Co-reporter:Eric G. Fuemmeler, Samuel N. Sanders, Andrew B. Pun, Elango Kumarasamy, Tao Zeng, Kiyoshi Miyata, Michael L. Steigerwald, X.-Y. Zhu, Matthew Y. Sfeir, Luis M. Campos, and Nandini Ananth
ACS Central Science 2016 Volume 2(Issue 5) pp:316
Publication Date(Web):May 5, 2016
DOI:10.1021/acscentsci.6b00063
Interest in materials that undergo singlet fission (SF) has been catalyzed by the potential to exceed the Shockley–Queisser limit of solar power conversion efficiency. In conventional materials, the mechanism of SF is an intermolecular process (xSF), which is mediated by charge transfer (CT) states and depends sensitively on crystal packing or molecular collisions. In contrast, recently reported covalently coupled pentacenes yield ∼2 triplets per photon absorbed in individual molecules: the hallmark of intramolecular singlet fission (iSF). However, the mechanism of iSF is unclear. Here, using multireference electronic structure calculations and transient absorption spectroscopy, we establish that iSF can occur via a direct coupling mechanism that is independent of CT states. We show that a near-degeneracy in electronic state energies induced by vibronic coupling to intramolecular modes of the covalent dimer allows for strong mixing between the correlated triplet pair state and the local excitonic state, despite weak direct coupling.
Co-reporter:Jessica R. Duke
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 21) pp:4219-4223
Publication Date(Web):October 1, 2015
DOI:10.1021/acs.jpclett.5b01957
Mapping variable ring polymer molecular dynamics (MV-RPMD) is an approximate quantum dynamics method based on imaginary-time path integrals for simulating electronically nonadiabatic photochemical processes. By employing a mapping protocol to transform from a discrete electronic state basis to continuous Cartesian phase-space variables, the method captures electronic state transitions coupled to nuclear motion using only classical MD trajectories. In this work, we extend the applicability of MV-RPMD to simulations of photoinduced excited electronic state dynamics in nonadiabatic systems with multiple avoided crossings. We achieve this by deriving a new electronic state population estimator in the phase space of electronic variables that is exact at equilibrium and numerically accurate in real time. Further, we introduce an efficient constraint protocol to initialize an MV-RPMD simulation to a particular electronic state. We numerically demonstrate the accuracy of this estimator and constraint technique in describing electronic state dynamics from an initial nonequilibrium state in six model systems, three of which describe photodissociation.
Co-reporter:Tao Zeng ; Roald Hoffmann
Journal of the American Chemical Society 2014 Volume 136(Issue 15) pp:5755-5764
Publication Date(Web):March 16, 2014
DOI:10.1021/ja500887a
We present a detailed study of pentacene monomer and dimer that serves to reconcile extant views of its singlet fission. We obtain the correct ordering of singlet excited-state energy levels in a pentacene molecule (E (S1) < E (D)) from multireference calculations with an appropriate active orbital space and dynamical correlation being incorporated. In order to understand the mechanism of singlet fission in pentacene, we use a well-developed diabatization scheme to characterize the six low-lying singlet states of a pentacene dimer that approximates the unit cell structure of crystalline pentacene. The local, single-excitonic diabats are not directly coupled with the important multiexcitonic state but rather mix through their mutual couplings with one of the charge-transfer configurations. We analyze the mixing of diabats as a function of monomer separation and pentacene rotation. By defining an oscillator strength measure of the coherent population of the multiexcitonic diabat, essential to singlet fission, we find this population can, in principle, be increased by small compression along a specific crystal direction.
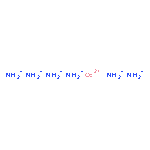
Co-reporter:Rachel L. Kenion and Nandini Ananth
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 37) pp:NaN26124-26124
Publication Date(Web):2016/08/31
DOI:10.1039/C6CP04882J
We present an atomistic simulation of the cobalt hexammine(II/III) self-exchange reaction using path integral (PI) methods. We construct a simple force field for the system in its reactant state that includes parameters for both atom–atom interactions, and interactions with an explicit transferring electron represented in the PI framework. We then calculate the outer sphere free energy barrier due to solvent reorganization from a PI molecular dynamics simulation and we obtain the dynamic transmission coefficient using ring polymer molecular dynamics. Combining these calculated values with literature values for the inner sphere reorganization energy, we obtain a reaction rate in good agreement with experimental measurements. The protocol introduced here circumvents the need for complex, system-specific force field parameterization along an assumed reaction coordinate making it sufficiently accurate, efficient, and broadly applicable to the study of both adiabatic and nonadiabatic charge transfer reactions in transition metal complexes.

![2,2'-Bipentacene, 6,6',13,13'-tetrakis[2-[tris(1-methylethyl)silyl]ethynyl]-](http://img.cochemist.com/ccimg/916000/915978-21-1.png)
![2,2'-Bipentacene, 6,6',13,13'-tetrakis[2-[tris(1-methylethyl)silyl]ethynyl]-](http://img.cochemist.com/ccimg/916000/915978-21-1_b.png)