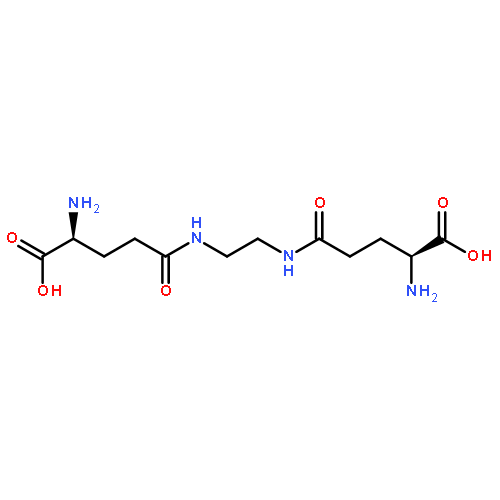
Co-reporter:Matthew J. Langton, Lorel M. Scriven, Nicholas H. Williams, and Christopher A. Hunter
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:15768-15768
Publication Date(Web):September 6, 2017
DOI:10.1021/jacs.7b07747
The on-demand delivery of drug molecules from nanoscale carriers with spatiotemporal control is a key challenge in modern medicine. Here we show that lipid bilayer vesicles (liposomes) can be triggered to release an encapsulated molecular cargo in response to an external control signal by employing an artificial transmembrane signal transduction mechanism. A synthetic signal transducer embedded in the lipid bilayer membrane acts as a switchable catalyst, catalyzing the formation of surfactant molecules inside the vesicle in response to a change in external pH. The surfactant permeabilizes the lipid bilayer membrane to facilitate release of an encapsulated hydrophilic cargo. In the absence of the pH control signal, the catalyst is inactive, and the cargo remains encapsulated within the vesicle.
Co-reporter:Yuan Chu, Nicholas H. Williams, and Alvan C. Hengge
Biochemistry August 1, 2017 Volume 56(Issue 30) pp:3923-3923
Publication Date(Web):July 5, 2017
DOI:10.1021/acs.biochem.7b00441
Catalytically promiscuous enzymes are an attractive frontier for biochemistry, because enzyme promiscuities not only plausibly explain enzyme evolution through the mechanism of gene duplication but also could provide an efficient route to changing the catalytic function of proteins by mimicking this evolutionary process. PP1γ is an effectively promiscuous phosphatase for the hydrolysis of both monoanionic and dianionic phosphate ester-based substrates. In addition to its native phosphate monoester substrate, PP1γ catalyzes the hydrolysis of aryl methylphosphonates, fluorophosphate esters, phosphorothioate esters, and phosphodiesters, with second-order rate accelerations that fall within the narrow range of 1011–1013. In contrast to the different transition states in the uncatalyzed hydrolysis reactions of these substrates, PP1γ catalyzes their hydrolysis through similar transition states. PP1γ does not catalyze the hydrolysis of a sulfate ester, which is unexpected. The PP1γ active site is tolerant of variations in the geometry of bound ligands, which permit the effective catalysis even of substrates whose steric requirements may result in perturbations to the positioning of the transferring group, both in the initial enzyme–substrate complex and in the transition state. The conservative mutation of arginine 221 to lysine results in a mutant that is a more effective catalyst toward monoanionic substrates. The surprising conversion of substrate preference lends support to the notion that mutations following gene duplication can result in an altered enzyme with different catalytic capabilities and preferences and may provide a pathway for the evolution of new enzymes.
Co-reporter:Anna Pabis;Nicholas H. Williams;Shina C. L. Kamerlin
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 35) pp:7308-7316
Publication Date(Web):2017/09/13
DOI:10.1039/C7OB01734K
Phosphoryl transfer reactions can proceed through several plausible mechanisms, and the potential for both solvent and substrate-assisted pathways (involving proton transfer to the phosphoryl oxygens) complicates both experimental and computational interpretations. To avoid this problem, we have used electronic structure calculations to probe the mechanisms of the reactions of pyridinio-N-phosphonates with pyridine. These compounds avoid the additional complexity introduced by proton transfer between the nucleophile and the leaving group, while also serving as a valuable model for biological P–N cleavage. Through a comparative study of a range of substrates of varying basicity, we demonstrate a unified concerted mechanism for the phosphoryl transfer reactions of these model compounds, proceeding through a dissociative transition state. Finally, a comparison of these transition states with previously characterized transition states for related compounds provides a more complete model for non-enzymatic phosphoryl transfer, which is a critical stepping stone to being able to fully understand phosphoryl transfer in biology.
