Co-reporter:Akinori Okano, Nicholas A. Isley, and Dale L. Boger
Chemical Reviews September 27, 2017 Volume 117(Issue 18) pp:11952-11952
Publication Date(Web):April 24, 2017
DOI:10.1021/acs.chemrev.6b00820
A review of efforts that have provided total syntheses of vancomycin and related glycopeptide antibiotics, their agylcons, and key analogues is provided. It is a tribute to developments in organic chemistry and the field of organic synthesis that not only can molecules of this complexity be prepared today by total synthesis but such efforts can be extended to the preparation of previously inaccessible key analogues that contain deep-seated structural changes. With the increasing prevalence of acquired bacterial resistance to existing classes of antibiotics and with the emergence of vancomycin-resistant pathogens (VRSA and VRE), the studies pave the way for the examination of synthetic analogues rationally designed to not only overcome vancomycin resistance but provide the foundation for the development of even more powerful and durable antibiotics.
Co-reporter:John C. Lukesh III, Daniel W. Carney, Huijun Dong, R. Matthew Cross, Vyom Shukla, Katharine K. Duncan, Shouliang Yang, Daniel M. Brody, Manuela M. Brütsch, Aleksandar Radakovic, and Dale L. Boger
Journal of Medicinal Chemistry September 14, 2017 Volume 60(Issue 17) pp:7591-7591
Publication Date(Web):August 31, 2017
DOI:10.1021/acs.jmedchem.7b00958
A series of 180 vinblastine 20′ amides were prepared in three steps from commercially available starting materials, systematically exploring a typically inaccessible site in the molecule enlisting a powerful functionalization strategy. Clear structure–activity relationships and a structural model were developed in the studies which provided many such 20′ amides that exhibit substantial and some even remarkable enhancements in potency, many that exhibit further improvements in activity against a Pgp overexpressing resistant cancer cell line, and an important subset of the vinblastine analogues that display little or no differential in activity against a matched pair of vinblastine sensitive and resistant (Pgp overexpressing) cell lines. The improvements in potency directly correlated with target tubulin binding affinity, and the reduction in differential functional activity against the sensitive and Pgp overexpressing resistant cell lines was found to correlate directly with an impact on Pgp-derived efflux.
Co-reporter:Anne F. Kornahrens, Armand B. Cognetta III, Daniel M. Brody, Megan L. Matthews, Benjamin F. Cravatt, and Dale L. Boger
Journal of the American Chemical Society May 24, 2017 Volume 139(Issue 20) pp:7052-7052
Publication Date(Web):May 12, 2017
DOI:10.1021/jacs.7b02985
The design and examination of 4,1,2-benzoxathiazin-3-one 1,1-dioxides as candidate serine hydrolase inhibitors are disclosed, and represent the synthesis and study of a previously unexplored heterocycle. This new class of activated cyclic carbamates provided selective irreversible inhibition of a small subset of serine hydrolases without release of a leaving group, does not covalently modify active site catalytic cysteine and lysine residues of other enzyme classes, and was found to be amenable to predictable structural modifications that modulate intrinsic reactivity or active site recognition. Even more remarkable and within the small pilot series of candidate inhibitors examined in an initial study, an exquisitely selective inhibitor for a poorly characterized serine hydrolase (PNPLA4, patatin-like phospholipase domain-containing protein 4) involved in adipocyte triglyceride homeostasis was discovered.
Co-reporter:Ryan E. Quiñones, Christopher M. Glinkerman, Kaicheng Zhu, and Dale L. Boger
Organic Letters July 7, 2017 Volume 19(Issue 13) pp:
Publication Date(Web):June 28, 2017
DOI:10.1021/acs.orglett.7b01543
Simple and direct nucleophilic addition of secondary amines, including imidazole, to 1,2,3-triazine under mild reaction conditions (THF, 25–65 °C, 12–48 h), requiring no additives, cleanly provides β-aminoenals 4 in good yields (21 examples, 31–79%). The reaction proceeds by amine nucleophilic addition to C4 of the 1,2,3-triazine, in situ loss of N2, and subsequent imine hydrolysis to provide 4.
Co-reporter:Dale L. Boger
The Journal of Organic Chemistry December 1, 2017 Volume 82(Issue 23) pp:11961-11961
Publication Date(Web):September 25, 2017
DOI:10.1021/acs.joc.7b02088
A Perspective of work in our laboratory on the examination of biologically active compounds, especially natural products, is presented. In the context of individual programs and along with a summary of our work, selected cases are presented that illustrate the impact single atom changes can have on the biological properties of the compounds. The examples were chosen to highlight single heavy atom changes that improve activity, rather than those that involve informative alterations that reduce or abolish activity. The examples were also chosen to illustrate that the impact of such single-atom changes can originate from steric, electronic, conformational, or H-bonding effects, from changes in functional reactivity, from fundamental intermolecular interactions with a biological target, from introduction of a new or altered functionalization site, or from features as simple as improvements in stability or physical properties. Nearly all the examples highlighted represent not only unusual instances of productive deep-seated natural product modifications and were introduced through total synthesis but are also remarkable in that they are derived from only a single heavy atom change in the structure.
Co-reporter:Oliver Allemann, R. Matthew Cross, Manuela M. Brütsch, Aleksandar Radakovic, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 14(Issue 14) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.bmcl.2017.05.058
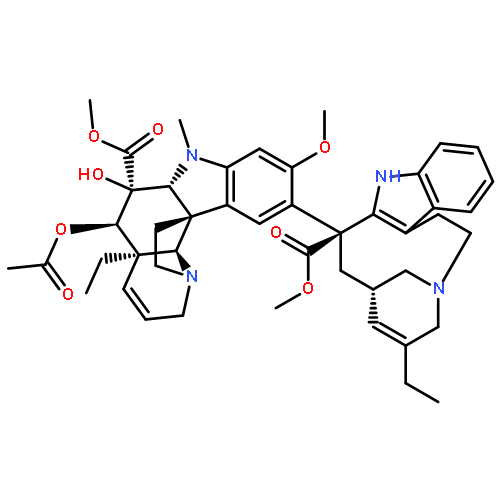
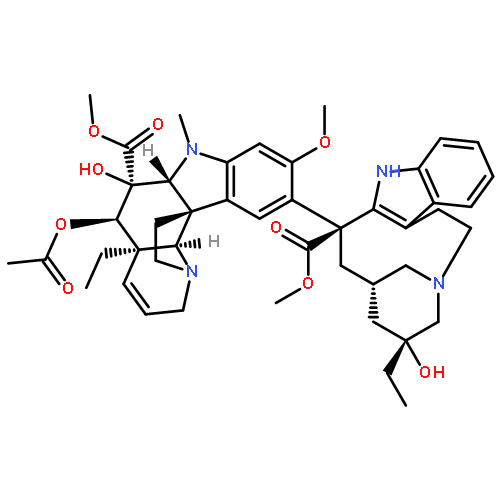
A key series of vinblastine analogs 7–13, which contain modifications to the C20′ ethyl group, was prepared with use of two distinct synthetic approaches that provide modifications of the C20′ side chain containing linear and cyclized alkyl groups or added functionalized substituents. Their examination revealed the unique nature of the improved properties of the synthetic vinblastine 6, offers insights into the origins of its increased tubulin binding affinity and 10-fold improved cell growth inhibition potency, and served to probe a small hydrophobic pocket anchoring the binding of vinblastine with tubulin. Especially noteworthy were the trends observed with substitution of the terminal carbon of the ethyl group that, with the exception of 9 (R = F vs H, equipotent), led to remarkably substantial reductions in activity (>10-fold): R = F (equipotent with H) > N3, CN (10-fold) > Me (50-fold) > Et (100-fold) > OH (inactive). This is in sharp contrast to the maintained (7) or enhanced activity (6) observed with its incorporation into a cyclic C20′/C15′-fused six-membered ring.Download high-res image (226KB)Download full-size image
Co-reporter:Shouliang Yang;Kuppusamy Sankar;Colin K. Skepper;Timothy J. Barker;John C. Lukesh III;Daniel M. Brody;Manuela M. Brütsch
Chemical Science (2010-Present) 2017 vol. 8(Issue 2) pp:1560-1569
Publication Date(Web):2017/01/30
DOI:10.1039/C6SC04146A
The total synthesis and evaluation of a key systematic series of vinblastines that incorporate the first deep-seated changes to the substituent at C4 are detailed. The synthetic approach features an expanded and redefined scope of a 1,3,4-oxadiazole [4 + 2]/[3 + 2] cycloaddition cascade in which electronically mismatched electron-deficient trisubstituted alkenes and unactivated trisubstituted alkenes were found to productively initiate the cycloaddition cascade with tethered electron-deficient 1,3,4-oxadiazoles. Such cycloaddition cascades were used to directly introduce altered C4 substituents, providing the basis for concise total syntheses of a series of C4 modified vindolines and their subsequent single-step incorporation into the corresponding synthetic vinblastines in routes as short as 8–12 steps. Evaluation of the synthetic vinblastines revealed a surprisingly large impact and role of the C4 substituent on activity even though it was previously not thought to intimately interact with the biological target tubulin. Only the introduction of a C4 methyl ester, a constitutional isomer of vinblastine in which the carbonyl carbon and ester oxygen of the C4 acetate are transposed, provided a synthetic vinblastine that matched the potency of the natural product. In contrast, even introduction of a C4 acetamide or N-methyl carboxamide, which incorporate single heavy atom exchanges (amide NH for ester oxygen) in vinblastine or the C4 methyl ester, provided compounds that were ≥10-fold less active than vinblastine. Other C4 acetate replacements, including a C4 amine, carboxylic acid, hydroxymethyl or acetoxymethyl group, led to even greater reductions in potency. Even replacement of the C4 acetoxy group or its equally active C4 methyl ester with an ethyl or isopropyl ester led to 10-fold or more reductions in activity. These remarkable trends in activity, which correlate with relative tubulin binding affinities, retrospectively may be ascribed to the role the substituent serves as a H-bond acceptor for α-tubulin Lys336 and Asn329 side chains at a site less tolerant of a H-bond donor, placing the methyl group of the C4 acetate or C4 methyl ester in a spatially restricted and well-defined hydrophobic half pocket created by a surrounding well-ordered loop. This remarkable impact of the C4 substituent, its stringency, and even the magnitude of its effect are extraordinary, and indicate that its presence was selected in Nature to enhance the effects of vinblastine and related natural products.
