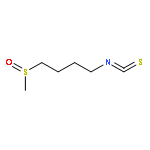
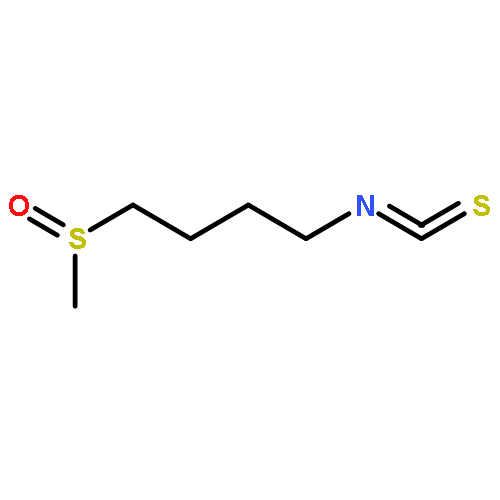
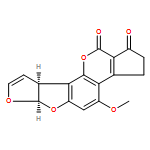
Co-reporter:Shiou-chi Chang, Uthpala I. Seneviratne, Jie Wu, Natalia Tretyakova, and John M. Essigmann
Chemical Research in Toxicology May 15, 2017 Volume 30(Issue 5) pp:1230-1230
Publication Date(Web):April 10, 2017
DOI:10.1021/acs.chemrestox.7b00064
The adverse effects of the human carcinogen 1,3-butadiene (BD) are believed to be mediated by its DNA-reactive metabolites such as 3,4-epoxybut-1-ene (EB) and 1,2,3,4-diepoxybutane (DEB). The specific DNA adducts responsible for toxic and mutagenic effects of BD, however, have yet to be identified. Recent in vitro polymerase bypass studies of BD-induced adenine (BD-dA) adducts show that DEB-induced N6,N6-DHB-dA (DHB = 2,3-dihydroxybutan-1,4-diyl) and 1,N6-γ-HMHP-dA (HMHP = 2-hydroxy-3-hydroxymethylpropan-1,3-diyl) adducts block replicative DNA polymerases but are bypassed by human polymerases η and κ, leading to point mutations and deletions. In contrast, EB-induced N6-HB-dA (HB = 2-hydroxy-3-buten-1-yl) does not block DNA synthesis and is nonmutagenic. In the present study, we employed a newly established in vivo lesion-induced mutagenesis/genotoxicity assay via next-generation sequencing to evaluate the in vivo biological consequences of S-N6-HB-dA, R,R-N6,N6-DHB-dA, S,S-N6,N6-DHB-dA, and R,S-1,N6-γ-HMHP-dA. In addition, the effects of AlkB-mediated direct reversal repair, MutM and MutY catalyzed base excision repair, and DinB translesion synthesis on the BD-dA adducts in bacterial cells were investigated. BD-dA adducts showed the expected inhibition of DNA replication in vivo but were not substantively mutagenic in any of the genetic environments investigated. This result is in contrast with previous in vitro observations and opens the possibility that E. coli repair and bypass systems other than the ones studied here are able to minimize the mutagenic properties of BD-dA adducts.
Co-reporter:Supawadee Chawanthayatham;Charles C. Valentine III;Bogdan I. Fedeles;Edward J. Fox;Lawrence A. Loeb;Stuart S. Levine;Stephen L. Slocum;Gerald N. Wogan;Robert G. Croy;John M. Essigmann
PNAS 2017 114 (15 ) pp:E3101-E3109
Publication Date(Web):2017-04-11
DOI:10.1073/pnas.1700759114
Aflatoxin B1 (AFB1) and/or hepatitis B and C viruses are risk factors for human hepatocellular carcinoma (HCC). Available evidence supports
the interpretation that formation of AFB1-DNA adducts in hepatocytes seeds a population of mutations, mainly G:C→T:A, and viral processes synergize to accelerate tumorigenesis,
perhaps via inflammation. Responding to a need for early-onset evidence predicting disease development, highly accurate duplex
sequencing was used to monitor acquisition of high-resolution mutational spectra (HRMS) during the process of hepatocarcinogenesis.
Four-day-old male mice were treated with AFB1 using a regimen that induced HCC within 72 wk. For analysis, livers were separated into tumor and adjacent cellular fractions.