Co-reporter:Fernanda Duarte, Alexandre Barrozo, Johan Åqvist, Nicholas H. Williams, and Shina C. L. Kamerlin
Journal of the American Chemical Society 2016 Volume 138(Issue 33) pp:10664-10673
Publication Date(Web):July 29, 2016
DOI:10.1021/jacs.6b06277
Despite the numerous experimental and theoretical studies on phosphate monoester hydrolysis, significant questions remain concerning the mechanistic details of these biologically critical reactions. In the present work we construct a linear free energy relationship for phosphate monoester hydrolysis to explore the effect of modulating leaving group pKa on the competition between solvent- and substrate-assisted pathways for the hydrolysis of these compounds. Through detailed comparative electronic-structure studies of methyl phosphate and a series of substituted aryl phosphate monoesters, we demonstrate that the preferred mechanism is dependent on the nature of the leaving group. For good leaving groups, a strong preference is observed for a more dissociative solvent-assisted pathway. However, the energy difference between the two pathways gradually reduces as the leaving group pKa increases and creates mechanistic ambiguity for reactions involving relatively poor alkoxy leaving groups. Our calculations show that the transition-state structures vary smoothly across the range of pKas studied and that the pathways remain discrete mechanistic alternatives. Therefore, while not impossible, a biological catalyst would have to surmount a significantly higher activation barrier to facilitate a substrate-assisted pathway than for the solvent-assisted pathway when phosphate is bonded to good leaving groups. For poor leaving groups, this intrinsic preference disappears.
Co-reporter:Emmanuel Y. Tirel ; Nicholas H. Williams
Chemistry - A European Journal 2015 Volume 21( Issue 19) pp:7053-7056
Publication Date(Web):
DOI:10.1002/chem.201500619
Abstract
Metal-ion complexes are the most effective artificial catalysts capable of cleaving phosphate diesters under mild aqueous conditions. A central strategy for making these complexes highly reactive has been to use ligand-based alcohols that are coordinated to the ion, providing an ionised nucleophile under neutral conditions but at the expense of deactivating it. We have created a highly reactive Zn complex that is 350-fold more reactive than an alcohol analogue by preventing the nucleophile binding to the metal ion. This strategy successfully delivers the benefits of efficient nucleophile delivery without strongly deactivating the metal ion Lewis acidity nor the oxyanion nucleophilicity. Varying the leaving group reveals that the transition state of the reaction is much further advanced than the reaction with hydroxide.
Co-reporter:Fernanda Duarte; Johan Åqvist; Nicholas H. Williams;Shina C. L. Kamerlin
Journal of the American Chemical Society 2014 Volume 137(Issue 3) pp:1081-1093
Publication Date(Web):November 7, 2014
DOI:10.1021/ja5082712
Understanding phosphoryl and sulfuryl transfer is central to many biochemical processes. However, despite decades of experimental and computational studies, a consensus concerning the precise mechanistic details of these reactions has yet to be reached. In this work we perform a detailed comparative theoretical study of the hydrolysis of p-nitrophenyl phosphate, methyl phosphate and p-nitrophenyl sulfate, all of which have served as key model systems for understanding phosphoryl and sulfuryl transfer reactions, respectively. We demonstrate the existence of energetically similar but mechanistically distinct possibilities for phosphate monoester hydrolysis. The calculated kinetic isotope effects for p-nitrophenyl phosphate provide a means to discriminate between substrate- and solvent-assisted pathways of phosphate monoester hydrolysis, and show that the solvent-assisted pathway dominates in solution. This preferred mechanism for p-nitrophenyl phosphate hydrolysis is difficult to find computationally due to the limitations of compressing multiple bonding changes onto a 2-dimensional energy surface. This problem is compounded by the need to include implicit solvation to at least microsolvate the system and stabilize the highly charged species. In contrast, methyl phosphate hydrolysis shows a preference for a substrate-assisted mechanism. For p-nitrophenyl sulfate hydrolysis there is only one viable reaction pathway, which is similar to the solvent-assisted pathway for phosphate hydrolysis, and the substrate-assisted pathway is not accessible. Overall, our results provide a unifying mechanistic framework that is consistent with the experimentally measured kinetic isotope effects and reconciles the discrepancies between theoretical and experimental models for these biochemically ubiquitous classes of reaction.