Co-reporter:Justin E. Sears and Dale L. Boger
Accounts of Chemical Research 2016 Volume 49(Issue 2) pp:241
Publication Date(Web):January 27, 2016
DOI:10.1021/acs.accounts.5b00510
A summary of the development and initial studies on the scope of a powerful tandem intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles is detailed and provides the foundation for its subsequent use in organic synthesis. Implemented with substrates in which both the initiating dienophile and subsequent dipolarophile are tethered to the 1,3,4-oxadiazoles, the studies expanded the scope of oxadiazoles that participate in the reaction cascade, permitted the use of differentiated dienophiles and dipolarophiles, extended their use to unsymmetrical dienophiles and dipolarophiles, provided exclusive control of the cycloaddition regioselectivities, and imposed exquisite control on the cycloaddition stereochemistry. As key reactivity and stereochemical features of the reactions were being defined, the cascade cycloaddition reaction was implemented in the total synthesis of a series of alkaloids including (−)-vindoline, (−)-vindorosine, the closely related natural products (+)-4-desacetoxyvindoline and (+)-4-desacetoxyvindorosine, natural minovine, (+)-N-methylaspidospermidine, (+)-spegazzinine, (−)-aspidospermine, and a number of key analogues. Most recently, it was used in the divergent total syntheses of (+)-fendleridine, (−)-kopsinine, (−)-kopsifoline D, and (−)-deoxoapodine, in which four different strategic bonds in four different classes of the hexacyclic alkaloids were formed from a common cascade cycloaddition intermediate. A large number of vindoline analogues were prepared by variations on the cascade cycloaddition reaction for single step incorporation into analogues of vinblastine. These structural changes to vindoline permitted both systematic alterations to the peripheral substituents as well as deep-seated changes to the core structure and embedded functionality of vinblastine not previously accessible. Although explored initially for accessing vindoline and vinblastine, the use of the cycloaddition cascade in the total synthesis of an impressive range of additional natural products illustrate the power of the methodology. Alternative tethering strategies for the cascade cycloaddition reaction, combined intramolecular and intermolecular variants of either the initiating Diels–Alder reaction or the subsequent carbonyl ylide 1,3-dipolar cycloaddition, an expanded examination of the tethered dipolarophile scope, and applications to additional natural product classes represent attractive areas for future work.
Co-reporter:Oliver Allemann; Manuela Brutsch; John C. LukeshIII; Daniel M. Brody
Journal of the American Chemical Society 2016 Volume 138(Issue 27) pp:8376-8379
Publication Date(Web):June 29, 2016
DOI:10.1021/jacs.6b04330
Many natural products, including vinblastine, have not been easily subjected to simplifications in their structures by synthetic means or modifications by late-stage semisynthetic derivatization in ways that enhance their biological potency. Herein, we detail a synthetic vinblastine that incorporates added benign complexity (ABC), which improves activity 10-fold, and is now accessible as a result of advances in the total synthesis of the natural product. The compound incorporates designed added molecular complexity but no new functional groups and maintains all existing structural and conformational features of the natural product. It constitutes a member of an analogue class presently inaccessible by semisynthetic derivatization of the natural product, by its late-stage functionalization, or by biosynthetic means. Rather, it was accessed by synthetic means, using an appropriately modified powerful penultimate single-step vindoline–catharanthine coupling strategy that proceeds with a higher diastereoselectivity than found for the natural product itself.
Co-reporter:Matthew D. Morin; Ying Wang; Brian T. Jones; Lijing Su; Murali M. R. P. Surakattula; Michael Berger; Hua Huang; Elliot K. Beutler; Hong Zhang; Bruce Beutler
Journal of Medicinal Chemistry 2016 Volume 59(Issue 10) pp:4812-4830
Publication Date(Web):April 6, 2016
DOI:10.1021/acs.jmedchem.6b00177
Herein, we report studies leading to the discovery of the neoseptins and a comprehensive examination of the structure–activity relationships (SARs) of this new class of small-molecule mouse Toll-like receptor 4 (mTLR4) agonists. The compounds in this class, which emerged from screening an α-helix mimetic library, stimulate the immune response, act by a well-defined mechanism (mouse TLR4 agonist), are easy to produce and structurally manipulate, exhibit exquisite SARs, are nontoxic, and elicit improved and qualitatively different responses compared to lipopolysaccharide, even though they share the same receptor.
Co-reporter:Prem B. Chanda, Kristopher E. Boyle, Daniel M. Brody, Vyom Shukla, Dale L. Boger
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 20) pp:4779-4786
Publication Date(Web):15 October 2016
DOI:10.1016/j.bmc.2016.04.050
The design, synthesis, and evaluation of methyl 1,2,8,8a-tetrahydrocyclopropa[c]imidazolo[4,5-e]indol-4-one-6-carboxylate (CImI) derivatives are detailed representing analogs of duocarmycin SA and yatakemycin containing an imidazole replacement for the fused pyrrole found in the DNA alkylation subunit.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Lijing Su;Ying Wang;Matthew D. Morin;Brian T. Jones;Murali M. R. P. Surakattula;Landon R. Whitby;Hua Huang;Hexin Shi;Jin Huk Choi;Kuan-wen Wang;Michael Berger;Eva Marie Y. Moresco;Xiaoming Zhan;Hong Zhang;Bruce Beutler
PNAS 2016 Volume 113 (Issue 7 ) pp:E884-E893
Publication Date(Web):2016-02-16
DOI:10.1073/pnas.1525639113
Structurally disparate molecules reportedly engage and activate Toll-like receptor (TLR) 4 and other TLRs, yet the interactions
that mediate binding and activation by dissimilar ligands remain unknown. We describe Neoseptins, chemically synthesized peptidomimetics
that bear no structural similarity to the established TLR4 ligand, lipopolysaccharide (LPS), but productively engage the mouse
TLR4 (mTLR4)/myeloid differentiation factor 2 (MD-2) complex. Neoseptin-3 activates mTLR4/MD-2 independently of CD14 and triggers
canonical myeloid differentiation primary response gene 88 (MyD88)- and Toll-interleukin 1 receptor (TIR) domain-containing
adaptor inducing IFN-beta (TRIF)-dependent signaling. The crystal structure mTLR4/MD-2/Neoseptin-3 at 2.57-Å resolution reveals
that Neoseptin-3 binds as an asymmetrical dimer within the hydrophobic pocket of MD-2, inducing an active receptor complex
similar to that induced by lipid A. However, Neoseptin-3 and lipid A form dissimilar molecular contacts to achieve receptor
activation; hence strong TLR4/MD-2 agonists need not mimic LPS.
Co-reporter:Christopher M. Glinkerman and Dale L. Boger
Journal of the American Chemical Society 2016 Volume 138(Issue 38) pp:
Publication Date(Web):August 29, 2016
DOI:10.1021/jacs.6b05438
Although it has been examined for decades, no general approach to catalysis of the inverse electron demand Diels–Alder reactions of heterocyclic azadienes has been introduced. Typically, additives such as Lewis acids lead to nonproductive consumption of the electron-rich dienophiles without productive activation of the electron-deficient heterocyclic azadienes. Herein, we report the first general method for catalysis of such cycloaddition reactions by using solvent hydrogen bonding of non-nucleophilic perfluoroalcohols, including hexafluoroisopropanol (HFIP) and trifluoroethanol (TFE), to activate the electron-deficient heterocyclic azadienes. Its use in promoting the cycloaddition of 1,2,3-triazine 4 with enamine 3 as the key step of a concise total synthesis of methoxatin is described.
Co-reporter:Daniel M. Brody;Manuela M. Brütsch;John C. Lukesh, III;Daniel W. Carney
PNAS 2016 Volume 113 (Issue 35 ) pp:9691-9698
Publication Date(Web):2016-08-30
DOI:10.1073/pnas.1611405113
Approaches to improving the biological properties of natural products typically strive to modify their structures to identify
the essential pharmacophore, or make functional group changes to improve biological target affinity or functional activity,
change physical properties, enhance stability, or introduce conformational constraints. Aside from accessible semisynthetic
modifications of existing functional groups, rarely does one consider using chemical synthesis to add molecular complexity
to the natural product. In part, this may be attributed to the added challenge intrinsic in the synthesis of an even more
complex compound. Herein, we report synthetically derived, structurally more complex vinblastines inaccessible from the natural
product itself that are a stunning 100-fold more active (IC50 values, 50–75 pM vs. 7 nM; HCT116), and that are now accessible because of advances in the total synthesis of the natural
product. The newly discovered ultrapotent vinblastines, which may look highly unusual upon first inspection, bind tubulin
with much higher affinity and likely further disrupt the tubulin head-to-tail α/β dimer–dimer interaction by virtue of the
strategic placement of an added conformationally well-defined, rigid, and extended C20′ urea along the adjacent continuing
protein–protein interface. In this case, the added molecular complexity was used to markedly enhance target binding and functional
biological activity (100-fold), and likely represents a general approach to improving the properties of other natural products
targeting a protein–protein interaction.
Co-reporter:Justin E. Sears and Dale L. Boger
Accounts of Chemical Research 2015 Volume 48(Issue 3) pp:653
Publication Date(Web):January 14, 2015
DOI:10.1021/ar500400w
Biologically active natural products composed of fascinatingly complex structures are often regarded as not amenable to traditional systematic structure–function studies enlisted in medicinal chemistry for the optimization of their properties beyond what might be accomplished by semisynthetic modification. Herein, we summarize our recent studies on the Vinca alkaloids vinblastine and vincristine, often considered as prototypical members of such natural products, that not only inspired the development of powerful new synthetic methodology designed to expedite their total synthesis but have subsequently led to the discovery of several distinct classes of new, more potent, and previously inaccessible analogues.With use of the newly developed methodology and in addition to ongoing efforts to systematically define the importance of each embedded structural feature of vinblastine, two classes of analogues already have been discovered that enhance the potency of the natural products >10-fold. In one instance, remarkable progress has also been made on the refractory problem of reducing Pgp transport responsible for clinical resistance with a series of derivatives made accessible only using the newly developed synthetic methodology. Unlike the removal of vinblastine structural features or substituents, which typically has a detrimental impact, the additions of new structural features have been found that can enhance target tubulin binding affinity and functional activity while simultaneously disrupting Pgp binding, transport, and functional resistance. Already analogues are in hand that are deserving of full preclinical development, and it is a tribute to the advances in organic synthesis that they are readily accessible even on a natural product of a complexity once thought refractory to such an approach.
