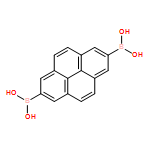
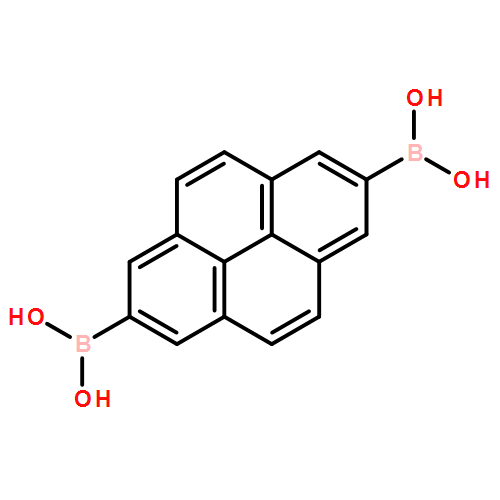
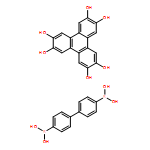
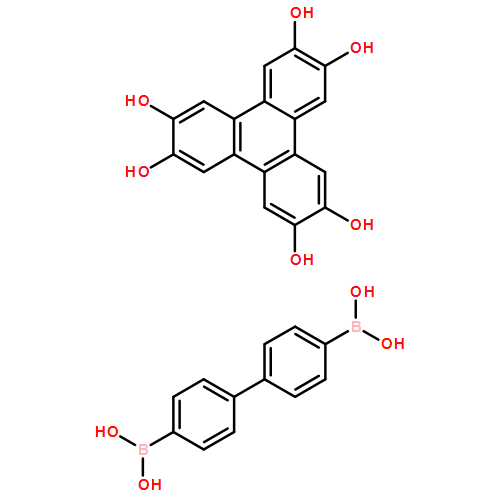
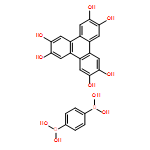
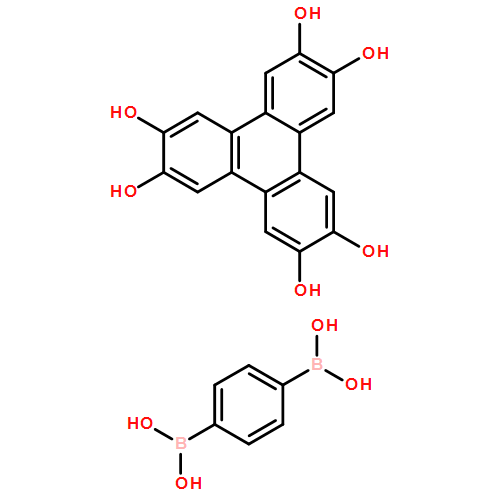
Co-reporter:Anton D. Chavez, Brian J. Smith, Merry K. Smith, Peter A. Beaucage, Brian H. Northrop, and William R. Dichtel
Chemistry of Materials 2016 Volume 28(Issue 14) pp:4884
Publication Date(Web):June 24, 2016
DOI:10.1021/acs.chemmater.6b01831
Co-reporter:Brian J. Smith, Anna C. Overholts, Nicky Hwang and William R. Dichtel
Chemical Communications 2016 vol. 52(Issue 18) pp:3690-3693
Publication Date(Web):01 Feb 2016
DOI:10.1039/C5CC10221A
We explore the crystallization of a high surface area imine-linked two-dimensional covalent organic framework (2D COF). The growth process reveals rapid initial formation of an amorphous network that subsequently crystallizes into the layered 2D network. The metastable amorphous polymer may be isolated and resubjected to growth conditions to form the COF. These experiments provide the first mechanistic insight into the mechanism of imine-linked 2D COF formation, which is distinct from that of boronate-ester linked COFs.
Co-reporter:Catherine R. DeBlase and William R. Dichtel
Macromolecules 2016 Volume 49(Issue 15) pp:5297-5305
Publication Date(Web):June 21, 2016
DOI:10.1021/acs.macromol.6b00891
Since their discovery in 2005, covalent organic frameworks (COFs) have attracted interest as potential materials for gas storage, catalysis, energy storage, and other applications because of their ability to periodically and reliably organize designed functionality into high surface area materials. Most of the first examples relied on boron-containing linkages, which suffer from hydrolytic and oxidative instability that limit their utility. In this Perspective, we describe the trend toward more robust linkages by highlighting the design, synthesis, and properties of several recent examples. The continued development of new COF chemistries, along with improved understanding of their formation and control of their final form, will provide a means to harness their molecularly precise solid-state structures for useful purposes.
Co-reporter:Ryan P. Bisbey, Catherine R. DeBlase, Brian J. Smith, and William R. Dichtel
Journal of the American Chemical Society 2016 Volume 138(Issue 36) pp:
Publication Date(Web):August 1, 2016
DOI:10.1021/jacs.6b04669
Two-dimensional covalent organic frameworks (2D COFs) are crystalline polymer networks whose modular 2D structures and permanent porosity motivate efforts to integrate them into sensing, energy storage, and optoelectronic devices. These applications require forming the material as a thin film instead of a microcrystalline powder, which has been achieved previously by including a substrate in the reaction mixture. This approach suffers from two key drawbacks: COF precipitates form concurrently and contaminate the film, and variable monomer and oligomer concentrations during the polymerization provide poor control over film thickness. Here we address these challenges by growing 2D COF thin films under continuous flow conditions. Initially homogeneous monomer solutions polymerize while pumped through heated tubing for a given residence time, after which they pass over a substrate. When the residence time and conditions are chosen judiciously, 2D COF powders form downstream of the substrate, and the chemical composition of the solution at the substrate remains constant. COF films grown in flow exhibit constant rates of mass deposition, enabling thickness control as well as access to thicker films than are available from previous static growth procedures. Notably, the crystallinity of COF films is observed only at longer residence times, suggesting that oligomeric and polymeric species play an important role in forming the 2D COF lattice. This approach, which we demonstrate for four different frameworks, is both a simple and powerful method to control the formation of COF thin films.