HRMS of cells surrounding the tumors revealed predominantly G:C→T:A mutations characteristic of AFB1 exposure. Importantly, 25% of all mutations were G→T in one trinucleotide context (CGC; the underlined G is the position of the mutation), which is also a hotspot mutation in human liver tumors whose incidence
correlates with AFB1 exposure. The technology proved sufficiently sensitive that the same distinctive spectrum was detected as early as 10 wk
after dosing, well before evidence of neoplasia. Additionally, analysis of tumor tissue revealed a more complex pattern than
observed in surrounding hepatocytes; tumor HRMS were a composite of the 10-wk spectrum and a more heterogeneous set of mutations
that emerged during tumor outgrowth. We propose that the 10-wk HRMS reflects a short-term mutational response to AFB1, and, as such, is an early detection metric for AFB1-induced liver cancer in this mouse model that will be a useful tool to reconstruct the molecular etiology of human hepatocarcinogenesis.
Co-reporter:Bogdan I. Fedeles;Emily Yau;Vipender Singh;Bret D. Freudenthal;Deyu Li;James C. Delaney;Shiou-chi Chang;John M. Essigmann;Samuel H. Wilson
PNAS 2015 Volume 112 (Issue 33 ) pp:E4571-E4580
Publication Date(Web):2015-08-18
DOI:10.1073/pnas.1507709112
During chronic inflammation, neutrophil-secreted hypochlorous acid can damage nearby cells inducing the genomic accumulation
of 5-chlorocytosine (5ClC), a known inflammation biomarker. Although 5ClC has been shown to promote epigenetic changes, it
has been unknown heretofore if 5ClC directly perpetrates a mutagenic outcome within the cell. The present work shows that
5ClC is intrinsically mutagenic, both in vitro and, at a level of a single molecule per cell, in vivo. Using biochemical and
genetic approaches, we have quantified the mutagenic and toxic properties of 5ClC, showing that this lesion caused C→T transitions
at frequencies ranging from 3–9% depending on the polymerase traversing the lesion. X-ray crystallographic studies provided
a molecular basis for the mutagenicity of 5ClC; a snapshot of human polymerase β replicating across a primed 5ClC-containing
template uncovered 5ClC engaged in a nascent base pair with an incoming dATP analog. Accommodation of the chlorine substituent
in the template major groove enabled a unique interaction between 5ClC and the incoming dATP, which would facilitate mutagenic
lesion bypass. The type of mutation induced by 5ClC, the C→T transition, has been previously shown to occur in substantial
amounts both in tissues under inflammatory stress and in the genomes of many inflammation-associated cancers. In fact, many
sequence-specific mutational signatures uncovered in sequenced cancer genomes feature C→T mutations. Therefore, the mutagenic
ability of 5ClC documented in the present study may constitute a direct functional link between chronic inflammation and the
genetic changes that enable and promote malignant transformation.
Co-reporter:Vipender Singh, Chunte Sam Peng, Deyu Li, Koyel Mitra, Katherine J. Silvestre, Andrei Tokmakoff, and John M. Essigmann
ACS Chemical Biology 2014 Volume 9(Issue 1) pp:227
Publication Date(Web):October 15, 2013
DOI:10.1021/cb400581f
Structural diversification of canonical nucleic acid bases and nucleotide analogues by tautomerism has been proposed to be a powerful on/off switching mechanism allowing regulation of many biological processes mediated by RNA enzymes and aptamers. Despite the suspected biological importance of tautomerism, attempts to observe minor tautomeric forms in nucleic acid or hybrid nucleic acid-ligand complexes have met with challenges due to the lack of sensitive methods. Here, a combination of spectroscopic, biochemical, and computational tools probed tautomerism in the context of an RNA aptamer-ligand complex; studies involved a model ligand, oxythiamine pyrophosphate (OxyTPP), bound to the thiamine pyrophosphate (TPP) riboswitch (an RNA aptamer) as well as its unbound nonphosphorylated form, oxythiamine (OxyT). OxyTPP, similarly to canonical heteroaromatic nucleic acid bases, has a pyrimidine ring that forms hydrogen bonding interactions with the riboswitch. Tautomerism was established using two-dimensional infrared (2D IR) spectroscopy, variable temperature FTIR and NMR spectroscopies, binding isotope effects (BIEs), and computational methods. All three possible tautomers of OxyT, including the minor enol tautomer, were directly identified, and their distributions were quantitated. In the bound form, BIE data suggested that OxyTPP existed as a 4′-keto tautomer that was likely protonated at the N1′-position. These results also provide a mechanistic framework for understanding the activation of riboswitch in response to deamination of the active form of vitamin B1 (or TPP). The combination of methods reported here revealing the fine details of tautomerism can be applied to other systems where the importance of tautomerism is suspected.
