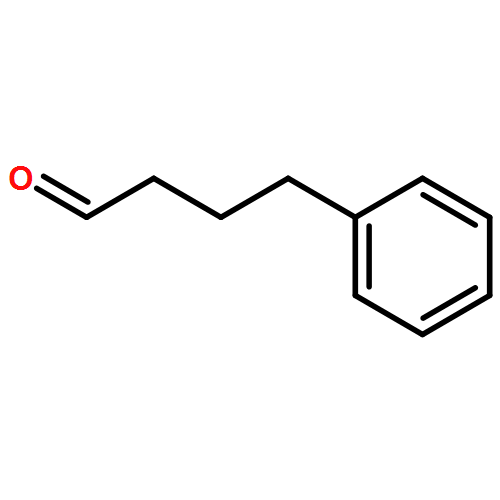
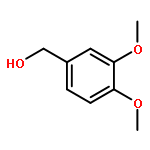
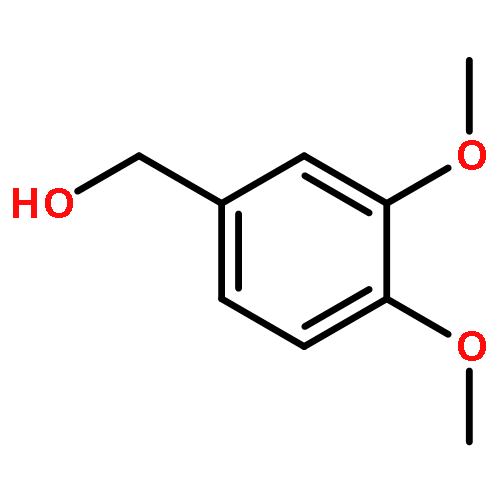
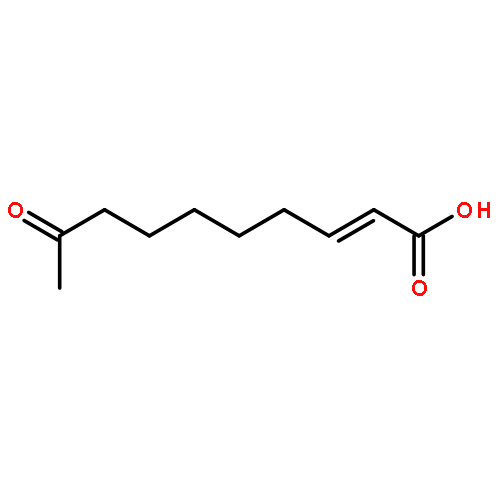
![4(3H)-Quinazolinone,7-bromo-6-chloro-3-[3-(3-hydroxy-2-piperidinyl)-2-oxopropyl]-, hydrobromide(1:1)](http://img.cochemist.com/ccimg/17400/17395-31-2.png)
![4(3H)-Quinazolinone,7-bromo-6-chloro-3-[3-(3-hydroxy-2-piperidinyl)-2-oxopropyl]-, hydrobromide(1:1)](http://img.cochemist.com/ccimg/17400/17395-31-2_b.png)

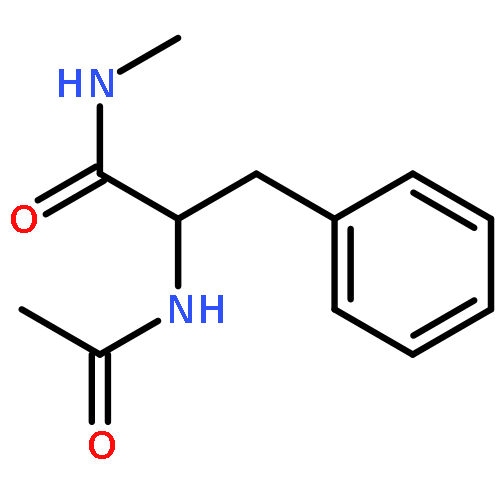




















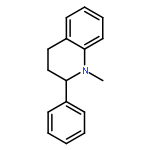
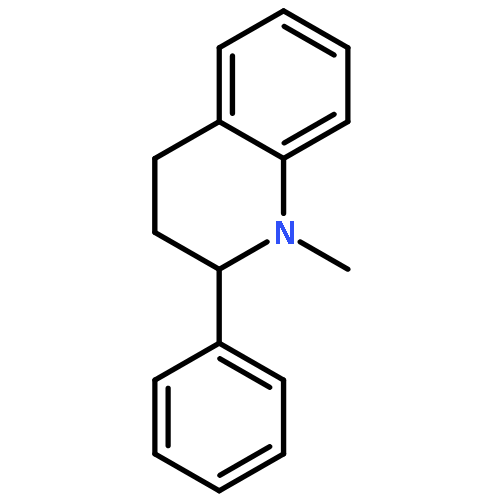

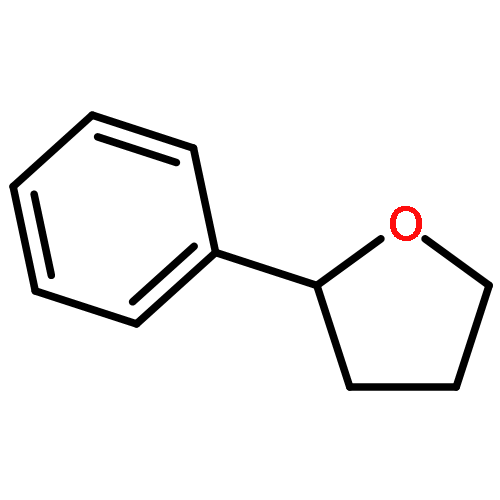










![2-(CYCLOHEXEN-1-YL)-N-[(4-METHOXYPHENYL)METHYL]ETHANAMINE](http://img.cochemist.com/ccimg/356600/356537-08-1.png)
![2-(CYCLOHEXEN-1-YL)-N-[(4-METHOXYPHENYL)METHYL]ETHANAMINE](http://img.cochemist.com/ccimg/356600/356537-08-1_b.png)







![2-METHYL-2-PROPANYL 4-OXO-3,4-DIHYDRO-1'H,2H-SPIRO[NAPHTHALENE-1,<WBR />4'-PIPERIDINE]-1'-CARBOXYLATE](http://img.cochemist.com/ccimg/35500/35418-07-6.png)
![2-METHYL-2-PROPANYL 4-OXO-3,4-DIHYDRO-1'H,2H-SPIRO[NAPHTHALENE-1,<WBR />4'-PIPERIDINE]-1'-CARBOXYLATE](http://img.cochemist.com/ccimg/35500/35418-07-6_b.png)






















![1H-Thieno[3,4-d]imidazole-4-pentanamide, hexahydro-2-oxo-N-2-propyn-1-yl-, (3aS,4S,6aR)-](/data/chemimg/103200/773888-45-2.png)
![1H-Thieno[3,4-d]imidazole-4-pentanamide, hexahydro-2-oxo-N-2-propyn-1-yl-, (3aS,4S,6aR)-](/data/chemimg/103200/773888-45-2_b.png)










![2-Tosyl-2,3,3a,4-tetrahydrocyclopenta[c]pyrrol-5(1H)-one](http://img.cochemist.com/ccimg/133900/133886-43-8.png)
![2-Tosyl-2,3,3a,4-tetrahydrocyclopenta[c]pyrrol-5(1H)-one](http://img.cochemist.com/ccimg/133900/133886-43-8_b.png)