Co-reporter:David J. Fortman; Jacob P. Brutman; Christopher J. Cramer; Marc A. Hillmyer
Journal of the American Chemical Society 2015 Volume 137(Issue 44) pp:14019-14022
Publication Date(Web):October 23, 2015
DOI:10.1021/jacs.5b08084
Vitrimers are polymer networks whose cross-links undergo associative exchange processes at elevated temperature, usually in the presence of an embedded catalyst. This design feature enables the reshaping of materials with mechanical properties similar to thermoset resins. Here we report a new class of vitrimers consisting of polyhydroxyurethanes (PHUs) derived from six-membered cyclic carbonates and amines. PHU networks relax stress and may be reprocessed at elevated temperature and pressure in the absence of an external catalyst. The as-synthesized networks exhibit tensile properties comparable to those of leading thermosets and recover ca. 75% of their as-synthesized values following reprocessing. Stress relaxation occurs through an associative process involving nucleophilic addition of free hydroxyl groups to the carbamate linkages and exhibits an Arrhenius activation energy of 111 ± 10 kJ/mol, which is lower than that observed for molecular model compounds (148 ± 7 kJ/mol). These findings suggest that transcarbamoylation is activated by mechanical stress, which we attribute, on the basis of DFT calculations, to the twisting of N lone pairs out of conjugation with the carbonyl π orbitals. PHU vitrimers are a promising new class of repairable networks because of their outstanding mechanical properties, avoidance of toxic isocyanate monomers, and catalyst-free repair processes.
Co-reporter:Deepti Gopalakrishnan and William R. Dichtel
Chemistry of Materials 2015 Volume 27(Issue 11) pp:3813
Publication Date(Web):May 5, 2015
DOI:10.1021/acs.chemmater.5b00857
Co-reporter:Chao Sun, Katherine L. Walker, Devin L. Wakefield, and William R. Dichtel
Chemistry of Materials 2015 Volume 27(Issue 12) pp:4499
Publication Date(Web):May 27, 2015
DOI:10.1021/acs.chemmater.5b01954
Graphene oxide (GO) has drawn interest for impermeable coatings, water purification, drug delivery vectors, and other applications that involve functionalizing its surface with molecular, polymeric, or biomolecular species. Existing GO functionalization strategies rely on covalent attachment to polar functionalities at its oxidized regions or weak nonspecific absorption to the graphitic regions. These modifications risk disrupting GO’s aqueous dispersibility and leave its hydrophobic patches available to disrupt protein structure and quench the emission of fluorophores. Here, we demonstrate a general strategy to functionalize GO noncovalently using tripodal binding motifs, which present three pyrene moieties that bind to the hydrophobic regions. Tripods immobilize the serine protease enzyme chymotrypsin (ChT) onto GO and preserve its native structure and activity. In contrast, unmodified GO is one of the strongest known ChT inhibitors, which we show to arise from interactions with both GO’s hydrophilic regions and hydrophobic regions. Furthermore, GO quenches the photoemission of many fluorescent probes, and its weak inherent photoemission is inconvenient for imaging via fluorescence microscopy. When presented on the GO surface through a tripod, the fluorescent dye Alexa Fluor 488 retains its fluorescence and allows the GO sheets to be imaged using a standard fluorescence microscope. As such, tripod-binding groups represent a useful strategy to functionalize GO with biomolecules and study its interactions with cells and living organisms.
Co-reporter:Brian J. Smith, Nicky Hwang, Anton D. Chavez, Jennifer L. Novotney and William R. Dichtel
Chemical Communications 2015 vol. 51(Issue 35) pp:7532-7535
Publication Date(Web):19 Mar 2015
DOI:10.1039/C5CC00379B
We examine the growth rates, activation energies, and hydrolytic stability of multiple 2D boronate ester covalent organic frameworks by turbidity measurements, observing a 200-fold range in stability. The rate-determining step in boronate ester 2D COF growth is not in-solution condensation, but rather interlayer polymer stacking through a nucleation–elongation process.
Co-reporter:Dr. Dan Lehnherr;Joaquin M. Alzola;Dr. Emil B. Lobkovsky;Dr. William R. Dichtel
Chemistry - A European Journal 2015 Volume 21( Issue 50) pp:
Publication Date(Web):
DOI:10.1002/chem.201585002
Co-reporter:John W. Colson;Jason A. Mann;Catherine R. DeBlase
Journal of Polymer Science Part A: Polymer Chemistry 2015 Volume 53( Issue 2) pp:378-384
Publication Date(Web):
DOI:10.1002/pola.27399
ABSTRACT
Two-dimensional covalent organic frameworks (COFs) are polymer networks that organize molecular building blocks into porous, layered structures of interest for organic optoelectronic and energy storage devices. Current synthetic methods produce these materials as either insoluble, microcrystalline powders or as oriented thin films on various substrates, including single-layer graphene (SLG). Under these conditions, COF thin films form on both the graphene-coated and bare regions of the substrate, suggesting uncontrolled nucleation processes that occur either in solution or nonselectively on different surfaces. Here, we describe modified polymerization conditions that provide COF films selectively on SLG. This finding enables COF films to be grown on lithographically patterned SLG substrates, which provide insight into the uniformity of film growth across the substrate and factors relevant to their nucleation and growth. The ability to grow COF films selectively on lithographically patterned SLG will facilitate their integration into devices. © 2014 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2015, 53, 378–384
Co-reporter:Catherine R. DeBlase, Kenneth Hernández-Burgos, Katharine E. Silberstein, Gabriel G. Rodríguez-Calero, Ryan P. Bisbey, Héctor D. Abruña, and William R. Dichtel