Co-reporter:Akinori Okano; Atsushi Nakayama; Kejia Wu; Erick A. Lindsey; Alex W. Schammel; Yiqing Feng; Karen C. Collins
Journal of the American Chemical Society 2015 Volume 137(Issue 10) pp:3693-3704
Publication Date(Web):March 9, 2015
DOI:10.1021/jacs.5b01008
Full details of studies are disclosed on the total syntheses of binding pocket analogues of vancomycin bearing the peripheral l-vancosaminyl-1,2-d-glucosyl disaccharide that contain changes to a key single atom in the residue-4 amide (residue-4 carbonyl O → S, NH, H2) designed to directly address the underlying molecular basis of resistance to vancomycin. Also disclosed are studies piloting the late-stage transformations conducted on the synthetically more accessible C-terminus hydroxymethyl aglycon derivatives and full details of the peripheral chlorobiphenyl functionalization of all of the binding-pocket-modified vancomycin analogues designed for dual d-Ala-d-Ala/d-Ala-d-Lac binding. Their collective assessment indicates that combined binding pocket and chlorobiphenyl peripherally modified analogues exhibit a remarkable spectrum of antimicrobial activity (VSSA, MRSA, and VanA and VanB VRE) and impressive potencies against both vancomycin-sensitive and vancomycin-resistant bacteria (MICs = 0.06–0.005 and 0.5–0.06 μg/mL for the amidine and methylene analogues, respectively) and likely benefit from two independent and synergistic mechanisms of action, only one of which is dependent on d-Ala-d-Ala/d-Ala-d-Lac binding. Such analogues are likely to display especially durable antibiotic activity that is not prone to rapidly acquired clinical resistance.
Co-reporter:Christopher M. Glinkerman and Dale L. Boger
Organic Letters 2015 Volume 17(Issue 16) pp:4002-4005
Publication Date(Web):July 14, 2015
DOI:10.1021/acs.orglett.5b01870
The examination of the cycloaddition reactions of 1,2,3-triazines 17–19, bearing electron-donating substituents at C5, are described. Despite the noncomplementary 1,2,3-triazine C5 substituents, amidines were found to undergo a powerful cycloaddition to provide 2,5-disubstituted pyrimidines in excellent yields (42–99%; EDG = SMe > OMe > NHAc). Even select ynamines and enamines were capable of cycloadditions with 17, but not 18 or 19, to provide trisubstituted pyridines in modest yields (37–40% and 33% respectively).
Co-reporter:Justin E. Sears, Timothy J. Barker, and Dale L. Boger
Organic Letters 2015 Volume 17(Issue 21) pp:5460-5463
Publication Date(Web):October 12, 2015
DOI:10.1021/acs.orglett.5b02818
It is reported that an allene dienophile can initiate a tandem intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles, that the intermediate cross-conjugated 1,3-dipole (a carbonyl ylide) can participate in an ensuing [3 + 2] dipolar cycloaddition in a remarkably effective manner, and that the reaction can be implemented to provide the core pentacyclic ring system of vindoline. Its discovery improves a previous total synthesis of (−)-vindoline and was used in a total synthesis of (+)-4-epi-vindoline and (+)-4-epi-vinblastine that additionally enlists an alternative series of late-stage transformations.
Co-reporter:Mika Uematsu, Daniel M. Brody, Dale L. Boger
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3101-3104
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.11.038
The preparation, characterization, and examination of the CBI-based 5-membered lactone 5 capable of serving as a prodrug or protein (antibody) conjugation reagent are disclosed along with its incorporation into the corresponding CC-1065 and duocarmycin analog 6, and the establishment of their properties.
Co-reporter:Kiyoun Lee, Dale L. Boger
Tetrahedron 2015 Volume 71(Issue 22) pp:3741-3746
Publication Date(Web):3 June 2015
DOI:10.1016/j.tet.2014.07.094
The total synthesis of (−)-kopsinine and its unnatural enantiomer is detailed, enlisting a late-stage SmI2-mediated transannular free radical conjugate addition reaction for construction of the core bicyclo[2.2.2]octane ring system with strategic C21–C2 bond formation. Key to the approach is assemblage of the underlying skeleton by an intramolecular [4+2]/[3+2] cycloaddition cascade of a 1,3,4-oxadiazole that provided the precursor C21 functionalized pentacyclic ring system 1 in a single step in which the C3 methyl ester found in the natural product served as a key 1,3,4-oxadiazole substituent, activating it for participation in the initiating Diels–Alder reaction and stabilizing the intermediate 1,3-dipole.
Co-reporter:Kiyoun Lee, Yam B. Poudel, Christopher M. Glinkerman, Dale L. Boger
Tetrahedron 2015 Volume 71(Issue 35) pp:5897-5905
Publication Date(Web):2 September 2015
DOI:10.1016/j.tet.2015.05.093
The total syntheses of dihydrolysergic acid and dihydrolysergol are detailed based on a Pd(0)-catalyzed intramolecular Larock indole cyclization for the preparation of the embedded tricyclic indole (ABC ring system) and a subsequent powerful inverse electron demand Diels–Alder reaction of 5-carbomethoxy-1,2,3-triazine with a ketone-derived enamine for the introduction of a functionalized pyridine, serving as the precursor for a remarkably diastereoselective reduction to the N-methylpiperidine D-ring. By design, the use of the same ketone-derived enamine and a set of related complementary heterocyclic azadiene [4+2] cycloaddition reactions permitted the late stage divergent preparation of a series of alternative heterocyclic derivatives not readily accessible by more conventional approaches.
Co-reporter:Akinori Okano ; Atsushi Nakayama ; Alex W. Schammel
Journal of the American Chemical Society 2014 Volume 136(Issue 39) pp:13522-13525
Publication Date(Web):September 11, 2014
DOI:10.1021/ja507009a
The total synthesis of two key analogues of vancomycin containing single-atom exchanges in the binding pocket (residue 4 amidine and thioamide) are disclosed as well as their peripherally modified (4-chlorobiphenyl)methyl (CBP) derivatives. Their assessment indicates that combined pocket amidine and CBP peripherally modified analogues exhibit a remarkable spectrum of antimicrobial activity (VSSA, MRSA, VanA and VanB VRE) and impressive potencies (MIC = 0.06–0.005 μg/mL) against both vancomycin-sensitive and -resistant bacteria and likely benefit from two independent and synergistic mechanisms of action. Like vancomycin, such analogues are likely to display especially durable antibiotic activity not prone to rapidly acquired clinical resistance.
Co-reporter:Adam S. Duerfeldt
Journal of the American Chemical Society 2014 Volume 136(Issue 5) pp:2119-2125
Publication Date(Web):January 12, 2014
DOI:10.1021/ja412298c
Total syntheses of (−)-pyrimidoblamic acid and P-3A are disclosed. Central to the convergent approach is a powerful inverse electron demand Diels–Alder reaction between substituted electron-deficient 1,2,3-triazines and a highly functionalized and chiral primary amidine, which forms the pyrimidine cores and introduces all necessary stereochemistry in a single step. Intrinsic in the convergent approach is the potential it provides for the late stage divergent synthesis of modified analogs bearing deep-seated changes in either the pyrimidine cores or the highly functionalized C2 side chain common to both natural products. The examination of the key cycloaddition reaction revealed that the inherent 1,2,3-triazine mode of cycloaddition (C4/N1 vs C5/N2) as well as the amidine regioselectivity were unaffected by introduction of two electron-withdrawing groups (−CO2R) at C4 and C6 of the 1,2,3-triazine even if C5 is unsubstituted (Me or H), highlighting the synthetic potential of the powerful pyrimidine synthesis.
Co-reporter:Kiyoun Lee
Journal of the American Chemical Society 2014 Volume 136(Issue 8) pp:3312-3317
Publication Date(Web):February 5, 2014
DOI:10.1021/ja500548e
Divergent total syntheses of (−)-kopsifoline D and (−)-deoxoapodine are detailed from a common pentacyclic intermediate 15, enlisting the late-stage formation of two different key strategic bonds (C21–C3 and C21–O–C6) unique to their hexacyclic ring systems that are complementary to its prior use in the total syntheses of kopsinine (C21–C2 bond formation) and (+)-fendleridine (C21–O–C19 bond formation). The combined efforts represent the total syntheses of members of four classes of natural products from a common intermediate functionalized for late-stage formation of four different key strategic bonds uniquely embedded in each natural product core structure. Key to the first reported total synthesis of a kopsifoline that is detailed herein was the development of a transannular enamide alkylation for late-stage formation of the C21–C3 bond with direct introduction of the reactive indolenine C2 oxidation state from a penultimate C21 functionalized Aspidosperma-like pentacyclic intermediate. Central to the assemblage of the underlying Apidosperma skeleton is a powerful intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of a 1,3,4-oxadiazole that provided the functionalized pentacyclic ring system 15 in a single step in which the C3 methyl ester found in the natural products served as a key 1,3,4-oxadiazole substituent, activating it for participation in the initiating Diels–Alder reaction and stabilizing the intermediate 1,3-dipole.
Co-reporter:Atsushi Nakayama, Akinori Okano, Yiqing Feng, James C. Collins, Karen C. Collins, Christopher T. Walsh, and Dale L. Boger
Organic Letters 2014 Volume 16(Issue 13) pp:3572-3575
Publication Date(Web):June 23, 2014
DOI:10.1021/ol501568t
Studies on the further development of the sequential glycosylations of the vancomycin aglycon catalyzed by the glycosyltransferases GtfE and GtfD and the observation of unusual, perhaps unexpected, aglycon substrate substituent effects on the rate and efficiency of the initial glycosylation reaction are reported.
Co-reporter:Erin D. Anderson, Adam S. Duerfeldt, Kaicheng Zhu, Christopher M. Glinkerman, and Dale L. Boger
Organic Letters 2014 Volume 16(Issue 19) pp:5084-5087
Publication Date(Web):September 15, 2014
DOI:10.1021/ol502436n
The scope of the [4 + 2] cycloaddition reactions of substituted 1,2,3-triazines, bearing noncomplementary substitution with electron-withdrawing groups at C4 and/or C6, is described. The studies define key electronic and steric effects of substituents impacting the reactivity, mode (C4/N1 vs C5/N2), and regioselectivity of the cycloaddition reactions of 1,2,3-triazines with amidines, enamines, and ynamines, providing access to highly functionalized heterocycles.