Co-reporter:Emmanuel Y. Tirel;Zoë Bellamy;Harry Adams;Vincent Lebrun;Ferna Duarte; Nicholas H. Williams
Angewandte Chemie International Edition 2014 Volume 53( Issue 31) pp:8246-8250
Publication Date(Web):
DOI:10.1002/anie.201400335
Abstract
Creating efficient artificial catalysts that can compete with biocatalysis has been an enduring challenge which has yet to be met. Reported herein is the synthesis and characterization of a series of zinc complexes designed to catalyze the hydrolysis of phosphate diesters. By introducing a hydrated aldehyde into the ligand we achieve turnover for DNA-like substrates which, combined with ligand methylation, increases reactivity by two orders of magnitude. In contrast to current orthodoxy and mechanistic explanations, we propose a mechanism where the nucleophile is not coordinated to the metal ion, but involves a tautomer with a more effective Lewis acid and more reactive nucleophile. This data suggests a new strategy for creating more efficient metal ion based catalysts, and highlights a possible mode of action for metalloenzymes.
Co-reporter:Fernanda Duarte, Ting Geng, Gaël Marloie, Adel O. Al Hussain, Nicholas H. Williams, and Shina Caroline Lynn Kamerlin
The Journal of Organic Chemistry 2014 Volume 79(Issue 7) pp:2816-2828
Publication Date(Web):November 26, 2013
DOI:10.1021/jo402420t
Sulfonate ester hydrolysis has been the subject of recent debate, with experimental evidence interpreted in terms of both stepwise and concerted mechanisms. In particular, a recent study of the alkaline hydrolysis of a series of benzene arylsulfonates (Babtie et al., Org. Biomol. Chem. 10, 2012, 8095) presented a nonlinear Brønsted plot, which was explained in terms of a change from a stepwise mechanism involving a pentavalent intermediate for poorer leaving groups to a fully concerted mechanism for good leaving groups and supported by a theoretical study. In the present work, we have performed a detailed computational study of the hydrolysis of these compounds and find no computational evidence for a thermodynamically stable intermediate for any of these compounds. Additionally, we have extended the experimental data to include pyridine-3-yl benzene sulfonate and its N-oxide and N-methylpyridinium derivatives. Inclusion of these compounds converts the Brønsted plot to a moderately scattered but linear correlation and gives a very good Hammett correlation. These data suggest a concerted pathway for this reaction that proceeds via an early transition state with little bond cleavage to the leaving group, highlighting the care that needs to be taken with the interpretation of experimental and especially theoretical data.
Co-reporter:Emmanuel Y. Tirel;Zoë Bellamy;Harry Adams;Vincent Lebrun;Ferna Duarte; Nicholas H. Williams
Angewandte Chemie 2014 Volume 126( Issue 31) pp:8385-8389
Publication Date(Web):
DOI:10.1002/ange.201400335
Abstract
Creating efficient artificial catalysts that can compete with biocatalysis has been an enduring challenge which has yet to be met. Reported herein is the synthesis and characterization of a series of zinc complexes designed to catalyze the hydrolysis of phosphate diesters. By introducing a hydrated aldehyde into the ligand we achieve turnover for DNA-like substrates which, combined with ligand methylation, increases reactivity by two orders of magnitude. In contrast to current orthodoxy and mechanistic explanations, we propose a mechanism where the nucleophile is not coordinated to the metal ion, but involves a tautomer with a more effective Lewis acid and more reactive nucleophile. This data suggests a new strategy for creating more efficient metal ion based catalysts, and highlights a possible mode of action for metalloenzymes.
Co-reporter:Osama El Zubir, Iain Barlow, Graham J. Leggett and Nicholas H. Williams
Nanoscale 2013 vol. 5(Issue 22) pp:11125-11131
Publication Date(Web):16 Sep 2013
DOI:10.1039/C3NR04701F
Nanoshaving, by tracing an atomic force microscope probe across a surface at elevated load, has been used to fabricate nanostructures in self-assembled monolayers of alkylphosphonates adsorbed at aluminium oxide surfaces. The simple process is implemented under ambient conditions. Because of the strong bond between the alkylphosphonates and the oxide surface, loads in excess of 400 nN are required to pattern the monolayer. Following patterning of octadecylphosphonate SAMs, adsorption of aminobutyl phosphonate yielded features as small as 39 nm. Shaving of monolayers of aryl azide-terminated alkylphosphonates, followed by attachment of polyethylene glycol to unmodified regions in a photochemical coupling reaction, yielded 102 nm trenches into which NeutrAvidin coated, dye-labelled, polymer nanospheres could be deposited, yielding bright fluorescence with little evidence of non-specific adsorption to other regions of the surface. Structures formed in alkylphosphonate films by nanoshaving were used to etch structures into the underlying metal. Because of the isotropic nature of the etch process, and the large grain size, some broadening was observed, but features 25–35 nm deep and 180 nm wide were fabricated.