ACS Nano 2015 Volume 9(Issue 3) pp:3178
Publication Date(Web):February 11, 2015
DOI:10.1021/acsnano.5b00184
Two-dimensional covalent organic frameworks (2D COFs) are ideally suited for organizing redox-active subunits into periodic, permanently porous polymer networks of interest for pseudocapacitive energy storage. Here we describe a method for synthesizing crystalline, oriented thin films of a redox-active 2D COF on Au working electrodes. The thickness of the COF film was controlled by varying the initial monomer concentration. A large percentage (80–99%) of the anthraquinone groups are electrochemically accessible in films thinner than 200 nm, an order of magnitude improvement over the same COF prepared as a randomly oriented microcrystalline powder. As a result, electrodes functionalized with oriented COF films exhibit a 400% increase in capacitance scaled to electrode area as compared to those functionalized with the randomly oriented COF powder. These results demonstrate the promise of redox-active COFs for electrical energy storage and highlight the importance of controlling morphology for optimal performance.Keywords: covalent organic framework; electrochemistry; energy storage; nanoporous materials; polymer films; supercapacitors; surface science;
Co-reporter:Dr. Dan Lehnherr;Joaquin M. Alzola;Dr. Emil B. Lobkovsky;Dr. William R. Dichtel
Chemistry - A European Journal 2015 Volume 21( Issue 50) pp:18122-18127
Publication Date(Web):
DOI:10.1002/chem.201503418
Abstract
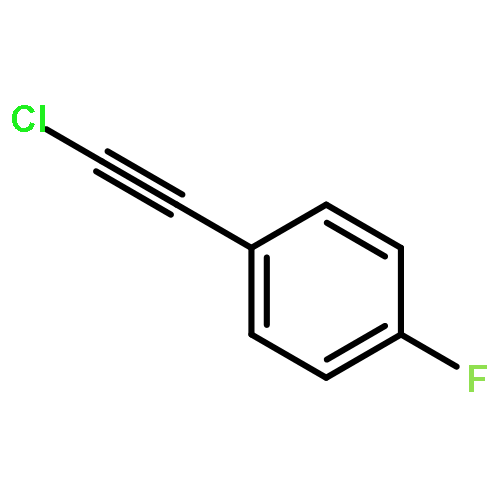
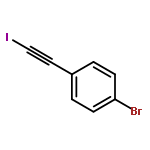
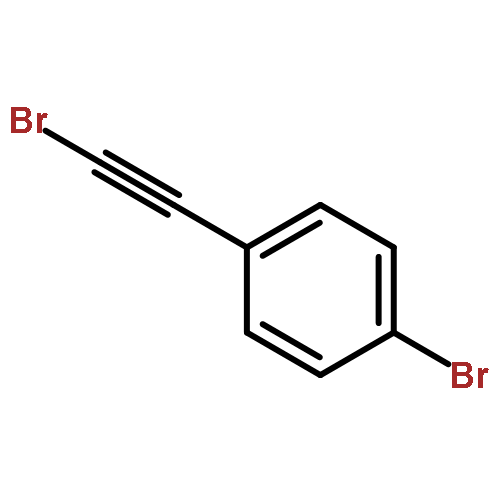
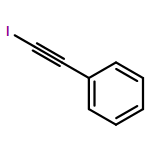
Independent control of halide substitution at six of the seven naphthalene positions of 2-arylnaphthalenes is achieved through the regioselective benzannulation of chloro-, bromo-, and iodoalkynes. The modularity of this approach is demonstrated through the preparation of 44 polyheterohalogenated naphthalene products, most of which are difficult to access through known naphthalene syntheses. The outstanding regioselectivity of the reaction is both predictable and proven unambiguously by single-crystal X-ray diffraction for many examples. This synthetic method enables the rapid preparation of complex aromatic systems poised for further derivatization using established cross-coupling methods. The power and versatility of this approach makes substituted naphthalenes highly attractive building blocks for new organic materials and diversity-oriented synthesis.
Co-reporter:Brian J. Smith
Journal of the American Chemical Society 2014 Volume 136(Issue 24) pp:8783-8789
Publication Date(Web):May 22, 2014
DOI:10.1021/ja5037868
Covalent organic frameworks (COFs) are periodic two- and three-dimensional (2D and 3D) polymer networks with high surface areas, low densities, and designed structures. Despite intense interest in framework materials, the nucleation and growth processes of COFs, and even of more established metal–organic frameworks (MOFs), are poorly understood. The kinetics of COF growth under varied reaction conditions provides mechanistic insight needed to improve their crystallinity and rationally synthesize new materials. Such kinetic measurements are unprecedented and difficult to perform on typical heterogeneous COF reaction mixtures. Here we synthesize 2D boronate ester-linked COF-5 under conditions in which the monomers are fully soluble. These homogeneous growth conditions provide equal or better material quality compared to any previous report and enable the first rigorous studies of the early stages of COF growth. COF-5 forms within minutes, and the precipitation rate is readily quantified from optical turbidity measurements. COF-5 formation follows an Arrhenius temperature dependence between 60–90 °C with an activation energy of 22–27 kcal/mol. The measured rate law includes a second order in both boronic acid and catechol moieties, and inverse second order in MeOH concentration. A competitive monofunctional catechol slows COF-5 formation but does not redissolve already precipitated COF, indicating both dynamic covalent bond formation and irreversible precipitation. Finally, stoichiometric H2O provides a 4-fold increase in crystallite domain areas, representing the first rational link between reaction conditions and material quality.
Co-reporter:Samuel J. Hein, Hasan Arslan, Ivan Keresztes, and William R. Dichtel
Organic Letters 2014 Volume 16(Issue 17) pp:4416-4419
Publication Date(Web):August 27, 2014
DOI:10.1021/ol501874s
Congested aromatic systems were prepared by benzannulating silyl-protected arylacetylenes. The silyl groups may be retained in the naphthalene products and transformed into iodides in high yield. The desirable attributes of this strategy, particularly its remarkable tolerance of sterically hindered alkynes, are showcased in the efficient synthesis of a congested, branched oligo(naphthalene). As such, benzannulations of diaryl and silyl-protected acetylenes show outstanding promise for accessing new aromatic architectures.
Co-reporter:Hasan Arslan, Katherine L. Walker, and William R. Dichtel
Organic Letters 2014 Volume 16(Issue 22) pp:5926-5929
Publication Date(Web):November 10, 2014
DOI:10.1021/ol502938y
Asao–Yamamoto benzannulations transform diarylalkynes into 2,3-diarylnaphthalenes, and regioselective variants of this reaction are of interest for synthesizing substituted polycyclic aromatic systems. It is shown that regioselective cycloadditions occur when one alkyne carbon preferentially stabilizes developing positive charge. Simple calculations of the relative energies of carbocations localized at each alkyne carbon of a substrate predict the regioselectivity, which is not eroded by bulky substituents, including 2,6-disubstituted aryl groups.