Co-reporter:Katerina Otrubova ; Benjamin F. Cravatt
Journal of Medicinal Chemistry 2014 Volume 57(Issue 3) pp:1079-1089
Publication Date(Web):January 23, 2014
DOI:10.1021/jm401820q
A series of α-ketooxazoles incorporating electrophiles at the C5 position of the pyridyl ring of 2 (OL-135) and related compounds were prepared and examined as inhibitors of fatty acid amide hydrolase (FAAH) that additionally target the cytosolic port Cys269. From this series, a subset of the candidate inhibitors exhibited time-dependent FAAH inhibition and noncompetitive irreversible inactivation of the enzyme, consistent with the targeted Cys269 covalent alkylation or addition, and maintained or enhanced the intrinsic selectivity for FAAH versus other serine hydrolases. A preliminary in vivo assessment demonstrates that these inhibitors raise endogenous brain levels of anandamide and other FAAH substrates upon intraperitoneal (i.p.) administration to mice, with peak levels achieved within 1.5–3 h, and that the elevations of the signaling lipids were maintained >6 h, indicating that the inhibitors effectively reach and remain active in the brain, inhibiting FAAH for a sustained period.
Co-reporter:Katharine K. Duncan, Katerina Otrubova, Dale L. Boger
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 9) pp:2763-2770
Publication Date(Web):1 May 2014
DOI:10.1016/j.bmc.2014.03.013
Co-reporter:Katerina Otrubova, Venkat Srinivasan, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 16) pp:3807-3813
Publication Date(Web):15 August 2014
DOI:10.1016/j.bmcl.2014.06.063
Two libraries of modestly reactive ureas containing either electron-deficient acyl anilines or acyl pyrazoles were prepared and are reported as screening libraries for candidate serine hydrolase inhibitors. Within each library is a small but powerful subset of compounds that serve as a chemotype fragment screening library capable of subsequent structural diversification. Elaboration of the pyrazole-based ureas provided remarkably potent irreversible inhibitors of fatty acid amide hydrolase (FAAH, apparent Ki = 100–200 pM) complementary to those previously disclosed enlisting electron-deficient aniline-based ureas.
Co-reporter:Mika Uematsu and Dale L. Boger
The Journal of Organic Chemistry 2014 Volume 79(Issue 20) pp:9699-9703
Publication Date(Web):September 23, 2014
DOI:10.1021/jo501839x
A short, asymmetric synthesis of a cyclic N-acyl O-amino phenol duocarmycin prodrug subject to reductive activation based on the simplified 1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) DNA alkylation subunit is described. A key element of the approach entailed treatment of iodo-epoxide 7, prepared by N-alkylation of 6 with (S)-glycidal 3-nosylate, with EtMgBr at room temperature to directly provide the optically pure alcohol 8 in 78% yield (99% ee) derived from an effective metal–halogen exchange and subsequent regioselective intramolecular 6-endo-tet cyclization. Following O-debenzylation, introduction of a protected N-methylhydroxamic acid, direct trannannular spirocyclization, and subsequent stereoelectronically controlled acid-catalyzed cleavage of the resulting cyclopropane (HCl), further improvements in a unique intramolecular cyclization with N–O bond formation originally introduced for formation of the reductively labile prodrug functionality are detailed.
Co-reporter:Steven P. Breazzano ; Yam B. Poudel
Journal of the American Chemical Society 2013 Volume 135(Issue 4) pp:1600-1606
Publication Date(Web):January 8, 2013
DOI:10.1021/ja3121394
Herein we report a systematic study of the Larock indole annulation designed to explore the scope and define the generality of its use in macrocyclization reactions, its use in directly accessing the chloropeptin I versus II DEF ring system as well as key unnatural isomers, its utility for both peptide-derived and more conventional carbon-chain based macrocycles, and its extension to intramolecular cyclizations with formation of common ring sizes. The studies define a powerful method complementary to the Stille or Suzuki cross-coupling reactions for the synthesis of cyclic or macrocyclic ring systems containing an embedded indole, tolerating numerous functional groups and incorporating various (up to 28-membered) ring sizes. As a result of the efforts to expand the usefulness and scope of the reaction, we also disclose a catalytic variant of the reaction, along with a powerful Pd2(dba)3-derived catalyst system, and an examination of the factors impacting reactivity and catalysis.
Co-reporter:Katerina Otrubova ; Monica Brown ; Michael S. McCormick ; Gye W. Han ; Scott T. O’Neal ; Benjamin F. Cravatt ; Raymond C. Stevens ; Aron H. Lichtman
Journal of the American Chemical Society 2013 Volume 135(Issue 16) pp:6289-6299
Publication Date(Web):April 12, 2013
DOI:10.1021/ja4014997
The design and characterization of α-ketoheterocycle fatty acid amide hydrolase (FAAH) inhibitors are disclosed that additionally and irreversibly target a cysteine (Cys269) found in the enzyme cytosolic port while maintaining the reversible covalent Ser241 attachment responsible for their rapid and initially reversible enzyme inhibition. Two α-ketooxazoles (3 and 4) containing strategically placed electrophiles at the C5 position of the pyridyl substituent of 2 (OL-135) were prepared and examined as inhibitors of FAAH. Consistent with the observed time-dependent noncompetitive inhibition, the cocrystal X-ray structure of 3 bound to a humanized variant of rat FAAH revealed that 3 was not only covalently bound to the active site catalytic nucleophile Ser241 as a deprotonated hemiketal, but also to Cys269 through the pyridyl C5-substituent, thus providing an inhibitor with dual covalent attachment in the enzyme active site. In vivo characterization of the prototypical inhibitors in mice demonstrates that they raise endogenous brain levels of FAAH substrates to a greater extent and for a much longer duration (>6 h) than the reversible inhibitor 2, indicating that the inhibitors accumulate and persist in the brain to completely inhibit FAAH for a prolonged period. Consistent with this behavior and the targeted irreversible enzyme inhibition, 3 reversed cold allodynia in the chronic constriction injury model of neuropathic pain in mice for a sustained period (>6 h) beyond that observed with the reversible inhibitor 2, providing effects that were unchanged over the 1–6 h time course monitored.
Co-reporter:Travis C. Turner, Kotaro Shibayama, and Dale L. Boger
Organic Letters 2013 Volume 15(Issue 5) pp:1100-1103
Publication Date(Web):February 19, 2013
DOI:10.1021/ol400135n
The regioselective intermolecular coupling reaction of vindoline with a wide range of substrates including β-ketoesters, β-diketones, β-ketoaldehydes, β-ketonitriles, malononitriles, and β-cyanoesters provides an opportunity for the synthesis of vinblastine analogues containing deep-seated changes in the upper velbanamine subunit. The transition-metal-free hypervalent iodine(III)-promoted intermolecular sp3/sp2 coupling, representing a special class of selective C–H activation with direct carbon–carbon bond formation, proceeds with generation of a quaternary center capable of incorporation of the vinblastine C16′ methyl ester and functionalized for subsequent divergent heterocycle introduction.
Co-reporter:Jian Xie, Amanda L. Wolfe, and Dale L. Boger
Organic Letters 2013 Volume 15(Issue 4) pp:868-870
Publication Date(Web):February 7, 2013
DOI:10.1021/ol303573f
The use of a powerful intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of an 1,3,4-oxadiazole in the divergent total synthesis of kopsinine (1), featuring an additional unique SmI2-promoted transannular cyclization reaction for formation of the bicyclo[2.2.2]octane central to its hexacyclic ring system, is detailed.
Co-reporter:Erica L. Campbell, Colin K. Skepper, Kuppusamy Sankar, Katharine K. Duncan, and Dale L. Boger
Organic Letters 2013 Volume 15(Issue 20) pp:5306-5309
Publication Date(Web):October 2, 2013
DOI:10.1021/ol402549n
A powerful tandem [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles initiated by a transannular [4 + 2] cycloaddition is detailed. An impressive four rings, four carbon–carbon bonds, and six stereocenters are set on each site of the newly formed central six-membered ring in a cascade thermal reaction that proceeds at temperatures as low as 80 °C. The resulting cycloadducts provide the basis for the synthesis of unique analogues of vinblastine containing metabolically benign deep-seated cyclic modifications at the C3/C4 centers of the vindoline-derived subunit of the natural product.
Co-reporter:Kristin D. Schleicher ; Yoshikazu Sasaki ; Annie Tam ; Daisuke Kato ; Katharine K. Duncan
Journal of Medicinal Chemistry 2013 Volume 56(Issue 2) pp:483-495
Publication Date(Web):December 19, 2012
DOI:10.1021/jm3014376
The total synthesis of a systematic series of vinblastine analogues that contain deep-seated structural modifications to the core ring system of the lower vindoline subunit is described. Complementary to the vindoline 6,5 DE ring system, compounds with 5,5, 6,6, and the reversed 5,6 membered DE ring systems were prepared. Both the natural cis and unnatural trans 6,6-membered ring systems proved accessible, with the latter representing a surprisingly effective class for analogue design. Following Fe(III)-promoted coupling with catharanthine and in situ oxidation to provide the corresponding vinblastine analogues, their evaluation provided unanticipated insights into how the structure of the vindoline subunit contributes to activity. Two potent analogues (81 and 44) possessing two different unprecedented modifications to the vindoline subunit core architecture were discovered that matched the potency of the comparison natural products and both lack the 6,7-double bond whose removal in vinblastine leads to a 100-fold drop in activity.
Co-reporter:Erick K. Leggans ; Katharine K. Duncan ; Timothy J. Barker ; Kristin D. Schleicher
Journal of Medicinal Chemistry 2013 Volume 56(Issue 3) pp:628-639
Publication Date(Web):December 17, 2012
DOI:10.1021/jm3015684
A systematic series of previously inaccessible key C20′ urea and thiourea derivatives of vinblastine were prepared from 20′-aminovinblastine that was made accessible through a unique Fe(III)/NaBH4-mediated alkene functionalization reaction of anhydrovinblastine. Their examination defined key structural features of the urea-based analogues that contribute to their properties and provided derivatives that match or exceed the potency of vinblastine by as much as 10-fold in cell-based functional assays, which is directly related to their relative tubulin binding affinity. In contrast to expectations based on apparent steric constraints of the tubulin binding site surrounding the vinblastine C20′ center depicted in an X-ray cocrystal structure, remarkably large C20′ urea derivatives are accommodated.