Co-reporter:Vipender Singh, Bogdan I. Fedeles, Deyu Li, James C. Delaney, Ivan D. Kozekov, Albena Kozekova, Lawrence J. Marnett, Carmelo J. Rizzo, and John M. Essigmann
Chemical Research in Toxicology 2014 Volume 27(Issue 9) pp:1619
Publication Date(Web):August 15, 2014
DOI:10.1021/tx5002817
The structurally related exocyclic guanine adducts α-hydroxypropano-dG (α-OH-PdG), γ-hydroxypropano-dG (γ-OH-PdG), and M1dG are formed when DNA is exposed to the reactive aldehydes acrolein and malondialdehyde (MDA). These lesions are believed to form the basis for the observed cytotoxicity and mutagenicity of acrolein and MDA. In an effort to understand the enzymatic pathways and chemical mechanisms that are involved in the repair of acrolein- and MDA-induced DNA damage, we investigated the ability of the DNA repair enzyme AlkB, an α-ketoglutarate/Fe(II) dependent dioxygenase, to process α-OH-PdG, γ-OH-PdG, and M1dG in both single- and double-stranded DNA contexts. By monitoring the repair reactions using quadrupole time-of-flight (Q-TOF) mass spectrometry, it was established that AlkB can oxidatively dealkylate γ-OH-PdG most efficiently, followed by M1dG and α-OH-PdG. The AlkB repair mechanism involved multiple intermediates and complex, overlapping repair pathways. For example, the three exocyclic guanine adducts were shown to be in equilibrium with open-ring aldehydic forms, which were trapped using (pentafluorobenzyl)hydroxylamine (PFBHA) or NaBH4. AlkB repaired the trapped open-ring form of γ-OH-PdG but not the trapped open-ring of α-OH-PdG. Taken together, this study provides a detailed mechanism by which three-carbon bridge exocyclic guanine adducts can be processed by AlkB and suggests an important role for the AlkB family of dioxygenases in protecting against the deleterious biological consequences of acrolein and MDA.
Co-reporter:Deyu Li;Bogdan I. Fedeles;Vipender Singh;Chunte Sam Peng;Katherine J. Silvestre;Allison K. Simi;Jeffrey H. Simpson;Andrei Tokmakoff;John M. Essigmann
PNAS 2014 111 (32 ) pp:E3252-E3259
Publication Date(Web):2014-08-12
DOI:10.1073/pnas.1405635111
Viral lethal mutagenesis is a strategy whereby the innate immune system or mutagenic pool nucleotides increase the error rate
of viral replication above the error catastrophe limit. Lethal mutagenesis has been proposed as a mechanism for several antiviral
compounds, including the drug candidate 5-aza-5,6-dihydro-2′-deoxycytidine (KP1212), which causes A-to-G and G-to-A mutations
in the HIV genome, both in tissue culture and in HIV positive patients undergoing KP1212 monotherapy. This work explored the
molecular mechanism(s) underlying the mutagenicity of KP1212, and specifically whether tautomerism, a previously proposed
hypothesis, could explain the biological consequences of this nucleoside analog. Establishing tautomerism of nucleic acid
bases under physiological conditions has been challenging because of the lack of sensitive methods. This study investigated
tautomerism using an array of spectroscopic, theoretical, and chemical biology approaches. Variable temperature NMR and 2D
infrared spectroscopic methods demonstrated that KP1212 existed as a broad ensemble of interconverting tautomers, among which
enolic forms dominated. The mutagenic properties of KP1212 were determined empirically by in vitro and in vivo replication
of a single-stranded vector containing a single KP1212. It was found that KP1212 paired with both A (10%) and G (90%), which
is in accord with clinical observations. Moreover, this mutation frequency is sufficient for pushing a viral population over
its error catastrophe limit, as observed before in cell culture studies. Finally, a model is proposed that correlates the
mutagenicity of KP1212 with its tautomeric distribution in solution.
Co-reporter:Deyu Li, Bogdan I. Fedeles, Nidhi Shrivastav, James C. Delaney, Xuedong Yang, Cintyu Wong, Catherine L. Drennan, and John M. Essigmann
Chemical Research in Toxicology 2013 Volume 26(Issue 8) pp:1182
Publication Date(Web):June 17, 2013
DOI:10.1021/tx400096m
The AlkB enzyme is an Fe(II)- and α-ketoglutarate-dependent dioxygenase that repairs DNA alkyl lesions by a direct reversal of damage mechanism as part of the adaptive response in E. coli. The reported substrate scope of AlkB includes simple DNA alkyl adducts, such as 1-methyladenine, 3-methylcytosine, 3-ethylcytosine, 1-methylguanine, 3-methylthymine, and N6-methyladenine, as well as more complex DNA adducts, such as 1,N6-ethenoadenine, 3,N4-ethenocytosine, and 1,N6-ethanoadenine. Previous studies have revealed, in a piecemeal way, that AlkB has an impressive repertoire of substrates. The present study makes two additions to this list, showing that alkyl adducts on the N2 position of guanine and N4 position of cytosine are also substrates for AlkB. Using high resolution ESI-TOF mass spectrometry, we show that AlkB has the biochemical capability to repair in vitro N2-methylguanine, N2-ethylguanine, N2-furan-2-yl-methylguanine, N2-tetrahydrofuran-2-yl-methylguanine, and N4-methylcytosine in ssDNA but not in dsDNA. When viewed together with previous work, the experimental data herein demonstrate that AlkB is able to repair all simple N-alkyl adducts occurring at the Watson–Crick base pairing interface of the four DNA bases, confirming AlkB as a versatile gatekeeper of genomic integrity under alkylation stress.