Co-reporter:Spencer D. Brucks, David N. Bunck, William R. Dichtel
Polymer 2014 Volume 55(Issue 1) pp:330-334
Publication Date(Web):14 January 2014
DOI:10.1016/j.polymer.2013.07.030
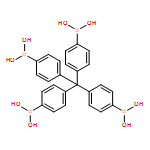
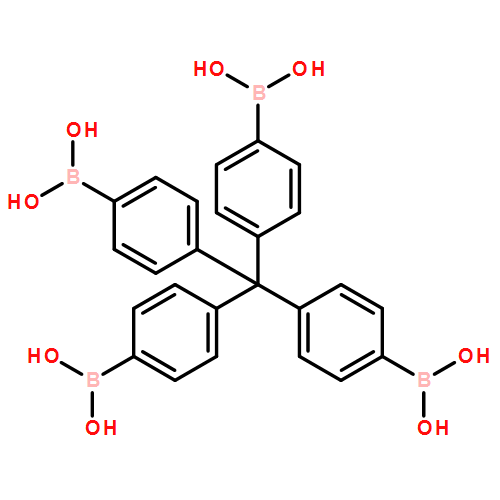
Co-crystallizing a monomer capable of forming a three-dimensional covalent organic framework (3D COF) with a truncated analog represents a robust strategy to functionalize the pores of these crystalline polymer networks. Here we elaborate this approach by demonstrating that monofunctional arylboronic acids serve as effective truncation/functionalization agents for COF-102, a boroxine-linked 3D network derived from the dehydration of a tetrahedral tetrakis(boronic acid) monomer. The COF-102 network forms under typical solvothermal conditions, even in the presence of a large excess of 4-tolylboronic acid, which is incorporated into the polymer's boroxine linkages up to a maximum loading level of ca. 33 mol%. This finding indicates the maximum truncation level for the COF-102 network and suggests that framework crystallization is irreversible. At high feed ratios of the monofunctional boronic acid, the isolated COF-102-tolyl powders are initially contaminated by significant amounts of tris(4-tolyl)boroxine, which is removed through a solution-based activation process to provide COF-102-tolyl samples with high functionalization density, long-range order, and permanent porosity. We also demonstrate the generality of this truncation study by evaluating several other readily available arylboronic acids, each of which are incorporated into the COF similarly. Together these findings demonstrate the simplicity and generality of this truncation/functionalization approach, as well as its fundamental limits.
Co-reporter:David N. Bunck and William R. Dichtel
Journal of the American Chemical Society 2013 Volume 135(Issue 40) pp:14952-14955
Publication Date(Web):September 23, 2013
DOI:10.1021/ja408243n
Co-reporter:Catherine R. DeBlase ; Katharine E. Silberstein ; Thanh-Tam Truong ; Héctor D. Abruña
Journal of the American Chemical Society 2013 Volume 135(Issue 45) pp:16821-16824
Publication Date(Web):October 22, 2013
DOI:10.1021/ja409421d
Two-dimensional covalent organic frameworks (2D COFs) are candidate materials for charge storage devices because of their micro- or mesoporosity, high surface area, and ability to predictably organize redox-active groups. The limited chemical and oxidative stability of established COF linkages, such as boroxines and boronate esters, precludes these applications, and no 2D COF has demonstrated reversible redox behavior. Here we describe a β-ketoenamine-linked 2D COF that exhibits reversible electrochemical processes of its anthraquinone subunits, excellent chemical stability to a strongly acidic electrolyte, and one of the highest surface areas of the imine- or enamine-linked 2D COFs. Electrodes modified with the redox-active COF show higher capacitance than those modified with a similar non-redox-active COF, even after 5000 charge–discharge cycles. These findings demonstrate the promise of using 2D COFs for capacitive storage.
Co-reporter:Deepti Gopalakrishnan
Journal of the American Chemical Society 2013 Volume 135(Issue 22) pp:8357-8362
Publication Date(Web):May 3, 2013
DOI:10.1021/ja402668e
1,3,5-Trinitroperhydro-1,3,5-triazine (RDX) is a principal component of plastic explosives used in acts of terrorism and within improvised explosive devices, among others. Approaches to detect RDX compatible with remote, “stand-off” sampling that do not require preconcentration strategies, such as the swabs commonly employed in airports, will benefit military and civilian security. Such detection remains a significant challenge because RDX is 103 less volatile than 1,3,5-trinitrotoluene (TNT), corresponding to a parts-per-trillion vapor pressure under ambient conditions. Therefore, while fluorescence quenching of conjugated polymers is sufficiently sensitive to detect TNT vapors, RDX vapor detection is undemonstrated. Here we report a cross-linked phenylene vinylene polymer network whose fluorescence is quenched by trace amounts of RDX introduced from solution or the vapor phase. Fluorescence quenching is reduced, but remains significant, when partially degraded RDX is employed, suggesting that the polymer responds to RDX itself. The polymer network also responds to TNT and PETN similarly introduced from solution or the vapor phase. Pure solvents, volatile amines, and the outgassed vapors from lipstick or sunscreen do not quench polymer fluorescence. The established success of TNT sensors based on fluorescence quenching makes this a material of interest for real-world explosive sensors and will motivate further interest in cross-linked polymers and framework materials for sensing applications.
Co-reporter:Hasan Arslan, Fernando J. Uribe-Romo, Brian J. Smith and William R. Dichtel
Chemical Science 2013 vol. 4(Issue 10) pp:3973-3978
Publication Date(Web):12 Jul 2013
DOI:10.1039/C3SC51212F
A rapid and efficient approach to prepare extended or partially fused hexabenzocoronene derivatives is described. The method is based on the sequential benzannulation and cyclodehydrogenation (Scholl oxidation) of simple diaryl alkynes. The benzannulation reaction proceeds efficiently on highly congested substrates and with complete regioselectivity. Scholl oxidation of the resulting oligo(arylene)s proceeds without rearrangements and provides either fully fused or specific partially fused polycyclic aromatic hydrocarbon products. The partially fused derivatives are a new class of contorted aromatic systems with high solubility, enhanced visible absorption, and reversible redox processes. The efficiency and specificity of the benzannulation and oxidation reactions are promising for accessing new classes of organic semiconductors and carbon nanostructures.