Co-reporter:Joseph R. Pinchman
Journal of Medicinal Chemistry 2013 Volume 56(Issue 10) pp:4116-4124
Publication Date(Web):April 25, 2013
DOI:10.1021/jm4004494
The selective functionalization of vancomycin aglycon derivatives through conversion of the E-ring aryl chloride to a reactive boronic acid and its use in the synthesis of a systematic series of vancomycin E-ring analogues are described. The series was used to examine the E-ring chloride impact in binding d-Ala-d-Ala and on antimicrobial activity. In contrast to the reduced activity of the unsubstituted E-ring derivatives, hydrophobic and relatively nonpolar substituents approach or match the chloro-substituted vancomycin and were insensitive to the electronic character of the substituent (e.g., Cl vs CN/OMe), whereas highly polar substituents fail to provide the enhancements. Moreover, the active permethylated vancomycin aglycon derivatives exhibit VanB VRE antimicrobial activity at levels that approach (typically within 2-fold) their activity against sensitive bacteria. The robust borylation reaction also enabled the functionalization of a minimally protected vancomycin aglycon (N-Boc-vancomycin aglycon) and provides a direct method for the preparation of previously inaccessible analogues.
Co-reporter:Amanda L. Wolfe ; Katharine K. Duncan ; Nikhil K. Parelkar ; Douglas Brown ; George A. Vielhauer
Journal of Medicinal Chemistry 2013 Volume 56(Issue 10) pp:4104-4115
Publication Date(Web):April 29, 2013
DOI:10.1021/jm400413r
Two novel cyclic N-acyl O-amino phenol prodrugs are reported as new members of a unique class of reductively cleaved prodrugs of the duocarmycin family of natural products. These prodrugs were explored with the expectation that they may be cleaved selectively within hypoxic tumor environments that have intrinsically higher concentrations of reducing nucleophiles and were designed to liberate the free drug without the release of an extraneous group. In vivo evaluation of the prodrug 6 showed that it exhibits extraordinary efficacy (T/C > 1500, L1210; 6/10 one year survivors), substantially exceeding that of the free drug, that its therapeutic window of activity is much larger, permitting a dosing ≥40-fold higher than the free drug, and yet that it displays a potency in vivo that approaches the free drug (within 3-fold). Clearly, the prodrug 6 benefits from either its controlled slow release of the free drug or its preferential intracellular reductive cleavage.
Co-reporter:Amanda L. Wolfe ; Katharine K. Duncan ; James P. Lajiness ; Kaicheng Zhu ; Adam S. Duerfeldt
Journal of Medicinal Chemistry 2013 Volume 56(Issue 17) pp:6845-6857
Publication Date(Web):August 14, 2013
DOI:10.1021/jm400665c
Two systematic series of increasingly hydrophilic derivatives of duocarmycin SA that feature the incorporation of ethylene glycol units (n = 1–5) into the methoxy substituents of the trimethoxyindole subunit are described. These derivatives exhibit progressively increasing water solubility along with progressive decreases in cell growth inhibitory activity and DNA alkylation efficiency with the incremental ethylene glycol unit incorporations. Linear relationships of cLogP with −log IC50 for cell growth inhibition and −log AE (AE = cell-free DNA alkylation efficiency) were observed, with the cLogP values spanning the productive range of 2.5–0.49 and the −log IC50 values spanning the range of 11.2–6.4, representing IC50 values that vary by a factor of 105 (0.008 to 370 nM). The results quantify the fundamental role played by the hydrophobic character of the compound in the expression of the biological activity of members in this class (driving the intrinsically reversible DNA alkylation reaction) and define the stunning magnitude of its effect.
Co-reporter:Timothy J. Barker, Katharine K. Duncan, Katerina Otrubova, and Dale L. Boger
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 10) pp:985-988
Publication Date(Web):September 9, 2013
DOI:10.1021/ml400281w
A series of disubstituted C20′-urea derivatives of vinblastine were prepared from 20′-aminovinblastine that was made accessible through a unique Fe(III)/NaBH4-mediated alkene functionalization reaction of anhydrovinblastine. Three analogues were examined across a panel of 15 human tumor cell lines, displaying remarkably potent cell growth inhibition activity (avg. IC50 = 200–300 pM), being 10–200-fold more potent than vinblastine (avg. IC50 = 6.1 nM). Significantly, the analogues also display further improved activity against the vinblastine-resistant HCT116/VM46 cell line that bears the clinically relevant overexpression of Pgp, exhibiting IC50 values on par with that of vinblastine against the sensitive HCT116 cell line, 100–200-fold greater than the activity of vinblastine against the resistant HCT116/VM46 cell line, and display a reduced 10–20-fold activity differential between the matched sensitive and resistant cell lines (vs 100-fold for vinblastine).Keywords: C20′-urea derivatives; chemotherapy; Vinblastine; vincristine;
Co-reporter:Joseph R. Pinchman, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 17) pp:4817-4819
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmcl.2013.06.080
A vancomycin aglycon analogue that possesses a reduced C-ring and an intact E-ring chloride was prepared and its antimicrobial activity towards Staphylococcus aureus and binding affinity to model cell wall ligands were established. Comparison of the derivative with a series of vancomycin aglycon analogues that possess and lack the chloro substituents on the aryl C- and E-rings defines the impact and further refines the role the C-ring chloride plays in promoting both target binding affinity and binding selectivity for d-Ala-d-Ala and its impact on antimicrobial activity.
Co-reporter:Landon R. Whitby and Dale L. Boger
Accounts of Chemical Research 2012 Volume 45(Issue 10) pp:1698
Publication Date(Web):July 16, 2012
DOI:10.1021/ar300025n
Transient protein–protein interactions (PPIs) are essential components in cellular signaling pathways as well as in important processes such as viral infection, replication, and immune suppression. The unknown or uncharacterized PPIs involved in such interaction networks often represent compelling therapeutic targets for drug discovery. To date, however, the main strategies for discovery of small molecule modulators of PPIs are typically limited to structurally characterized targets.Recent developments in molecular scaffolds that mimic the side chain display of peptide secondary structures have yielded effective designs, but few screening libraries of such mimetics are available to interrogate PPI targets. We initiated a program to prepare a comprehensive small molecule library designed to mimic the three major recognition motifs that mediate PPIs (α-helix, β-turn, and β-strand). Three libraries would be built around templates designed to mimic each such secondary structure and substituted with all triplet combinations of groups representing the 20 natural amino acid side chains. When combined, the three libraries would contain a member capable of mimicking the key interaction and recognition residues of most targetable PPIs.In this Account, we summarize the results of the design, synthesis, and validation of an 8000 member α-helix mimetic library and a 4200 member β-turn mimetic library. We expect that the screening of these libraries will not only provide lead structures against α-helix- or β-turn-mediated protein–protein or peptide–receptor interactions, even if the nature of the interaction is unknown, but also yield key insights into the recognition motif (α-helix or β-turn) and identify the key residues mediating the interaction. Consistent with this expectation, the screening of the libraries against p53/MDM2 and HIV-1 gp41 (α-helix mimetic library) or the opioid receptors (β-turn mimetic library) led to the discovery of library members expected to mimic the known endogenous ligands. These efforts led to the discovery of high-affinity α-helix mimetics (Ki = 0.7 μM) against HIV-1 gp41 as well as high-affinity and selective β-turn mimetics (Ki = 80 nM) against the κ-opioid receptor. The results suggest that the use of such comprehensive libraries of peptide secondary structure mimetics, built around effective molecular scaffolds, constitutes a powerful method of interrogating PPIs. These structures provide small molecule modulators of PPI networks for therapeutic target validation, lead compound discovery, and the identification of modulators of biological processes for further study.
Co-reporter:Akinori Okano ; Robert C. James ; Joshua G. Pierce ; Jian Xie
Journal of the American Chemical Society 2012 Volume 134(Issue 21) pp:8790-8793
Publication Date(Web):May 8, 2012
DOI:10.1021/ja302808p
Development of a general Ag(I)-promoted reaction for the conversion of thioamides to amidines is disclosed. This reaction was employed to prepare a key series of vancomycin aglycon residue 4 substituted amidines that were used to clarify their interaction with model ligands of peptidoglycan precursors and explore their resulting impact on antimicrobial properties.
Co-reporter:Hiroaki Gotoh ; Justin E. Sears ; Albert Eschenmoser
Journal of the American Chemical Society 2012 Volume 134(Issue 32) pp:13240-13243
Publication Date(Web):August 2, 2012
DOI:10.1021/ja306229x
A definition of the scope of aromatic substrates that participate with catharanthine in an Fe(III)-mediated coupling reaction, an examination of the key structural features of catharanthine required for participation in the reaction, and the development of a generalized indole functionalization reaction that bears little structural relationship to catharanthine itself are detailed. In addition to providing insights into the mechanism of the Fe(III)-mediated coupling reaction of catharanthine with vindoline suggesting the reaction conducted in acidic aqueous buffer may be radical mediated, the studies provide new opportunities for the preparation of previously inaccessible vinblastine analogs and define powerful new methodology for the synthesis of indole-containing natural and unnatural products.
Co-reporter:Timothy J. Barker
Journal of the American Chemical Society 2012 Volume 134(Issue 33) pp:13588-13591
Publication Date(Web):August 3, 2012
DOI:10.1021/ja3063716
A powerful Fe(III)/NaBH4-mediated free radical hydrofluorination of unactivated alkenes is disclosed using Selectfluor reagent as a source of fluorine and resulting in exclusive Markovnikov addition. In contrast to the traditional and unmanageable free radical hydrofluorination of alkenes, the Fe(III)/NaBH4-mediated reaction is conducted under exceptionally mild reaction conditions (0 °C, 5 min, CH3CN/H2O). The reaction can be conducted open to the air and with water as a cosolvent and demonstrates an outstanding substrate scope and functional group tolerance.
Co-reporter:Erick K. Leggans, Timothy J. Barker, Katharine K. Duncan, and Dale L. Boger
Organic Letters 2012 Volume 14(Issue 6) pp:1428-1431
Publication Date(Web):February 28, 2012
DOI:10.1021/ol300173v
An Fe(III)/NaBH4-mediated reaction for the functionalization of unactivated alkenes is described defining the alkene substrate scope, establishing the exclusive Markovnikov addition, exploring a range of free radical traps, examining the Fe(III) salt and initiating hydride source, introducing H2O–cosolvent mixtures, and exploring catalytic variants. Its use led to the preparation of a novel, potent, and previously inaccessible C20′-vinblastine analogue.
Co-reporter:James P. Lajiness, Wanlong Jiang, and Dale L. Boger
Organic Letters 2012 Volume 14(Issue 8) pp:2078-2081
Publication Date(Web):April 5, 2012
DOI:10.1021/ol300599p
Divergent total syntheses of (+)-spegazzinine (1) and (−)-aspidospermine (2) and their extensions to the synthesis of C19-epi-aspidospermine and C3-epi-spegazzinine are detailed, confirming the relative stereochemistry and establishing the absolute configuration of (+)-spegazzinine. A powerful intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of a 1,3,4-oxadiazole provided the pentacyclic skeleton and all the requisite stereochemistry of the natural products in a single reaction that forms three rings, four C–C bonds, and five stereocenters.