Co-reporter:Anthony J. Kirby, Michelle Medeiros, José R. Mora, Pedro S. M. Oliveira, Almahdi Amer, Nicholas H. Williams, and Faruk Nome
The Journal of Organic Chemistry 2013 Volume 78(Issue 4) pp:1343-1353
Publication Date(Web):February 1, 2013
DOI:10.1021/jo302498g
Notwithstanding its half-life of 70 years at 25 °C, the spontaneous hydrolysis of the anion of di-2-pyridyl phosphate (DPP) is thousands of times faster (ca. 3000 at 100 °C, over 10000-fold at 25 °C) than expected for a diester with leaving groups of pKa 9.09. The kinetic parameters do not permit a conclusive choice between five possible mechanisms considered, but the combination of kinetics and calculational evidence supports a single-step, concerted, SN2(P) mechanism involving the attack of solvent water on phosphorus assisted by intramolecular catalysis by a (weakly basic) pyridine nitrogen acting as a general base. Catalysis is relatively efficient for this mechanism, with an estimated effective molarity (EM) of the general base of >15 M, consistent with the absence of catalysis by typical buffers. Further new results confirm that varying the nonleaving group has minimal effect on the rate of spontaneous diester hydrolysis, in striking contrast to the major effect on the corresponding reaction of triesters: though protonation of one nitrogen of DPP– increases the rate of hydrolysis by 6 orders of magnitude, in line with expectation.
Co-reporter:Osama El Zubir, Iain Barlow, Ehtsham Ul-Haq, Hairul A. Tajuddin, Nicholas H. Williams, and Graham J. Leggett
Langmuir 2013 Volume 29(Issue 4) pp:1083-1092
Publication Date(Web):December 17, 2012
DOI:10.1021/la303746e
A series of aryl azide terminated thiols and phosphonic acids has been synthesized, and used to prepare self-assembled monolayers on (respectively) gold and aluminum oxide surfaces. The rates of photoactivation were determined using contact angle measurement and X-ray photoelectron spectroscopy (XPS). The behavior of a diazirine functionalized aryl thiol was also studied. The rates of activation were found to be similar for all five adsorbates. However, the extent of photochemical coupling of a primary amine was significantly greater for the aryl azides than for the diazirine. A range of primary amines was successfully coupled to all of the azides with high yield. Little difference in reactivity was observed following perfluorination of the aromatic ring. Micrometer-scale patterns were fabricated by carrying out exposures of the aryl azide terminated SAMs through a mask submerged under a film of primary amine. Contrasting amines could be introduced to unreacted regions in a subsequent maskless step. A scanning near-field optical microscope was used to fabricate nanopatterns. Exposure of the azides to irradiation at 325 nm in air enabled selective deactivation of azides. The surrounding surface was functionalized with a primary amine in a maskless process; when a protein-resistant oligo(ethylene glycol) functionalized amine was used it was possible to produce protein nanopatterns, by adsorbing protein to features defined using near-field exposure.