Co-reporter:Deyu Li ; James C. Delaney ; Charlotte M. Page ; Xuedong Yang ; Alvin S. Chen ; Cintyu Wong ; Catherine L. Drennan ;John M. Essigmann
Journal of the American Chemical Society 2012 Volume 134(Issue 21) pp:8896-8901
Publication Date(Web):April 18, 2012
DOI:10.1021/ja3010094
The DNA and RNA repair protein AlkB removes alkyl groups from nucleic acids by a unique iron- and α-ketoglutarate-dependent oxidation strategy. When alkylated adenines are used as AlkB targets, earlier work suggests that the initial target of oxidation can be the alkyl carbon adjacent to N1. Such may be the case with ethano-adenine (EA), a DNA adduct formed by an important anticancer drug, BCNU, whereby an initial oxidation would occur at the carbon adjacent to N1. In a previous study, several intermediates were observed suggesting a pathway involving adduct restructuring to a form that would not hinder replication, which would match biological data showing that AlkB almost completely reverses EA toxicity in vivo. The present study uses more sensitive spectroscopic methodology to reveal the complete conversion of EA to adenine; the nature of observed additional putative intermediates indicates that AlkB conducts a second oxidation event in order to release the two-carbon unit completely. The second oxidation event occurs at the exocyclic carbon adjacent to the N6 atom of adenine. The observation of oxidation of a carbon at N6 in EA prompted us to evaluate N6-methyladenine (m6A), an important epigenetic signal for DNA replication and many other cellular processes, as an AlkB substrate in DNA. Here we show that m6A is indeed a substrate for AlkB and that it is converted to adenine via its 6-hydroxymethyl derivative. The observation that AlkB can demethylate m6A in vitro suggests a role for AlkB in regulation of important cellular functions in vivo.
Co-reporter:Eunsuk Kim, Peter T. Rye, John M. Essigmann, Robert G. Croy
Journal of Inorganic Biochemistry 2009 Volume 103(Issue 2) pp:256-261
Publication Date(Web):February 2009
DOI:10.1016/j.jinorgbio.2008.10.013
A strategy is described for the re-design of DNA damaging platinum(II) complexes to afford elevated toxicity towards cancer cells expressing the estrogen receptor (ER). Two platinum-based toxicants are described in which a DNA damaging warhead, [Pt(en)Cl2] (en, ethylenediamine), is tethered to either of two functional groups. The first agent, [6-(2-amino-ethylamino)-hexyl]-carbamic acid 2-[6-(7α-estra-1,3,5,(10)-triene)-hexylamino]-ethyl ester platinum(II) dichloride ((Est-en)PtCl2), terminates in a ligand for the ER. The second agent is a control compound lacking the steroid; this compound, N-[6-(2-amino-ethylamino)-hexyl]-benzamide platinum(II) dichloride ((Bz-en)PtCl2)), terminates in a benzamide moiety, which lacks affinity for the ER. Using a competitive binding assay, Est-en had 28% relative binding affinity (RBA) for the ER as compared to 17β-estradiol. After covalent binding to a synthetic DNA duplex 16-mer, the compound retained its affinity for the ER; specificity of the binding event was demonstrated by the ability of free 17β-estradiol as a competitor to disrupt the DNA adduct-ER complex. The (Est-en)PtCl2 compound showed higher toxicity against the ER positive ovarian cancer cell line CAOV3 than did the control compound. (Est-en)PtCl2 was also more toxic to the ER positive breast cancer line, MCF-7, than to an ER negative line, MDA-MB231.
Co-reporter:JamesC. Delaney Dr.;Jianmin Gao Dr.;Haibo Liu Dr.;Nidhi Shrivastav;JohnM. Essigmann Dr.;EricT. Kool Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 25) pp:4524-4527
Publication Date(Web):
DOI:10.1002/anie.200805683
Co-reporter:JamesC. Delaney Dr.;Jianmin Gao Dr.;Haibo Liu Dr.;Nidhi Shrivastav;JohnM. Essigmann Dr.;EricT. Kool Dr.