Co-reporter:Amanda L. Wolfe ; Katharine K. Duncan ; Nikhil K. Parelkar ; Scott J. Weir ; George A. Vielhauer
Journal of Medicinal Chemistry 2012 Volume 55(Issue 12) pp:5878-5886
Publication Date(Web):May 31, 2012
DOI:10.1021/jm300330b
A unique heterocyclic carbamate prodrug of seco-CBI-indole2 that releases no residual byproduct is reported as a new member of a class of hydrolyzable prodrugs of the duocarmycin and CC-1065 family of natural products. The prodrug was designed to be activated by hydrolysis of a carbamate releasing the free drug without the cleavage release of a traceable extraneous group. Unlike prior carbamate prodrugs examined that are rapidly cleaved in vivo, the cyclic carbamate was found to be exceptionally stable to hydrolysis under both chemical and biological conditions providing a slow, sustained release of the exceptionally potent free drug. An in vivo evaluation of the prodrug found that its efficacy exceeded that of the parent drug, that its therapeutic window of efficacy versus toxicity is much larger than the parent drug, and that its slow free drug release permitted the safe and efficacious use of doses 150-fold higher than the parent compound.
Co-reporter:Robert C. James, Joshua G. Pierce, Akinori Okano, Jian Xie, and Dale L. Boger
ACS Chemical Biology 2012 Volume 7(Issue 5) pp:797
Publication Date(Web):February 13, 2012
DOI:10.1021/cb300007j
The glycopeptide antibiotics are the most important class of drugs used in the treatment of resistant bacterial infections including those caused by methicillin-resistant Staphylococcus aureus (MRSA). After more than 50 years of clinical use, the emergence of glycopeptide-resistant Gram-positive pathogens such as vancomycin-resistant enterococci (VRE) and vancomycin-resistant Staphylococcus aureus (VRSA) presents a serious global challenge to public health at a time few new antibiotics are being developed. This has led to renewed interest in the search for additional effective treatments including the development of new derivatives of the glycopeptide antibiotics. General approaches have been explored for modifying glycopeptide antibiotics, typically through the derivatization of the natural products themselves or more recently through chemical total synthesis. In this Perspective, we consider recent efforts to redesign glycopeptide antibiotics for the treatment of resistant microbial infections, including VRE and VRSA, and examine their future potential for providing an even more powerful class of antibiotics that are even less prone to bacterial resistance.
Co-reporter:Katerina Otrubova and Dale L. Boger
ACS Chemical Neuroscience 2012 Volume 3(Issue 5) pp:340
Publication Date(Web):December 20, 2011
DOI:10.1021/cn2001206
A summary of the initial discovery and characterization of the enzyme fatty acid amide hydrolase (FAAH), and the subsequent advancement of an important class of competitive, reversible, potent, and selective inhibitors is presented. Initially explored using substrate-inspired inhibitors bearing electrophilic carbonyls, the examination of α-ketoheterocyle-based inhibitors of FAAH with the benefit of a unique activity-based protein-profiling (ABPP)-based proteome-wide selectivity assay, a powerful in vivo biomarker-based in vivo screen, and subsequent retrospective X-ray cocrystal structures with the enzyme, is summarized. These efforts defined the impact of the central activating heterocycle and its key substituents, provided key simplifications in the C2 acyl side chain and clear interpretations for the unique role and subsequent optimization of the central activating heterocycle, and established the basis for the recent further conformational constraints in the C2 acyl side chain, providing potent, long-acting, orally active FAAH inhibitors.Keywords: FAAH; Fatty acid amide hydrolase; pain; sleep; α-ketoheterocycles
Co-reporter:Landon R. Whitby, Kristopher E. Boyle, Lifeng Cai, Xiaoqian Yu, Miriam Gochin, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 8) pp:2861-2865
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmcl.2012.02.062
The evaluation of a comprehensive α-helix mimetic library for binding the gp41 NHR hydrophobic pocket recognizing an intramolecular CHR α-helix provided a detailed depiction of structural features required for binding and led to the discovery of small molecule inhibitors (Ki 0.6–1.3 μM) that not only match or exceed the potency of those disclosed over the past decade, but that also exhibit effective activity in a cell–cell fusion assay (IC50 5–8 μM).The evaluation of a comprehensive α-helix mimetic library for binding the gp41 NHR hydrophobic pocket recognizing an intramolecular CHR α-helix provided a detailed depiction of structural features required for binding and led to the discovery of small molecule inhibitors (Ki 0.6–1.3 μM) that not only match or exceed the potency of those disclosed over the past decade, but that also exhibit effective activity in a cell–cell fusion assay (IC50 5–8 μM).
Co-reporter:Mauro Mileni ; Joie Garfunkle ; Cyrine Ezzili ; Benjamin F. Cravatt ; Raymond C. Stevens
Journal of the American Chemical Society 2011 Volume 133(Issue 11) pp:4092-4100
Publication Date(Web):February 28, 2011
DOI:10.1021/ja110877y
Two cocrystal X-ray structures of the exceptionally potent α-ketoheterocycle inhibitor 1 (Ki = 290 pM) bound to a humanized variant of rat fatty acid amide hydrolase (FAAH) are disclosed, representing noncovalently and covalently bound states of the same inhibitor with the enzyme. Key to securing the structure of the noncovalently bound state of the inhibitor was the inclusion of fluoride ion in the crystallization conditions that is proposed to bind the oxyanion hole precluding inhibitor covalent adduct formation with stabilization of the tetrahedral hemiketal. This permitted the opportunity to detect important noncovalent interactions stabilizing the binding of the inhibitor within the FAAH active site independent of the covalent reaction. Remarkably, noncovalently bound 1 in the presence of fluoride appears to capture the active site in the same “in action” state with the three catalytic residues Ser241−Ser217−Lys142 occupying essentially identical positions observed in the covalently bound structure of 1, suggesting that this technique of introducing fluoride may have important applications in structural studies beyond inhibiting substrate or inhibitor oxyanion hole binding. Key insights to emerge from the studies include the observations that noncovalently bound 1 binds in its ketone (not gem diol) form, that the terminal phenyl group in the acyl side chain of the inhibitor serves as the key anchoring interaction overriding the intricate polar interactions in the cytosolic port, and that the role of the central activating heterocycle is dominated by its intrinsic electron-withdrawing properties. These two structures are also briefly compared with five X-ray structures of α-ketoheterocycle-based inhibitors bound to FAAH recently disclosed.
Co-reporter:Landon R. Whitby ; Yoshio Ando ; Vincent Setola ; Peter K. Vogt ; Bryan L. Roth
Journal of the American Chemical Society 2011 Volume 133(Issue 26) pp:10184-10194
Publication Date(Web):May 24, 2011
DOI:10.1021/ja201878v
The design and synthesis of a β-turn mimetic library as a key component of a small-molecule library targeting the major recognition motifs involved in protein–protein interactions is described. Analysis of a geometric characterization of 10 245 β-turns in the protein data bank (PDB) suggested that trans-pyrrolidine-3,4-dicarboxamide could serve as an effective and synthetically accessible library template. This was confirmed by initially screening select compounds against a series of peptide-activated GPCRs that recognize a β-turn structure in their endogenous ligands. This validation study was highlighted by identification of both nonbasic and basic small molecules with high affinities (Ki = 390 and 23 nM, respectively) for the κ-opioid receptor (KOR). Consistent with the screening capabilities of collaborators and following the design validation, the complete library was assembled as 210 mixtures of 20 compounds, providing a total of 4200 compounds designed to mimic all possible permutations of 3 of the 4 residues in a naturally occurring β-turn. Unique to the design and because of the C2 symmetry of the template, a typical 20 × 20 × 20-mix (8000 compounds prepared as 400 mixtures of 20 compounds) needed to represent 20 variations in the side chains of three amino acid residues reduces to a 210 × 20-mix, thereby simplifying the library synthesis and subsequent screening. The library was prepared using a solution-phase synthetic protocol with liquid–liquid or liquid–solid extractions for purification and conducted on a scale that insures its long-term availability for screening campaigns. Screening the library against the human opioid receptors (KOR, MOR, and DOR) identified not only the activity of library members expected to mimic the opioid receptor peptide ligands but also additional side-chain combinations that provided enhanced receptor binding selectivities (>100-fold) and affinities (as low as Ki = 80 nM for KOR). A key insight to emerge from the studies is that the phenol of Tyr in endogenous ligands bearing the H-Tyr-Pro-Trp/Phe-Phe-NH2 β-turn is important for MOR binding but may not be important for KOR (accommodated, but not preferred) and that the resulting selectivity for KOR observed with its removal can be increased by replacing the phenol OH with a chlorine substituent, further enhancing KOR affinity.
Co-reporter:Steven P. Breazzano
Journal of the American Chemical Society 2011 Volume 133(Issue 45) pp:18495-18502
Publication Date(Web):October 13, 2011
DOI:10.1021/ja208570q
Recently, we reported the first total synthesis of chloropeptin II (1, complestatin), the more strained and challenging of the two naturally occurring chloropeptins. Central to the design of the approach and by virtue of a single-step, acid-catalyzed ring expansion rearrangement of chloropeptin II to chloropeptin I, the route also provided a total synthesis of chloropeptin I. Herein, we report a complementary and divergent oxidation of chloropeptin II (1, complestatin) to either complestatin A (2, neuroprotectin A) or complestatin B (3, neuroprotectin B), providing the first synthesis of the natural products and establishing their remaining stereochemical assignments. Key to the approach to complestatin A (2, neuroprotectin A) was the development of two different single-step indole oxidations (HCl–DMSO and NBS, THF–H2O) that avoid the rearrangement of chloropeptin II (1) to chloropeptin I (4), providing the 2-oxindole 2 in superb yields (93% and 82%). With a mechanistic understanding of features that impact the latter oxidation and an appreciation of the intrinsic reactivity of the chloropeptin II indole, its modification (NCS, THF–H2O; Cs2CO3, DMF–H2O) provided a two-step, single-pot oxidation of chloropeptin II (1) to afford directly the 3-hydroxy-2-oxindole complestatin B (3, neuroprotectin B). Extensive studies conducted on the fully functionalized synthetic DEF ring system of chloropeptin II were key to the unambiguous assignment of the stereochemistry as well as the exploration and subsequent development of the mild oxidation conditions used in the synthesis of complestatin A and B.