Co-reporter:Dr. Heidi Korhonen;Dr. Satu Mikkola; Nicholas H. Williams
Chemistry - A European Journal 2012 Volume 18( Issue 2) pp:659-670
Publication Date(Web):
DOI:10.1002/chem.201100721
Abstract
The cleavage and isomerisation of uridine 3′-alkylphosphates was studied in the presence of a dinuclear Zn2+complex, 3. The rate acceleration of the cleavage by 1 mM3 is approximately 106-fold under neutral conditions. Most remarkably, the complex also promotes the isomerisation of phosphodiester bonds, although the rate-enhancement is more modest: under neutral conditions complex 3 (1 mM) catalyses isomerisation by about 500-fold. The observation of this reaction shows that the reactions of these substrates catalysed by 3 proceed through a stepwise mechanism involving an intermediate phosphorane. A βlg value of −0.92 was determined for the 3-promoted cleavage reaction, and modest kinetic solvent deuterium isotope effects ranging from 1.5 to 2.8 were observed. Isomerisation was less sensitive to the nature of the esterifying group, with a β value of −0.5, and the kinetic solvent deuterium isotope effects were less than 1.5. Most of these characteristics of the 3-promoted cleavage are very similar to those for the cleavage of nucleoside 3′-phosphotriesters. These data are explained by a mechanism in which the complex primarily acts as an electrophilic catalyst neutralising the charge on the phosphate and stabilising an intermediate phosphorane, with general acid catalysis promoting the cleavage reaction. In contrast to the behaviour of triesters, isomerisation is significantly slower than cleavage; this suggests that the changes in geometry that occur during isomerisation lead to a much less stable complex between 3 and the phosphorane intermediate.
Co-reporter: Anthony J. Kirby;Michelle Medeiros;Pedro S. M. Oliveira;Dr. Elisa S. Orth;Dr. Tiago A. S. Brão;Eduardo H. Werlind;Almahdi Amer;Dr. Nicholas H. Williams; Faruk Nome
Chemistry - A European Journal 2011 Volume 17( Issue 52) pp:14996-15004
Publication Date(Web):
DOI:10.1002/chem.201101926
Abstract
The high rate of spontaneous hydrolysis of tris-2-pyridyl phosphate (TPP) is explained by the activating effects of the non-leaving (“spectator”) groups on POAr cleavage, and not by intramolecular catalysis. Previous work on phosphate-transfer reactions has concentrated on the contributions to reactivity of the nucleophile and the leaving group, but our results make clear that the effects of the non-leaving groups on phosphorus can be equally significant. Rate measurements for three series of phosphate triesters showed that sensitivities to the non-leaving groups are substantial for spontaneous hydrolysis reactions, although significantly smaller for reactions with good nucleophiles. There are clear differences between triaryl and dialkyl aryl triesters in sensitivities to leaving and non-leaving groups with the more reactive triaryl systems showing lower values for both βLG and βNLG. Intramolecular catalysis of the hydrolysis of TPP by the neighbouring pyridine nitrogens is insignificant, primarily because of their low basicity.
Co-reporter:Guoqiang Feng ; Eric A. Tanifum ; Harry Adams ; Alvan C. Hengge ;Nicholas H. Williams
Journal of the American Chemical Society 2009 Volume 131(Issue 35) pp:12771-12779
Publication Date(Web):August 12, 2009
DOI:10.1021/ja904134n
Reactivities of five phosphonate esters each coordinated to a dinuclear Co(III) complex were investigated ([Co2(tacn)2(OH)2{O2P(Me)OAr}]3+; tacn = 1,4,7-triazacyclononane; substituent = m-F, p-NO2 (1a); p-NO2 (1b); m-NO2 (1c); p-Cl (1d); unsubstituted (1e)). Hydrolysis of the phosphonate esters in 1a to 1e is specific base catalyzed and takes place by intramolecular oxide attack on the bridging phosphonate. These data define a Brønsted βlg of −1.12, considerably more negative than that of the hydrolysis of the uncomplexed phosphonates (−0.69). For 1b, the kinetic isotope effects in the leaving group are 18klg = 1.0228 and 15k = 1.0014, at the nonbridging phosphoryl oxygens 18knonbridge = 0.9954, and at the nucleophilic oxygen18knuc = 1.0105. The KIEs and the βlg data point to a transition state for the alkaline hydrolysis of 1b that is similar to that of a phosphate monoester complex with the same leaving group, rather than the isoelectronic diester complex. The data from these model systems parallel the observation that in protein phosphatase-1, which has an active site that resembles the structures of these complexes, the catalyzed hydrolysis of aryl methylphosphonates and aryl phosphates are much more similar to one another than the uncomplexed hydrolysis reactions of the two substrates.