Co-reporter:David N. Bunck and William R. Dichtel
Chemical Communications 2013 vol. 49(Issue 24) pp:2457-2459
Publication Date(Web):11 Feb 2013
DOI:10.1039/C3CC40358K
We demonstrate a tandem truncation–functionalization strategy using the 3D covalent organic framework, COF-102. Allyl groups incorporated within the pore walls were subjected to thiol–ene coupling conditions, achieving high conversions and maintaining the crystallinity and permanent porosity of the parent framework.
Co-reporter:Thomas Alava, Jason A. Mann, Cécile Théodore, Jaime J. Benitez, William R. Dichtel, Jeevak M. Parpia, and Harold G. Craighead
Analytical Chemistry 2013 Volume 85(Issue 5) pp:2754
Publication Date(Web):January 31, 2013
DOI:10.1021/ac303268z
Graphene’s suite of useful properties makes it of interest for use in biosensors. However, graphene interacts strongly with hydrophobic components of biomolecules, potentially altering their conformation and disrupting their biological activity. We have immobilized the protein Concanavalin A onto a self-assembled monolayer of multivalent tripodal molecules on single-layer graphene. We used a quartz crystal microbalance (QCM) to show that tripod-bound Concanavalin A retains its affinity for polysaccharides containing α-d-glucopyrannosyl groups as well as for the α-d-mannopyranosyl groups located on the cell wall of Bacillus subtilis. QCM measurements on unfunctionalized graphene indicate that adsorption of Concanavalin A onto graphene is accompanied by near-complete loss of these functions, suggesting that interactions with the graphene surface induce deleterious structural changes to the protein. Given that Concanavalin A’s tertiary structure is thought to be relatively robust, these results suggest that other proteins might also be denatured upon adsorption onto graphene, such that the graphene–biomolecule interface must be considered carefully. Multivalent tripodal binding groups address this challenge by anchoring proteins without loss of function and without disrupting graphene’s desirable electronic structure.
Co-reporter:Jason A. Mann and William R. Dichtel
ACS Nano 2013 Volume 7(Issue 8) pp:7193
Publication Date(Web):July 16, 2013
DOI:10.1021/nn402599x
Graphene is an atomically thin, transparent, and conductive electrode material of interest for sensors and energy conversion and storage devices, among others. Fully realizing its potential will require robust and general methods to anchor active functionality onto its pristine basal plane. Such strategies should not utilize covalent bond formation, which disrupts the graphene’s π-electron system, from which most of its desirable properties arise. We recently introduced a tripodal binding motif, which forms robust monolayers on graphene capable of immobilizing active proteins and preventing their denaturation. Here we describe structure–property relationships for a series of tripod binding groups with “feet” of different sizes. Each derivative adsorbs strongly (ΔGads ≈ −39 kJ mol–1) to graphene’s basal plane, yet the resulting monolayers exhibit kinetic stabilities that vary over 2 orders of magnitude and molecular densities that vary by a factor of 2. This study identifies phenanthrene as a superior anchor relative to pyrene on the basis of its increased monolayer density and similar kinetic stability. We also demonstrate that varying the length of the methylene linkers between the feet and tripodal core does not affect binding substantially. These results represent the first demonstration of structure–property relationships in the assembly of molecular adsorbates on graphene and provide a paradigm for designing effective graphene binding motifs.Keywords: cobalt bis-terpyridine; electrochemistry; functionalization; graphene electrode; monolayer; noncovalent; self-assembly
Co-reporter:Jason A. Mann and William R. Dichtel
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 16) pp:2649-2657
Publication Date(Web):July 25, 2013
DOI:10.1021/jz4010448
The desirable properties and increased availability of single- and few-layer graphene have motivated interest in interfacing functional molecules and materials to its high surface area basal plane. The surface of pristine graphene lacks chemical functionality to allow for covalent modification without interrupting its continuous π-orbital system. In contrast, noncovalent functionalization does not suffer from these drawbacks and offers a means to tune graphene properties and incorporate molecular recognition or other active elements. In this Perspective, we describe emerging strategies to interface molecular compounds and polymers to graphene as well as the enhanced properties and new functions that they impart.
Co-reporter:David N. Bunck ;Dr. William R. Dichtel
Chemistry - A European Journal 2013 Volume 19( Issue 3) pp:818-827
Publication Date(Web):
DOI:10.1002/chem.201203145
Abstract
Framework materials have attracted intense interest for gas storage, separations, catalysis, and other applications as a consequence of their periodicity, high specific surface area, and rational synthesis. Cocrystallizing multiple monomers with identical linking chemistry represents an emerging route to access materials with increased complexity and advanced functions. This Concept Article highlights three strategies for framework synthesis that employ mixtures of monomers with 1) identical linking geometries, 2) different linking geometries, or 3) in which one monomer is truncated with respect to the other. These approaches offer a diverse toolbox to modify framework topology, incorporate active functionality, and rationally control crystallite size and morphology.