Angewandte Chemie 2009 Volume 121( Issue 25) pp:4594-4597
Publication Date(Web):
DOI:10.1002/ange.200805683
Co-reporter:James C. Delaney and John M. Essigmann
Chemical Research in Toxicology 2008 Volume 21(Issue 1) pp:232
Publication Date(Web):December 12, 2007
DOI:10.1021/tx700292a
The genome and its nucleotide precursor pool are under sustained attack by radiation, reactive oxygen and nitrogen species, chemical carcinogens, hydrolytic reactions, and certain drugs. As a result, a large and heterogeneous population of damaged nucleotides forms in all cells. Some of the lesions are repaired, but for those that remain, there can be serious biological consequences. For example, lesions that form in DNA can lead to altered gene expression, mutation, and death. This perspective examines systems developed over the past 20 years to study the biological properties of single DNA lesions.
Co-reporter:Sarah Delaney, James C. Delaney and John M. Essigmann
Chemical Research in Toxicology 2007 Volume 20(Issue 11) pp:1718
Publication Date(Web):October 18, 2007
DOI:10.1021/tx700273u
DNA-damaging agents usually produce a vast collection of lesions within the genome. Analysis of these lesions from the structural and biological viewpoints is often complicated by the reality that some of the lesions are chemically fragile, leading to an even larger set of secondary and tertiary products. In an effort to deconvolute complex DNA-damage spectra, a strategy is presented whereby an oligonucleotide containing a specific target for chemical reaction is allowed to react with a DNA-damaging agent. A large collection of HPLC-resolvable modified oligonucleotides is generated, and chromatographically distinct members of the set are then individually characterized using chemical, spectroscopic, biochemical, and genetic probes. The biological component of this “chemical–biological fingerprinting” tool is the use of polymerase bypass in vivo in cells having defined replication status and quantitative and qualitative patterns of lesion-directed mutagenesis, as key properties that complement physical analysis of modified DNA. This approach was applied to the complex product spectrum generated by peroxynitrite in the presence of CO2; peroxynitrite is a powerful oxidizing and nitrating agent generated as part of immune response. An oligonucleotide containing the primary oxidation product, 7,8-dihydro-8-oxoguanine (8-oxoGua), which is highly susceptible to further oxidation and/or nitration, was treated with peroxynitrite. Using mass spectrometry, coelution with authentic standards, sensitivity to piperidine, recognition and strand cleavage by the DNA repair enzyme MutM, and mutagenicity and genotoxicity in vivo, a matrix was created that defined the properties of the secondary DNA lesions formed when 3-morpholinosydnonimine (SIN-1) delivered a low, constant flux of peroxynitrite to an oligonucleotide containing 8-oxoGua. Two lesions were identified as the diastereomers of spiroiminodihydantoin (Sp), which had been observed previously in nucleoside-based experiments employing SIN-1. A third lesion, triazine, was tentatively identified. However, in addition to these lesions, a number of secondary lesions were generated that had chemical–biological fingerprints inconsistent with that of any known 8-oxoGua-derived lesion described to date. In vitro experiments showed that while some of these newly characterized secondary lesions were removed from DNA by MutM, others were in fact very poor substrates for this repair enzyme. These 8-oxoGua-derived lesions also showed varying degrees of sensitivity to piperidine. Furthermore, all of the secondary lesions observed in this work were potently mutagenic and genotoxic in Escherichia coli. Therefore, while 8-oxoGua itself is nontoxic and only mildly mutagenic in repair-proficient cells, peroxynitrite reveals the promutagenic potential and triggers the covert nature of this DNA lesion.
Co-reporter:James C. Delaney;Catherine L. Drennan;Lauren E. Frick;Cintyu Wong;John M. Essigmann
PNAS 2007 Volume 104 (Issue 3 ) pp:755-760
Publication Date(Web):2007-01-16
DOI:10.1073/pnas.0607377104
1,N6-ethanoadenine (EA) forms through the reaction of adenine in DNA with the antitumor agent 1,3-bis(2-chloroethyl)-1-nitrosourea,
a chemotherapeutic used to combat various brain, head, and neck tumors. Previous studies of the toxic and mutagenic properties
of the DNA adduct EA have been limited to in vitro experiments using mammalian polymerases and have revealed the lesion to be both miscoding and genotoxic. This work explores
lesion bypass and mutagenicity of EA replicated in vivo and demonstrates that EA is neither toxic nor mutagenic in wild-type Escherichia coli. Although the base excision repair glycosylase enzymes of both humans and E. coli possess a weak ability to act on the lesion in vitro, an in vivo repair pathway has not yet been demonstrated. Here we show that an enzyme mechanistically unrelated to DNA glycosylases,
the adaptive response protein AlkB, is capable of acting on EA via its canonical mechanism of oxidative dealkylation. The
reaction alleviates the unrepaired adduct's potent toxicity through metabolism at the C8 position (attached to N1 of adenine),
producing a nontoxic and weakly mutagenic N6 adduct. AlkB is shown here to be a geno-protective agent that reduces the toxicity of DNA damage by converting the primary
adduct to a less toxic secondary product.