Co-reporter:Claire McWhirter ; Elizabeth A. Lund ; Eric A. Tanifum ; Guoqiang Feng ; Qaiser I. Sheikh ; Alvan C. Hengge ;Nicholas H. Williams
Journal of the American Chemical Society 2008 Volume 130(Issue 41) pp:13673-13682
Publication Date(Web):September 18, 2008
DOI:10.1021/ja803612z
The reaction catalyzed by the protein phosphatase-1 (PP1) has been examined by linear free energy relationships and kinetic isotope effects. With the substrate 4-nitrophenyl phosphate (4NPP), the reaction exhibits a bell-shaped pH-rate profile for kcat/KM indicative of catalysis by both acidic and basic residues, with kinetic pKa values of 6.0 and 7.2. The enzymatic hydrolysis of a series of aryl monoester substrates yields a Brønsted βlg of −0.32, considerably less negative than that of the uncatalyzed hydrolysis of monoester dianions (−1.23). Kinetic isotope effects in the leaving group with the substrate 4NPP are 18(V/K)bridge = 1.0170 and 15(V/K) = 1.0010, which, compared against other enzymatic KIEs with and without general acid catalysis, are consistent with a loose transition state with partial neutralization of the leaving group. PP1 also efficiently catalyzes the hydrolysis of 4-nitrophenyl methylphosphonate (4NPMP). The enzymatic hydrolysis of a series of aryl methylphosphonate substrates yields a Brønsted βlg of −0.30, smaller than the alkaline hydrolysis (−0.69) and similar to the βlg measured for monoester substrates, indicative of similar transition states. The KIEs and the βlg data point to a transition state for the alkaline hydrolysis of 4NPMP that is similar to that of diesters with the same leaving group. For the enzymatic reaction of 4NPMP, the KIEs are indicative of a transition state that is somewhat looser than the alkaline hydrolysis reaction and similar to the PP1-catalyzed monoester reaction. The data cumulatively point to enzymatic transition states for aryl phosphate monoester and aryl methylphosphonate hydrolysis reactions that are much more similar to one another than the nonenzymatic hydrolysis reactions of the two substrates.
Co-reporter:Charles J. M. Stirling, L. Johan Fundin and Nicholas H. Williams
Chemical Communications 2007 (Issue 17) pp:1748-1750
Publication Date(Web):27 Mar 2007
DOI:10.1039/B700469A
Nonylresorcinarene, in contrast to methylresorcinarene and nonylpyrogallene, exists in three isolable phases: a layer structure, a hexamer and an amorphous phase; the three are in thermal equilibrium.
Co-reporter:Harmen P. Dijkstra Dr.;Jordan J. Hutchinson;Christopher A. Hunter ;Haiyuan Qin Dr.;Salvador Tomas Dr.;Simon J. Webb Dr.;Nicholas H. Williams Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 25) pp:
Publication Date(Web):19 JUN 2007
DOI:10.1002/chem.200601723
A synthetic transmembrane receptor that is capable of transmitting binding information across a lipid bilayer membrane is reported. The binding event is based on aggregation of the receptor triggered by copper(II) complexation to ethylenediamine functionalities. By labelling the receptor with fluorescent dansyl groups, the copper(II) binding event could be monitored by measuring the extent of fluorescence quenching. Comparing the receptor with a control receptor lacking the transmembrane linkage revealed that the transmembrane receptor binds copper(II) ions more tightly than the non-spanning control receptor at low copper(II) concentrations. Since the intrinsic binding to copper(II) is the same for both receptors, this effect was attributed to synergy between the connected interior and exterior binding sides of the transmembrane receptor. Thus, this is the first reported artificial signalling event in which binding of a messenger on one side of the membrane leads to a cooperative binding event on the opposite side of the membrane, resembling biological signalling systems and helping us to get a better understanding of the requirements for more effective artificial signalling systems.
Co-reporter:Guoqiang Feng, Juan C. Mareque-Rivas and Nicholas H. Williams
Chemical Communications 2006 (Issue 17) pp:1845-1847
Publication Date(Web):03 Apr 2006
DOI:10.1039/B514328D
Introducing ligand based hydrogen bond donors to increase the activity of a mononuclear Zn(II) complex for catalysing phosphate ester cleavage can be a more effective strategy than making the dinuclear analogue.
Co-reporter:Guoqiang Feng Dr.;Daniela Natale;Ravi Prabaharan Dr.;Juan C. Mareque-Rivas Dr.;Nicholas H. Williams Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 42) pp:
Publication Date(Web):29 SEP 2006
DOI:10.1002/anie.200602532
A combined attack: Hydrogen-bonding interactions with double Lewis acid activation generate a dinuclear ZnII complex that is exceptionally effective for binding monoanionic phosphate diesters in water and for catalyzing phosphodiester transesterifications. The complex catalyzes the hydrolytic cleavage of RNA-like activated, artificial substrates and nonactivated, natural substrates with similar efficiencies.