Co-reporter:Jason A. Mann;Dr. Thomas Alava;Dr. Harold G. Craighead;Dr. William R. Dichtel
Angewandte Chemie International Edition 2013 Volume 52( Issue 11) pp:3177-3180
Publication Date(Web):
DOI:10.1002/anie.201209149
Co-reporter:Joaquín Rodríguez-López ; Nicole L. Ritzert ; Jason A. Mann ; Cen Tan ; William R. Dichtel ;Héctor D. Abruña
Journal of the American Chemical Society 2012 Volume 134(Issue 14) pp:6224-6236
Publication Date(Web):March 12, 2012
DOI:10.1021/ja2106724
The surface diffusion of a cobalt bis-terpyridine, Co(tpy)2-containing tripodal compound (1·2PF6), designed to noncovalently adsorb to graphene through three pyrene moieties, has been studied by scanning electrochemical microscopy (SECM) on single-layer graphene (SLG). An initial boundary approach was designed in which picoliter droplets (radii ∼15–50 μm) of the tripodal compound were deposited on an SLG electrode, yielding microspots in which a monolayer of the tripodal molecules is initially confined. The time evolution of the electrochemical activity of these spots was detected at the aqueous phosphate buffer/SLG interface by SECM, in both generation/collection (G/C) and feedback modes. The tripodal compound microspots exhibit differential reactivity with respect to the underlying graphene substrate in two different electrochemical processes. For example, during the oxygen reduction reaction, adsorbed 1·2PF6 tripodal molecules generate more H2O2 than the bare graphene surface. This product was detected with spatial and temporal resolution using the SECM tip. The tripodal compound also mediates the oxidation of a Fe(II) species, generated at the SECM tip, under conditions in which SLG shows slow interfacial charge transfer. In each case, SECM images, obtained at increasing times, show a gradual decrease in the electrochemical response due to radial diffusion of the adsorbed molecules outward from the microspots onto the unfunctionalized areas of the SLG surface. This response was fit to a simple surface diffusion model, which yielded excellent agreement between the two experiments for the effective diffusion coefficients: Deff = 1.6 (±0.9) × 10–9 cm2/s and Deff = 1.5 (±0.6) × 10–9 cm2/s for G/C and feedback modes, respectively. Control experiments ruled out alternative explanations for the observed behavior, such as deactivation of the Co(II/III) species or of the SLG, and verified that the molecules do not diffuse when confined to obstructed areas. The noncovalent nature of the surface functionalization, together with the surface reactivity and mobility of these molecules, provides a means to couple the superior electronic properties of graphene to compounds with enhanced electrochemical performance, a key step toward developing dynamic electrode surfaces for sensing, electrocatalysis, and electronic applications.
Co-reporter:David N. Bunck ;Dr. William R. Dichtel
Angewandte Chemie International Edition 2012 Volume 51( Issue 8) pp:1885-1889
Publication Date(Web):
DOI:10.1002/anie.201108462
Co-reporter:Dr. Eric L. Spitler;John W. Colson;Dr. Ferno J. Uribe-Romo;Dr. Arthur R. Woll;Marissa R. Giovino;Abraham Saldivar;Dr. William R. Dichtel
Angewandte Chemie International Edition 2012 Volume 51( Issue 11) pp:2623-2627
Publication Date(Web):
DOI:10.1002/anie.201107070
Co-reporter:Hasan Arslan;Jonathan D. Saathoff;David N. Bunck;Dr. Paulette Clancy;Dr. William R. Dichtel
Angewandte Chemie International Edition 2012 Volume 51( Issue 48) pp:12051-12054
Publication Date(Web):
DOI:10.1002/anie.201206964
Co-reporter:David N. Bunck ;Dr. William R. Dichtel
Angewandte Chemie 2012 Volume 124( Issue 8) pp:1921-1925
Publication Date(Web):
DOI:10.1002/ange.201108462
Co-reporter:Dr. Eric L. Spitler;John W. Colson;Dr. Ferno J. Uribe-Romo;Dr. Arthur R. Woll;Marissa R. Giovino;Abraham Saldivar;Dr. William R. Dichtel
Angewandte Chemie 2012 Volume 124( Issue 11) pp:2677-2681
Publication Date(Web):
DOI:10.1002/ange.201107070
Co-reporter:Hasan Arslan;Jonathan D. Saathoff;David N. Bunck;Dr. Paulette Clancy;Dr. William R. Dichtel
Angewandte Chemie 2012 Volume 124( Issue 48) pp:12217-12220
Publication Date(Web):
DOI:10.1002/ange.201206964
Co-reporter:Jason A. Mann ; Joaquín Rodríguez-López ; Héctor D. Abruña
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17614-17617
Publication Date(Web):October 11, 2011
DOI:10.1021/ja208239v
Single-layer graphene is a newly available conductive material ideally suited for forming well-defined interfaces with electroactive compounds. Aromatic moieties typically interact with the graphene surface to maximize van der Waals interactions, predisposing most compounds to lie flat on its basal plane. Here we describe a tripodal motif that binds multivalently to graphene through three pyrene moieties and projects easily varied functionality away from the surface. The thermodynamic and kinetic binding parameters of a tripod bearing a redox-active Co(II) bis-terpyridyl complex were investigated electrochemically. The complex binds strongly to graphene and forms monolayers with a molecular footprint of 2.3 nm2 and a ΔGads = −38.8 ± 0.2 kJ mol–1. Its monolayers are stable in fresh electrolyte for more than 12 h and desorb from graphene 1000 times more slowly than model compounds bearing a single aromatic binding group. Differences in the heterogeneous rate constants of electron transfer between the two compounds suggest that the tripod projects its redox couple away from the graphene surface.
Co-reporter:Eric L. Spitler ; Brian T. Koo ; Jennifer L. Novotney ; John W. Colson ; Fernando J. Uribe-Romo ; Gregory D. Gutierrez ; Paulette Clancy
Journal of the American Chemical Society 2011 Volume 133(Issue 48) pp:19416-19421
Publication Date(Web):October 20, 2011
DOI:10.1021/ja206242v
Two-dimensional layered covalent organic frameworks (2D COFs) organize π-electron systems into ordered structures ideal for exciton and charge transport and exhibit permanent porosity available for subsequent functionalization. A 2D COF with the largest pores reported to date was synthesized by condensing 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) and 4,4′-diphenylbutadiynebis(boronic acid) (DPB). The COF was prepared as both a high surface area microcrystalline powder as well as a vertically oriented thin film on a transparent single-layer graphene/fused silica substrate. Complementary molecular dynamics and density functional theory calculations provide insight into the interlayer spacing of the COF and suggest that adjacent layers are horizontally offset by 1.7–1.8 Å, in contrast to the eclipsed AA stacking typically proposed for these materials.