Co-reporter:Jian Xie ; Akinori Okano ; Joshua G. Pierce ; Robert C. James ; Simon Stamm ; Christine M. Crane
Journal of the American Chemical Society 2011 Volume 134(Issue 2) pp:1284-1297
Publication Date(Web):December 20, 2011
DOI:10.1021/ja209937s
The total synthesis of [Ψ[C(═S)NH]Tpg4]vancomycin aglycon (8) and its unique AgOAc-promoted single-step conversion to [Ψ[C(═NH)NH]Tpg4]vancomycin aglycon (7), conducted on a fully deprotected substrate, are disclosed. The synthetic approach not only permits access to 7, but it also allows late-stage access to related residue 4 derivatives, alternative access to [Ψ[CH2NH]Tpg4]vancomycin aglycon (6) from a common late-stage intermediate, and provides authentic residue 4 thioamide and amidine derivatives of the vancomycin aglycon that will facilitate ongoing efforts on their semisynthetic preparation. In addition to early stage residue 4 thioamide introduction, allowing differentiation of one of seven amide bonds central to the vancomycin core structure, the approach relied on two aromatic nucleophilic substitution reactions for formation of the 16-membered diaryl ethers in the CD/DE ring systems, an effective macrolactamization for closure of the 12-membered biaryl AB ring system, and the defined order of CD, AB, and DE ring closures. This order of ring closures follows their increasing ease of thermal atropisomer equilibration, permitting the recycling of any newly generated unnatural atropisomer under progressively milder thermal conditions where the atropoisomer stereochemistry already set is not impacted. Full details of the evaluation of 7 and 8 along with several related key synthetic compounds containing the core residue 4 amidine and thioamide modifications are reported. The binding affinity of compounds containing the residue 4 amidine with the model d-Ala-d-Ala ligand 2 was found to be only 2–3 times less than the vancomycin aglycon (5), and this binding affinity is maintained with the model d-Ala-d-Lac ligand 4, representing a nearly 600-fold increase in affinity relative to the vancomycin aglycon. Importantly, the amidines display effective dual, balanced binding affinity for both ligands (Ka2/4 = 0.9–1.05), and they exhibit potent antimicrobial activity against VanA resistant bacteria (E. faecalis, VanA VRE) at a level accurately reflecting these binding characteristics (MIC = 0.3–0.6 μg/mL), charting a rational approach forward in the development of antibiotics for the treatment of vancomycin-resistant bacterial infections. In sharp contrast, 8 and related residue 4 thioamides failed to bind either 2 or 4 to any appreciable extent, do not exhibit antimicrobial activity, and serve to further underscore the remarkable behavior of the residue 4 amidines.
Co-reporter:Erin D. Anderson
Journal of the American Chemical Society 2011 Volume 133(Issue 31) pp:12285-12292
Publication Date(Web):July 7, 2011
DOI:10.1021/ja204856a
A systematic study of the inverse electron demand Diels–Alder reactions of 1,2,3-triazines is disclosed, including an examination of the impact of a C5 substituent. Such substituents were found to exhibit a remarkable impact on the cycloaddition reactivity of the 1,2,3-triazine without altering, and perhaps even enhancing, the intrinsic cycloaddition regioselectivity. The study revealed not only that the reactivity may be predictably modulated by a C5 substituent (R = CO2Me > Ph > H) but also that the impact is of a magnitude to convert 1,2,3-triazine (1) and its modest cycloaddition scope into a heterocyclic azadiene system with a reaction scope that portends extensive synthetic utility, expanding the range of participating dienophiles. Significantly, the studies define a now powerful additional heterocyclic azadiene, complementary to the isomeric 1,2,4-triazines and 1,3,5-triazines, capable of dependable participation in inverse electron demand Diels–Alder reactions, extending the number of complementary heterocyclic ring systems accessible with implementation of the methodology.
Co-reporter:Jian Xie ; Joshua G. Pierce ; Robert C. James ; Akinori Okano
Journal of the American Chemical Society 2011 Volume 133(Issue 35) pp:13946-13949
Publication Date(Web):August 8, 2011
DOI:10.1021/ja207142h
The emergence of bacteria resistant to vancomycin, often the antibiotic of last resort, poses a major health problem. Vancomycin-resistant bacteria sense a glycopeptide antibiotic challenge and remodel their cell wall precursor peptidoglycan terminus from d-Ala-d-Ala to d-Ala-d-Lac, reducing the binding of vancomycin to its target 1000-fold and accounting for the loss in antimicrobial activity. Here, we report [Ψ[C(═NH)NH]Tpg4]vancomycin aglycon designed to exhibit the dual binding to d-Ala-d-Ala and d-Ala-d-Lac needed to reinstate activity against vancomycin-resistant bacteria. Its binding to a model d-Ala-d-Ala ligand was found to be only 2-fold less than vancomycin aglycon and this affinity was maintained with a model d-Ala-d-Lac ligand, representing a 600-fold increase relative to vancomycin aglycon. Accurately reflecting these binding characteristics, it exhibits potent antimicrobial activity against vancomycin-resistant bacteria (MIC = 0.31 μg/mL, VanA VRE). Thus, a complementary single atom exchange in the vancomycin core structure (O → NH) to counter the single atom exchange in the cell wall precursors of resistant bacteria (NH → O) reinstates potent antimicrobial activity and charts a rational path forward for the development of antibiotics for the treatment of vancomycin-resistant bacterial infections.
Co-reporter:Cyrine Ezzili ; Mauro Mileni ; Nicholas McGlinchey ; Jonathan Z. Long ; Steven G. Kinsey ; Dustin G. Hochstatter ; Raymond C. Stevens ; Aron H. Lichtman ; Benjamin F. Cravatt ; Edward J. Bilsky
Journal of Medicinal Chemistry 2011 Volume 54(Issue 8) pp:2805-2822
Publication Date(Web):March 23, 2011
DOI:10.1021/jm101597x
A series of α-ketooxazoles containing conformational constraints in the C2 acyl side chain of 2 (OL-135) were examined as inhibitors of fatty acid amide hydrolase (FAAH). Only one of the two possible enantiomers displayed potent FAAH inhibition (S vs R enantiomer), and their potency is comparable or improved relative to 2, indicating that the conformational restriction in the C2 acyl side chain is achievable. A cocrystal X-ray structure of the α-ketoheterocycle 12 bound to a humanized variant of rat FAAH revealed its binding details, confirmed that the (S)-enantiomer is the bound active inhibitor, shed light on the origin of the enantiomeric selectivity, and confirmed that the catalytic Ser241 is covalently bound to the electrophilic carbonyl as a deprotonated hemiketal. Preliminary in vivo characterization of the inhibitors 12 and 14 is reported demonstrating that they raise brain anandamide levels following either intraperitoneal (ip) or oral (po) administration indicative of effective in vivo FAAH inhibition. Significantly, the oral administration of 12 caused dramatic accumulation of anandamide in the brain, with peak levels achieved between 1.5 and 3 h, and these elevations were maintained over 9 h. Additional studies of these two representative members of the series (12 and 14) in models of thermal hyperalgesia and neuropathic pain are reported, including the demonstration that 12 administered orally significantly attenuated mechanical (>6 h) and cold (>9 h) allodynia for sustained periods consistent with its long-acting effects in raising the endogenous concentration of anandamide.
Co-reporter:Hiroaki Gotoh, Katharine K. Duncan, William M. Robertson, and Dale L. Boger
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 12) pp:948
Publication Date(Web):October 18, 2011
DOI:10.1021/ml200236a
A study on the impact of catharanthine C10 and C12 indole substituents on the biomimetic Fe(III)-mediated coupling with vindoline led to the discovery and characterization of two new and substantially more potent derivatives, 10′-fluorovinblastine and 10′-fluorovincristine. In addition to defining a pronounced and unanticipated substituent effect on the biomimetic coupling, fluorine substitution at C10′, which minimally alters the natural products, was found to uniquely enhance the activity 8-fold against both sensitive (IC50 = 800 pM, HCT116) and vinblastine-resistant tumor cell lines (IC50 = 80 nM, HCT166/VM46). As depicted in the X-ray structure of vinblastine bound to tubulin, this site resides at one end of the upper portion of the T-shaped conformation of the tubulin-bound molecule, suggesting that the 10′-fluorine substituent makes critical contacts with the protein at a hydrophobic site uniquely sensitive to steric interactions.Keywords: 10′-Fluorovinblastine; 10′-fluorovincristine; synthesis; vinblastine; vinca alkaloids; vincristine
Co-reporter:Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 17) pp:4944
Publication Date(Web):1 September 2011
DOI:10.1016/j.bmcl.2011.08.013
Co-reporter:Christopher P. Burke ; Nadia Haq
Journal of the American Chemical Society 2010 Volume 132(Issue 7) pp:2157-2159
Publication Date(Web):January 28, 2010
DOI:10.1021/ja9097252
A total synthesis of phostriecin (2), previously known as sultriecin (1), its structural reassignment as a phosphate versus sulfate monoester, and the assignment of its relative and absolute stereochemistry are disclosed herein. Key elements of the work, which provided first the originally assigned sulfate monoester 1 and then the reassigned and renamed phosphate monoester 2, relied on diagnostic 1H NMR spectroscopic properties of the natural product for the assignment of relative and absolute stereochemistry as well as the subsequent structural reassignment, and a convergent asymmetric total synthesis to provide the unequivocal authentic materials. Key steps of the synthetic approach include a Brown allylation for diastereoselective introduction of the C9 stereochemistry, an asymmetric CBS reduction to establish the lactone C5-stereochemistry, diastereoselective oxidative ring expansion of an α-hydroxyfuran to access the pyran lactone precursor, and single-step installation of the sensitive Z,Z,E-triene unit through a chelation-controlled cuprate addition with installation of the C11 stereochemistry. The approach allows ready access to analogues that can now be used to probe important structural features required for protein phosphatase 2A inhibition, the mechanism of action defined herein.