Co-reporter:Gottfried K. Schroeder;Chetan Lad;Paul Wyman;Nicholas H. Williams;Richard Wolfenden
PNAS 2006 Volume 103 (Issue 11 ) pp:4052-4055
Publication Date(Web):2006-03-14
DOI:10.1073/pnas.0510879103
Phosphodiester linkages, including those that join the nucleotides of DNA, are highly resistant to spontaneous hydrolysis.
The rate of water attack at the phosphorus atom of phosphodiesters is known only as an upper limit, based on the hydrolysis
of the dimethyl phosphate anion. That reaction was found to proceed at least 99% by C–O cleavage, at a rate suggesting an
upper limit of 10−15 s−1 for P–O cleavage of phosphodiester anions at 25°C. To evaluate the rate enhancement produced by P–O cleaving phosphodiesterases
such as staphylococcal nuclease, we decided to establish the actual value of the rate constant for P–O cleavage of a simple
phosphodiester anion. In dineopentyl phosphate, C–O cleavage is sterically precluded so that hydrolysis occurs only by P–O
cleavage. Measurements at elevated temperatures indicate that the dineopentyl phosphate anion undergoes hydrolysis in water
with a t
1/2 of 30,000,000 years at 25°C, furnishing an indication of the resistance of the internucleotide linkages of DNA to water attack
at phosphorus. These results imply that staphylococcal nuclease (k
cat = 95 s−1) enhances the rate of phosphodiester hydrolysis by a factor of ≈1017. In alkaline solution, thymidylyl-3′-5′-thymidine (TpT) has been reported to decompose 105-fold more rapidly than does dineopentyl phosphate. We find however that TpT and thymidine decompose at similar rates and
with similar activation parameters, to a similar set of products, at pH 7 and in 1 M KOH. We infer that the decomposition
of TpT is initiated by the breakdown of thymidine, not by phosphodiester hydrolysis.
Co-reporter:Guoqiang Feng Dr.;Daniela Natale;Ravi Prabaharan Dr.;Juan C. Mareque-Rivas Dr.;Nicholas H. Williams Dr.
Angewandte Chemie 2006 Volume 118(Issue 42) pp:
Publication Date(Web):29 SEP 2006
DOI:10.1002/ange.200602532
Gemeinsamer Angriff: Wasserstoffbrücken und eine Lewis-Säure-Aktivierung führen zur Bildung eines zweikernigen ZnII-Komplexes, der in Wasser monoanionische Phosphatdiester bindet und deren Umesterung katalysiert. Der Komplex katalysiert die hydrolytische Spaltung RNA-artiger aktivierter, künstlicher Substrate und nichtaktivierter, natürlicher Substrate mit ähnlicher Effizienz.
Co-reporter:Michela Padovani;Nicholas H. Williams;Paul Wyman
Journal of Physical Organic Chemistry 2004 Volume 17(Issue 6‐7) pp:472-477
Publication Date(Web):25 MAY 2004
DOI:10.1002/poc.770
The TrpnCo(III)(OH)(OH2)-promoted hydrolysis of a range of methyl aryl phosphate diesters was investigated at 37°C and I = 0.1 M (NaClO4). The pH–rate profile confirms that the aqua-hydroxy form of the complex is the only kinetically significant ionic form. At pH 6.9, all the reactions are first order in both diester and Co(III) complex. Plotting the second-order rate constant for Co(III) complex-promoted hydrolysis against the pKa of the leaving aryloxy group revealed a bent LFER indicating a change in rate-limiting step. This is discussed in terms of either a change from rate-limiting hydrolysis to rate-limiting binding or the presence of a phosphorane intermediate. Copyright © 2004 John Wiley & Sons, Ltd.
Co-reporter:Marcello Forconi;Nicholas H. Williams Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 5) pp:
Publication Date(Web):7 MAR 2002
DOI:10.1002/1521-3773(20020301)41:5<849::AID-ANIE849>3.0.CO;2-7
An intramolecular OH group can become much more effective in promoting phosphate hydrolysis when combined with multiple interactions in a model for the metallophosphatase active site (see picture; tacn=1,4,7-triazacyclononane).
Co-reporter:Marcello Forconi;Nicholas H. Williams Dr.