Co-reporter:Tae Woo Kim;James C. Delaney;John M. Essigmann;Eric T. Kool;
Proceedings of the National Academy of Sciences 2005 102(44) pp:15803-15808
Publication Date(Web):October 25, 2005
DOI:10.1073/pnas.0505113102
We describe the use of a series of gradually expanded thymine nucleobase analogs in probing steric effects in DNA polymerase
efficiency and fidelity. In these nonpolar compounds, the base size was increased incrementally over a 1.0-Å range by use
of variably sized atoms (H, F, Cl, Br, and I) to replace the oxygen molecules of thymine. Kinetics studies with DNA Pol I
(Klenow fragment, exonuclease-deficient) in vitro showed that replication efficiency opposite adenine increased through the series, reaching a peak at the chlorinated compound.
Efficiency then dropped markedly as a steric tightness limit was apparently reached. Importantly, fidelity also followed this
trend, with the fidelity maximum at dichlorotoluene, the largest compound that fits without apparent repulsion. The fidelity
at this point approached that of wild-type thymine. Surprisingly, the maximum fidelity and efficiency was found at a base
pair size significantly larger than the natural size. Parallel bypass and mutagenesis experiments were then carried out in vivo with a bacterial assay for replication. The cellular results were virtually the same as those seen in solution. The results
provide direct evidence for the importance of a tight steric fit on DNA replication fidelity. In addition, the results suggest
that even high-fidelity replicative enzymes have more steric room than necessary, possibly to allow for an evolutionarily
advantageous mutation rate.
Co-reporter:James C. Delaney;John M. Essigmann
PNAS 2004 Volume 101 (Issue 39 ) pp:14051-14056
Publication Date(Web):2004-09-28
DOI:10.1073/pnas.0403489101
AlkB repairs 1-alkyladenine and 3-methylcytosine lesions in DNA by directly reversing the base damage. Although repair studies
with randomly alkylated substrates have been performed, the miscoding nature of these and related individually alkylated bases
and the suppression of mutagenesis by AlkB within cells have not yet been explored. Here, we address the miscoding potential
of 1-methyldeoxyadenosine (m1A), 3-methyldeoxycytidine (m3C), 3-ethyldeoxycytidine (e3C), 1-methyldeoxyguanosine (m1G), and
3-methyldeoxythymidine (m3T) by synthesizing single-stranded vectors containing each alkylated base, followed by vector passage
through Escherichia coli. In SOS–, AlkB-deficient cells, m1A was only 1% mutagenic; however, m3C and e3C were 30% mutagenic, rising to 70% in SOS+ cells. In contrast, the mutagenicity of m1G and m3T in AlkB– cells dropped slightly when SOS polymerases were expressed (m1G from 80% to 66% and m3T from 60% to 53%). Mutagenicity was
abrogated for m1A, m3C, and e3C in wild-type (AlkB+) cells, whereas m3T mutagenicity was only partially reduced. Remarkably, m1G mutagenicity was also eliminated in AlkB+ cells, establishing it as a natural AlkB substrate. All lesions were blocks to replication in AlkB-deficient cells. The m1A,
m3C, and e3C blockades were completely removed in wild-type cells; the m1G blockade was partially removed and that for m3T
was unaffected by the presence of AlkB. All lesions demonstrated enhanced bypass when SOS polymerases were induced. This work
provides direct evidence that AlkB suppresses both genotoxicity and mutagenesis by physiologically realistic low doses of
1-alkylpurine and 3-alkylpyrimidine DNA damage in vivo.