Co-reporter:Eric L. Spitler, Marissa R. Giovino, Sarah L. White and William R. Dichtel
Chemical Science 2011 vol. 2(Issue 8) pp:1588-1593
Publication Date(Web):09 Jun 2011
DOI:10.1039/C1SC00260K
Three boronate ester-linked covalent organic frameworks (COFs) were synthesized using a new approach that employs polyfunctional boronic acid and acetonide-protected catechol reactants in the presence of the Lewis acid catalyst BF3·OEt2. This transformation avoids the use of unstable and insoluble polyfunctional catechols. The COF-5 and COF-10 hexagonal lattices were obtained from a triphenylene tris(acetonide) and the appropriate diboronic acid linker, whereas a square Ni phthalocyanine COF was prepared from the appropriate Ni phthalocyanine tetra(acetonide). The powder X-ray diffraction, infrared spectra, and measured surface areas of these materials matched or exceeded previously reported values. A mechanistic study of this transformation revealed that the dehydrative trimerization of boronic acids to boroxines and the formation of a nonproductive aryl boronic acid–BF3 complex strongly affect the rate of boronate ester formation. Crossover experiments employing substituted boronate ester derivatives suggest that esterhydrolysis is the most likely exchange mechanism during COF formation under BF3·OEt2-catalyzed conditions.
Co-reporter:John W. Colson;Arthur R. Woll;Arnab Mukherjee;Mark P. Levendorf;Eric L. Spitler;Virgil B. Shields;Michael G. Spencer;Jiwoong Park
Science 2011 Volume 332(Issue 6026) pp:228-231
Publication Date(Web):08 Apr 2011
DOI:10.1126/science.1202747
Microporous covalent organic frameworks, which usually form as insoluble powders, grow as crystalline films on graphene.
Co-reporter:Brian J. Smith, Anna C. Overholts, Nicky Hwang and William R. Dichtel
Chemical Communications 2016 - vol. 52(Issue 18) pp:NaN3693-3693
Publication Date(Web):2016/02/01
DOI:10.1039/C5CC10221A
We explore the crystallization of a high surface area imine-linked two-dimensional covalent organic framework (2D COF). The growth process reveals rapid initial formation of an amorphous network that subsequently crystallizes into the layered 2D network. The metastable amorphous polymer may be isolated and resubjected to growth conditions to form the COF. These experiments provide the first mechanistic insight into the mechanism of imine-linked 2D COF formation, which is distinct from that of boronate-ester linked COFs.
Co-reporter:Brian J. Smith, Nicky Hwang, Anton D. Chavez, Jennifer L. Novotney and William R. Dichtel
Chemical Communications 2015 - vol. 51(Issue 35) pp:NaN7535-7535
Publication Date(Web):2015/03/19
DOI:10.1039/C5CC00379B
We examine the growth rates, activation energies, and hydrolytic stability of multiple 2D boronate ester covalent organic frameworks by turbidity measurements, observing a 200-fold range in stability. The rate-determining step in boronate ester 2D COF growth is not in-solution condensation, but rather interlayer polymer stacking through a nucleation–elongation process.
Co-reporter:David N. Bunck and William R. Dichtel
Chemical Communications 2013 - vol. 49(Issue 24) pp:NaN2459-2459
Publication Date(Web):2013/02/11
DOI:10.1039/C3CC40358K
We demonstrate a tandem truncation–functionalization strategy using the 3D covalent organic framework, COF-102. Allyl groups incorporated within the pore walls were subjected to thiol–ene coupling conditions, achieving high conversions and maintaining the crystallinity and permanent porosity of the parent framework.
Co-reporter:Eric L. Spitler, Marissa R. Giovino, Sarah L. White and William R. Dichtel
Chemical Science (2010-Present) 2011 - vol. 2(Issue 8) pp:NaN1593-1593
Publication Date(Web):2011/06/09
DOI:10.1039/C1SC00260K
Three boronate ester-linked covalent organic frameworks (COFs) were synthesized using a new approach that employs polyfunctional boronic acid and acetonide-protected catechol reactants in the presence of the Lewis acid catalyst BF3·OEt2. This transformation avoids the use of unstable and insoluble polyfunctional catechols. The COF-5 and COF-10 hexagonal lattices were obtained from a triphenylene tris(acetonide) and the appropriate diboronic acid linker, whereas a square Ni phthalocyanine COF was prepared from the appropriate Ni phthalocyanine tetra(acetonide). The powder X-ray diffraction, infrared spectra, and measured surface areas of these materials matched or exceeded previously reported values. A mechanistic study of this transformation revealed that the dehydrative trimerization of boronic acids to boroxines and the formation of a nonproductive aryl boronic acid–BF3 complex strongly affect the rate of boronate ester formation. Crossover experiments employing substituted boronate ester derivatives suggest that esterhydrolysis is the most likely exchange mechanism during COF formation under BF3·OEt2-catalyzed conditions.
Co-reporter:Hasan Arslan, Fernando J. Uribe-Romo, Brian J. Smith and William R. Dichtel
Chemical Science (2010-Present) 2013 - vol. 4(Issue 10) pp:NaN3978-3978
Publication Date(Web):2013/07/12
DOI:10.1039/C3SC51212F
A rapid and efficient approach to prepare extended or partially fused hexabenzocoronene derivatives is described. The method is based on the sequential benzannulation and cyclodehydrogenation (Scholl oxidation) of simple diaryl alkynes. The benzannulation reaction proceeds efficiently on highly congested substrates and with complete regioselectivity. Scholl oxidation of the resulting oligo(arylene)s proceeds without rearrangements and provides either fully fused or specific partially fused polycyclic aromatic hydrocarbon products. The partially fused derivatives are a new class of contorted aromatic systems with high solubility, enhanced visible absorption, and reversible redox processes. The efficiency and specificity of the benzannulation and oxidation reactions are promising for accessing new classes of organic semiconductors and carbon nanostructures.