Angewandte Chemie 2002 Volume 114(Issue 5) pp:
Publication Date(Web):7 MAR 2002
DOI:10.1002/1521-3757(20020301)114:5<877::AID-ANGE877>3.0.CO;2-K
Eine intramolekulare OH-Gruppe kann die Phosphathydrolyse viel effektiver beschleunigen, wenn sie mit einem Modell für das aktive Zentrum der Metallophosphatasen kombiniert wird (siehe Bild; tacn=1,4,7-Triazacyclononan).
Co-reporter:Patrick Barton Dr.;Christopher A. Hunter ;Timothy J. Potter;Simon J. Webb Dr.;Nicholas H. Williams Dr.
Angewandte Chemie 2002 Volume 114(Issue 20) pp:
Publication Date(Web):18 OCT 2002
DOI:10.1002/1521-3757(20021018)114:20<4034::AID-ANGE4034>3.0.CO;2-V
Kommunikation durch Wände: Ein synthetisches System für die Signalübertragung durch eine vesikuläre Doppelmembran hindurch wurde entwickelt. Hierbei überquert der externe sekundäre Botenstoff (violett) die Membran nicht selbst, sondern initiiert die Freisetzung eines weiteren sekundären Botenstoffs (rot) im Vesikelinneren.
Co-reporter:Patrick Barton Dr.;Christopher A. Hunter ;Timothy J. Potter;Simon J. Webb Dr.;Nicholas H. Williams Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 20) pp:
Publication Date(Web):18 OCT 2002
DOI:10.1002/1521-3773(20021018)41:20<3878::AID-ANIE3878>3.0.CO;2-F
Talking through walls: A synthetic system has been developed for transducing a signal across a vesicular bilayer membrane. The external messenger (purple) does not cross the membrane itself, but instigates the release of a secondary messenger (red) on the interior of the vesicle.
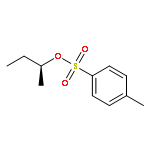
Co-reporter:Nicholas H. Williams and Paul Wyman
Chemical Communications 2001 (Issue 14) pp:1268-1269
Publication Date(Web):21 Jun 2001
DOI:10.1039/B103317B
The rate of attack of hydroxide on dialkyl phosphate diesters
is far slower than previously estimated, allowing us to estimate the
stability of the diester link in DNA and showing that ethylene phosphate is
1011 fold more reactive towards attack by hydroxide than an
acyclic diester (1000 fold more than previously estimated).
Co-reporter:Nicholas H. Williams and Paul Wyman
Organic & Biomolecular Chemistry 2001 (Issue 11) pp:2068-2073
Publication Date(Web):02 Oct 2001
DOI:10.1039/B106374J
The hydrolysis of methyl aryl phosphate diesters coordinated to a dinuclear Co(III) complex ([Co2(tame)2(OH)2{O2P(OAr)(OMe)}]3+; tame = 1,1,1-tris(aminomethyl)ethane; 2) has been studied in aqueous solution at 25 °C. Hydrolysis of the phosphate diester is base catalysed and occurs 30 to 60 fold faster than in analogous complexes where tame is replaced by 1,4,7-triazacyclononane (tacn) (1). The second order rate constants for base catalysed hydrolysis of 2 are highly sensitive to the basicity of the aryloxy leaving group with βlg
=
−1.29 ± 0.03. This leaving group dependence is similar to that of 1 (βlg
=
−1.38 ± 0.01),
showing that the ligand affects reactivity without greatly altering the transition state at phosphorus. The slight decrease in βlg is consistent with previous rationalisations of this high sensitivity. Dimethyl phosphate coordinated to both types of complex (3, tame; 4, tacn) only dissociates from the complex, with no hydrolysis. Base catalysed dissociation is slower with tame (3 20 fold slower than 4) but the pH independent reaction is faster (3 10 fold faster than 4). These data suggest that the reactivity and turnover properties of these dinuclear complexes may be tuned rationally and independently.
Co-reporter:Charles J. M. Stirling, L. Johan Fundin and Nicholas H. Williams
Chemical Communications 2007(Issue 17) pp:NaN1750-1750
Publication Date(Web):2007/03/27
DOI:10.1039/B700469A
Nonylresorcinarene, in contrast to methylresorcinarene and nonylpyrogallene, exists in three isolable phases: a layer structure, a hexamer and an amorphous phase; the three are in thermal equilibrium.