Co-reporter:James C. Delaney;Paul T. Henderson;Sandra A. Helquist;Juan C. Morales;John M. Essigmann;Eric T. Kool
PNAS 2003 100 (8 ) pp:4469-4473
Publication Date(Web):2003-04-15
DOI:10.1073/pnas.0837277100
We report studies testing the importance of Watson–Crick hydrogen bonding, base-pair geometry, and steric effects during DNA
replication in living bacterial cells. Nonpolar DNA base shape mimics of thymine and adenine (abbreviated F and Q, respectively)
were introduced into Escherichia coli by insertion into a phage genome followed by transfection of the vector into bacteria. Genetic assays showed that these two
base mimics were bypassed with moderate to high efficiency in the cells and with very high efficiency under damage-response
(SOS induction) conditions. Under both sets of conditions, the T-shape mimic (F) encoded genetic information in the bacteria
as if it were thymine, directing incorporation of adenine opposite it with high fidelity. Similarly, the A mimic (Q) directed
incorporation of thymine opposite itself with high fidelity. The data establish that Watson–Crick hydrogen bonding is not
necessary for high-fidelity replication of a base pair in vivo. The results suggest that recognition of DNA base shape alone serves as the most powerful determinant of fidelity during
transfer of genetic information in a living organism.
Co-reporter:Maryann E. Smela;Michelle L. Hamm;Paul T. Henderson;Constance M. Harris;Thomas M. Harris;John M. Essigmann;
Proceedings of the National Academy of Sciences 2002 99(10) pp:6655-6660
Publication Date(Web):2002-05-14
DOI:10.1073/pnas.102167699
A G to T mutation has been observed at the third position of codon 249 of the p53 tumor-suppressor gene in over 50% of the
hepatocellular carcinoma cases associated with high exposure to aflatoxin B1 (AFB1). Hypotheses have been put forth that AFB1, in concert with hepatitis B virus (HBV), may play a role in the formation of, and/or the selection for, this mutation. The
primary DNA adduct of AFB1 is 8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 (AFB1-N7-Gua), which is converted naturally to two secondary lesions, an apurinic site and an AFB1-formamidopyrimidine (AFB1-FAPY) adduct. AFB1-FAPY is detected at near maximal levels in rat DNA days to weeks after AFB1 exposure, underscoring its high persistence in vivo. The present study reveals two striking properties of this DNA adduct: (i) AFB1-FAPY was found to cause a G to T mutation frequency in Escherichia coli approximately 6 times higher than that of AFB1-N7-Gua, and (ii) one proposed rotamer of AFB1-FAPY is a block to replication, even when the efficient bypass polymerase MucAB is used by the cell. Taken together, these
characteristics make the FAPY adduct the prime candidate for both the genotoxicity of aflatoxin, because mammalian cells also
have similar bypass mechanisms for combating DNA damage, and the mutagenicity that ultimately may lead to liver cancer.
Co-reporter:Nirachara Techapiesancharoenkij, Jeannette L.A. Fiala, Panida Navasumrit, Robert G. Croy, Gerald N. Wogan, John D. Groopman, Mathuros Ruchirawat, John M. Essigmann
Toxicology and Applied Pharmacology (1 January 2015) Volume 282(Issue 1) pp:52-60
Publication Date(Web):1 January 2015
DOI:10.1016/j.taap.2014.11.004
•This study revealed sulforaphane (SF)-deregulated gene sets in aflatoxin B1 (AFB1)-treated rat livers.•SF redirects biochemical networks toward lipid biosynthesis in AFB1-dosed rats.•SF enhanced gene sets that would be expected to favor cell repair and regeneration.Aflatoxin B1 (AFB1) is one of the major risk factors for liver cancer globally. A recent study showed that sulforaphane (SF), a potent inducer of phase II enzymes that occurs naturally in widely consumed vegetables, effectively induces hepatic glutathione S-transferases (GSTs) and reduces levels of hepatic AFB1-DNA adducts in AFB1-exposed Sprague Dawley rats. The present study characterized the effects of SF pre-treatment on global gene expression in the livers of similarly treated male rats. Combined treatment with AFB1 and SF caused reprogramming of a network of genes involved in signal transduction and transcription. Changes in gene regulation were observable 4 h after AFB1 administration in SF-pretreated animals and may reflect regeneration of cells in the wake of AFB1-induced hepatotoxicity. At 24 h after AFB1 administration, significant induction of genes that play roles in cellular lipid metabolism and acetyl-CoA biosynthesis was detected in SF-pretreated AFB1-dosed rats. Induction of this group of genes may indicate a metabolic shift toward glycolysis and fatty acid synthesis to generate and maintain pools of intermediate molecules required for tissue repair, cell growth and compensatory hepatic cell proliferation. Collectively, gene expression data from this study provide insights into molecular mechanisms underlying the protective effects of SF against AFB1 hepatotoxicity and hepatocarcinogenicity, in addition to the chemopreventive activity of this compound as a GST inducer.