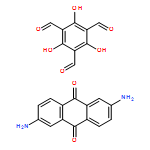
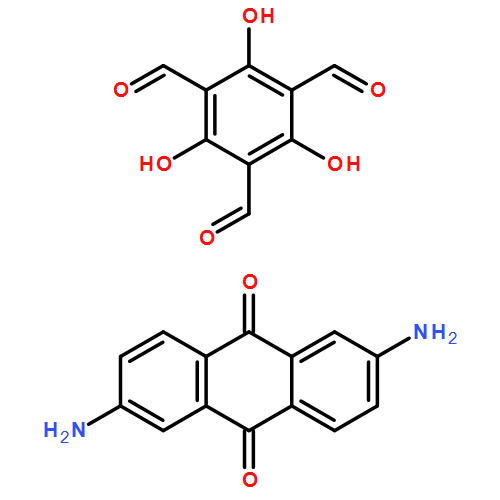
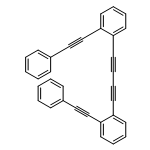
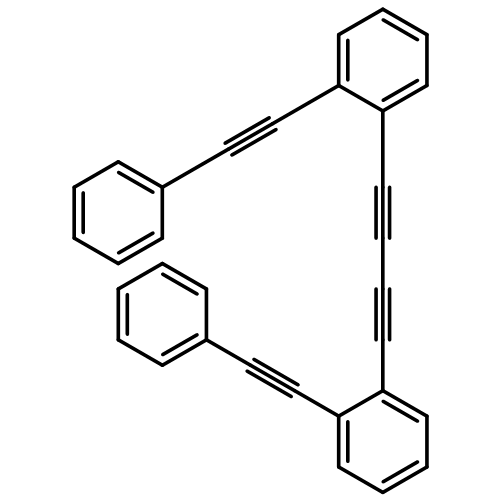
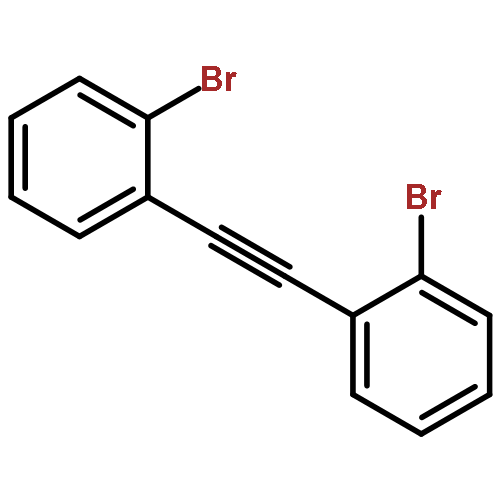
Co-reporter:Dan Lehnherr, Chen Chen, Zahra Pedramrazi, Catherine R. DeBlase, Joaquin M. Alzola, Ivan Keresztes, Emil B. Lobkovsky, Michael F. Crommie and William R. Dichtel
Chemical Science (2010-Present) 2016 - vol. 7(Issue 10) pp:NaN6364-6364
Publication Date(Web):2016/07/08
DOI:10.1039/C6SC02520J
A Cu-catalyzed benzannulation reaction transforms ortho(arylene ethynylene) oligomers into ortho-arylenes. This approach circumvents iterative Suzuki cross-coupling reactions previously used to assemble hindered ortho-arylene backbones. These derivatives form helical folded structures in the solid-state and in solution, as demonstrated by X-ray crystallography and solution-state NMR analysis. DFT calculations of misfolded conformations are correlated with variable-temperature 1H and EXSY NMR to reveal that folding is cooperative and more favorable in halide-substituted naphthalenes. Helical ortho-arylene foldamers with specific aromatic sequences organize functional π-electron systems into arrangements ideal for ambipolar charge transport and show preliminary promise for the surface-mediated synthesis of structurally defined graphene nanoribbons.
.jpg)








![Silane, [(4-methoxyphenyl)ethynyl]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/144400/144304-05-2.png)
![Silane, [(4-methoxyphenyl)ethynyl]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/144400/144304-05-2_b.png)
![1,3-DIOXAN-2-ONE, 5,5'-[OXYBIS(METHYLENE)]BIS[5-ETHYL-](http://img.cochemist.com/ccimg/84100/84056-46-2.png)
![1,3-DIOXAN-2-ONE, 5,5'-[OXYBIS(METHYLENE)]BIS[5-ETHYL-](http://img.cochemist.com/ccimg/84100/84056-46-2_b.png)
![Benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetrone, 2,7-bis(4-aminophenyl)-](/data/chemimg/3749600/49546-11-4.png)
![Benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetrone, 2,7-bis(4-aminophenyl)-](/data/chemimg/3749600/49546-11-4_b.png)


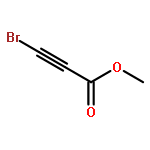
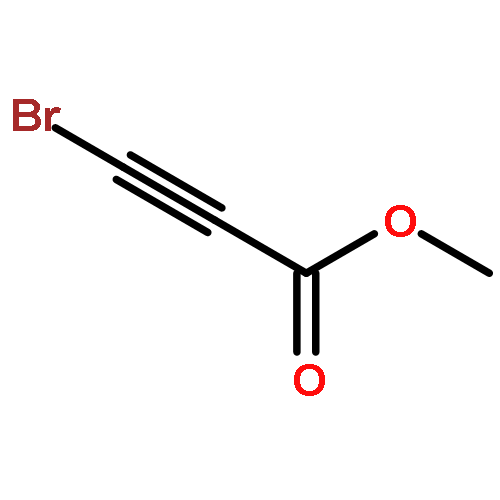
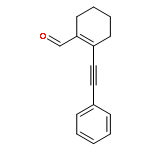
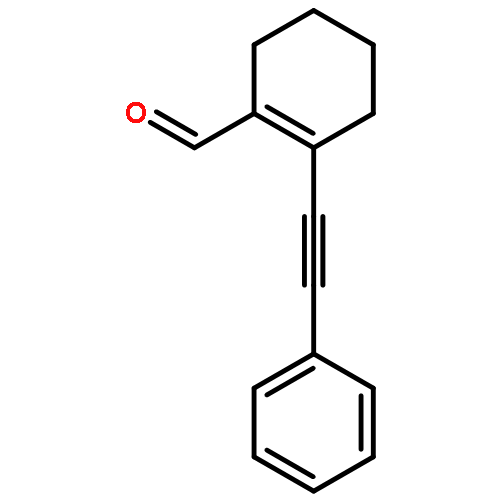









![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)