.jpg)
![2-[6-[(11s,13s,17s)-17-hydroxy-13-methyl-3-oxo-2,6,7,8,11,12,14,15,16,17-decahydro-1h-cyclopenta[a]phenanthren-11-yl]hexylamino]ethyl N-[3-[4-[bis(2-chloroethyl)amino]phenyl]propyl]carbamate](http://img.cochemist.com/ccimg/865100/865070-37-7.png)
![2-[6-[(11s,13s,17s)-17-hydroxy-13-methyl-3-oxo-2,6,7,8,11,12,14,15,16,17-decahydro-1h-cyclopenta[a]phenanthren-11-yl]hexylamino]ethyl N-[3-[4-[bis(2-chloroethyl)amino]phenyl]propyl]carbamate](http://img.cochemist.com/ccimg/865100/865070-37-7_b.png)
![(6aS,8R,9R,9aR)-8-(2-amino-6-oxo-3,6-dihydro-9H-purin-9-yl)-9-hydroxy-4-methoxy-2,3,6a,8,9,9a-hexahydrocyclopenta[c]furo[3',2':4,5]furo[2,3-h]chromene-1,11-dione](http://img.cochemist.com/ccimg/80000/79982-94-8.png)
![(6aS,8R,9R,9aR)-8-(2-amino-6-oxo-3,6-dihydro-9H-purin-9-yl)-9-hydroxy-4-methoxy-2,3,6a,8,9,9a-hexahydrocyclopenta[c]furo[3',2':4,5]furo[2,3-h]chromene-1,11-dione](http://img.cochemist.com/ccimg/80000/79982-94-8_b.png)





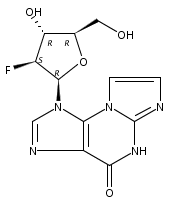
![1H-Imidazo[2,1-b]purin-4(5H)-one](http://img.cochemist.com/ccimg/63000/62962-42-9.png)
![1H-Imidazo[2,1-b]purin-4(5H)-one](http://img.cochemist.com/ccimg/63000/62962-42-9_b.png)
![1,4-DIHYDROIMIDAZO[1,2-A]PURIN-9-ONE](http://img.cochemist.com/ccimg/56300/56287-13-9.png)
![1,4-DIHYDROIMIDAZO[1,2-A]PURIN-9-ONE](http://img.cochemist.com/ccimg/56300/56287-13-9_b.png)
![1H-Imidazo[2,1-i]purine, 7,8-dihydro-](http://img.cochemist.com/ccimg/773800/773787-37-4.png)
![1H-Imidazo[2,1-i]purine, 7,8-dihydro-](http://img.cochemist.com/ccimg/773800/773787-37-4_b.png)
![Formamide,N-(2,6-diamino-1,4-dihydro-4-oxo-5-pyrimidinyl)-N-[(6aS,8R,9R,9aR)-1,2,3,6a,8,9,9a,11-octahydro-9-hydroxy-4-methoxy-1,11-dioxocyclopenta[c]furo[3',2':4,5]furo[2,3-h][1]benzopyran-8-yl]-](http://img.cochemist.com/ccimg/65400/65386-83-6.png)
![Formamide,N-(2,6-diamino-1,4-dihydro-4-oxo-5-pyrimidinyl)-N-[(6aS,8R,9R,9aR)-1,2,3,6a,8,9,9a,11-octahydro-9-hydroxy-4-methoxy-1,11-dioxocyclopenta[c]furo[3',2':4,5]furo[2,3-h][1]benzopyran-8-yl]-](http://img.cochemist.com/ccimg/65400/65386-83-6_b.png)











![Imidazo[1,2-c]pyrimidin-5(6H)-one](http://img.cochemist.com/ccimg/55700/55662-66-3.png)
![Imidazo[1,2-c]pyrimidin-5(6H)-one](http://img.cochemist.com/ccimg/55700/55662-66-3_b.png)
![3H-Imidazo[2,1-i]purine](http://img.cochemist.com/ccimg/13900/13875-63-3.png)
![3H-Imidazo[2,1-i]purine](http://img.cochemist.com/ccimg/13900/13875-63-3_b.png)






![Thiazolium,3-[(3,4-dihydro-2-methyl-4-oxo-5-pyrimidinyl)methyl]-5-(2-hydroxyethyl)-4-methyl-](http://img.cochemist.com/ccimg/200/136-16-3.png)
![Thiazolium,3-[(3,4-dihydro-2-methyl-4-oxo-5-pyrimidinyl)methyl]-5-(2-hydroxyethyl)-4-methyl-](http://img.cochemist.com/ccimg/200/136-16-3_b.png)