Co-reporter:James P. Lajiness
Journal of the American Chemical Society 2010 Volume 132(Issue 39) pp:13936-13940
Publication Date(Web):September 14, 2010
DOI:10.1021/ja106986f
The synthesis of 1,2,10,11-tetrahydro-9H-cyclobuta[c]benzo[e]indol-4-one (17, CbBI), which contains a deep-seated fundamental structural modification in the CC-1065 and duocarmycin alkylation subunit consisting of the incorporation of a ring-expanded fused cyclobutane (vs cyclopropane), its chemical and structural characterization, and its incorporation into a key analogue of the natural products are detailed. The approach to the preparation of CbBI was based on a precedented (Ar-3′ and Ar-5′) but previously unknown Ar-4′ spirocyclization of a phenol onto a tethered alkyl halide to form the desired cyclobutane. The conditions required for the implementation of the Ar-4′ spirocyclization indicate that the entropy of activation substantially impacts the rate of reaction relative to that for the much more facile Ar-3′ spirocyclization, while the higher enthalpy of activation slows the reaction relative to an Ar-5′ spirocyclization. The characterization of the CbBI-based agents revealed their exceptional stability and exquisite reaction regioselectivity, and a single-crystal X-ray structure analysis of N-Boc-CbBI (13) revealed their structural origins. The reaction regioselectivity may be attributed to the stereoelectronic alignment of the two available cyclobutane bonds with the cyclohexadienone π-system, which resides in the bond that extends to the less substituted cyclobutane carbon for 13. The remarkable stability of N-Boc-CbBI (which is stable even at pH 1) relative to N-Boc-CBI containing a cyclopropane (t1/2 = 133 h at pH 3) may be attributed to a combination of the increased extent of vinylogous amide conjugation, the nonoptimal geometric alignment of the cyclobutane with the activating cyclohexadienone, and the intrinsic but modestly lower strain energy (1.8 kcal/mol) of a cyclobutane versus a cyclopropane.
Co-reporter:Erica L. Campbell ; Andrea M. Zuhl ; Christopher M. Liu
Journal of the American Chemical Society 2010 Volume 132(Issue 9) pp:3009-3012
Publication Date(Web):February 11, 2010
DOI:10.1021/ja908819q
A total synthesis of the Aspidosperma alkaloids (+)-fendleridine and (+)-1-acetylaspidoalbidine is detailed, providing access to both enantiomers of the natural products and establishing their absolute configuration. Central to the synthetic approach is a powerful intramolecular [4+2]/[3+2] cycloaddition cascade of a 1,3,4-oxadiazole in which the pentacyclic skeleton and all the stereochemistry of the natural products are assembled in a reaction that forms three rings, four C−C bonds, and five stereogenic centers including three contiguous quaternary centers, and introduces the correct oxidation state at C19 in a single synthetic operation. The final tetrahydrofuran bridge is subsequently installed in one step, enlisting an intramolecular alcohol addition to an iminium ion generated by nitrogen-assisted opening of the cycloadduct oxido bridge, with a modification that permits release of useful functionality (a ketone) at the cleavage termini.
Co-reporter:Daisuke Kato ; Yoshikazu Sasaki
Journal of the American Chemical Society 2010 Volume 132(Issue 11) pp:3685-3687
Publication Date(Web):February 26, 2010
DOI:10.1021/ja910695e
A concise asymmetric total synthesis of (−)-vindoline (1) is detailed based on a tandem intramolecular [4+2]/[3+2] cycloaddition cascade of a 1,3,4-oxadiazole inspired by the natural product structure, in which the tether linking the initiating dienophile and oxadiazole bears a chiral substituent that controls the facial selectivity of the initiating Diels−Alder reaction and sets absolute stereochemistry of the remaining six stereocenters in the cascade cycloadduct. This key reaction introduces three rings and four C−C bonds central to the pentacyclic ring system setting all six stereocenters and introducing essentially all the functionality found in the natural product in a single step. Implementation of the approach also required the development of a unique ring expansion reaction to provide a six-membered ring suitably functionalized for introduction of the Δ(6, 7)-double bond found in the core structure of vindoline and defined our use of a protected hydroxymethyl group as the substituent used to control the stereochemical course of the cycloaddition cascade.
Co-reporter:Hiroyuki Shimamura ; Steven P. Breazzano ; Joie Garfunkle ; F. Scott Kimball ; John D. Trzupek
Journal of the American Chemical Society 2010 Volume 132(Issue 22) pp:7776-7783
Publication Date(Web):May 17, 2010
DOI:10.1021/ja102304p
Full details of the initial development and continued examination of a powerful intramolecular palladium(0)-mediated indole annulation for macrocyclization closure of the strained 16-membered biaryl ring system found in complestatin (1, chloropeptin II) and the definition of factors impacting its intrinsic atropodiastereoselectivity are described. Its examination and use in an alternative, second-generation total synthesis of complestatin are detailed in which the order of the macrocyclization reactions was reversed from our first-generation total synthesis. In this approach and with the ABCD biaryl ether ring system in place, the key Larock cyclization was conducted with substrate 36 (containing four phenols, five secondary amides, one carbamate, and four labile aryl chlorides) and provided the product 37 (56%) exclusively as a single atropisomer (>20:1, detection limits) possessing the natural (R)-configuration. In this instance, the complexity of the substrate and the reverse macrocyclization order did not diminish the atropodiastereoselectivity; rather, it provided an improvement over the 4:1 selectivity that was observed with the analogous substrate used to provide the isolated DEF ring system in our first-generation approach. Just as significant, the atroposelectivity represents a complete reversal of the diasteroselectivity observed with analogous macrocyclizations conducted using a Suzuki biaryl coupling.
Co-reporter:Porino Va ; Erica L. Campbell ; William M. Robertson
Journal of the American Chemical Society 2010 Volume 132(Issue 24) pp:8489-8495
Publication Date(Web):June 2, 2010
DOI:10.1021/ja1027748
A remarkably concise seven- to eight-step total synthesis of a systematic series of key vinblastine derivatives is detailed and used to characterize the importance and probe the role of the C5 ethyl substituent (R = H, Me, Pr, CH═CH2, C≡CH, CH2OH, and CHO vs Et). The analogues, which bear deep-seated structural changes accessible only by total synthesis, were prepared using a powerful intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles ideally suited for use in the assemblage of the vindoline-derived lower subunit followed by their incorporation into the vinblastine analogues through the use of a single-step biomimetic coupling with catharanthine. The evaluation of the series revealed that the tubulin binding site surrounding this C5 substituent is exquisitely sensitive to the presence (Et > H, 10-fold), size (Me ≤ Et > Pr, 10-fold), shape (Et > CH═CH2 and C≡CH, >4-fold), and polarity (Et > CHO > CH2OH, >10−20-fold) of this substituent and that on selected occasions only a C5 methyl group may provide analogues that approach the activity observed with the naturally occurring C5 ethyl group.
Co-reporter:Paresma R. Patel
Journal of the American Chemical Society 2010 Volume 132(Issue 25) pp:8527-8529
Publication Date(Web):June 8, 2010
DOI:10.1021/ja103933n
The first intramolecular thermal reactions of cyclopropenone ketals are reported and the work examined substrates tethered to an electron-deficient olefin bearing a single electron-withdrawing substituent. Whereas the intermolecular variants of the reactions provide only the products of an endo-selective [1 + 2] cycloaddition or a carbonyl addition reaction of a thermally generated π-delocalized singlet vinylcarbene, the intramolecular variants provide either [1 + 2] or [3 + 2] cycloadducts in reactions that depend on the reaction conditions, the alkene-activating substituent, and the nature of the tethering. In addition to providing key mechanistic insights into the thermal [3 + 2] cycloaddition reaction for such substrates, they were also found to proceed under conditions that reflect the ease and regioselectivity of the cyclopropenone ketal cleavage for π-delocalized singlet vinylcarbene generation. The most effective combination of structural features that impact the reactivity was observed with substrates bearing an aldehyde- or ketone-substituted electron-deficient olefin and incorporating an aryl cyclopropenone ketal substituent built into the linking tether. Simply warming a solution of such substrates in toluene at 80−100 °C directly provided the [3 + 2] cycloadducts in excellent yields (60−88%) under mild thermal reaction conditions.
Co-reporter:James S. Oakdale and Dale L. Boger
Organic Letters 2010 Volume 12(Issue 5) pp:1132-1134
Publication Date(Web):February 11, 2010
DOI:10.1021/ol100146b
Two complementary concise total syntheses of lycogarubin C (1) and lycogalic acid (2, aka chromopyrrolic acid) are detailed utilizing a 1,2,4,5-tetrazine → 1,2-diazine → pyrrole Diels−Alder strategy and enlisting acetylenic dienophiles.
Co-reporter:Paresma R. Patel and Dale L. Boger
Organic Letters 2010 Volume 12(Issue 15) pp:3540-3543
Publication Date(Web):July 8, 2010
DOI:10.1021/ol101375a
The first intramolecular cycloaddition reactions of cyclopropenone ketals with tethered electron-deficient, electron-rich, and neutral 1-substituted dienes are reported, constituting inverse electron demand, normal, and neutral Diels−Alder reactions, that provide exclusively the exo [4 + 2] cycloaddition products without the intervention of [1 + 2], [3 + 2], or [3 + 4] cycloadducts in reactions whose courses do not depend on the reaction conditions, the diene activating substituent, or the nature of the tethering.
Co-reporter:Mauro Mileni ; Joie Garfunkle ; Cyrine Ezzili ; F. Scott Kimball ; Benjamin F. Cravatt ; Raymond C. Stevens
Journal of Medicinal Chemistry 2010 Volume 53(Issue 1) pp:230-240
Publication Date(Web):November 19, 2009
DOI:10.1021/jm9012196
Three cocrystal X-ray structures of the α-ketoheterocycle inhibitors 3−5 bound to a humanized variant of fatty acid amide hydrolase (FAAH) are disclosed and comparatively discussed alongside those of 1 (OL-135) and its isomer 2. These five X-ray structures systematically probe each of the three active site regions key to substrate or inhibitor binding: (1) the conformationally mobile acyl chain-binding pocket and membrane access channel responsible for fatty acid amide substrate and inhibitor acyl chain binding, (2) the atypical active site catalytic residues and surrounding oxyanion hole that covalently binds the core of the α-ketoheterocycle inhibitors captured as deprotonated hemiketals mimicking the tetrahedral intermediate of the enzyme-catalyzed reaction, and (3) the cytosolic port and its uniquely important imbedded ordered water molecules and a newly identified anion binding site. The detailed analysis of their key active site interactions and their implications on the interpretation of the available structure−activity relationships are discussed providing important insights for future design.
Co-reporter:Christine M. Crane ; Joshua G. Pierce ; Siegfried S. F. Leung ; Julian Tirado-Rives ; William L. Jorgensen
Journal of Medicinal Chemistry 2010 Volume 53(Issue 19) pp:7229-7235
Publication Date(Web):September 20, 2010
DOI:10.1021/jm100946e
A select series of methyl ether derivatives of vancomcyin aglycon were prepared and examined for antimicrobial activity against vancomycin-sensitive Staphylococcus aureus and vancomycin-resistant Enterococci faecalis as well as their binding affinity for d-Ala-d-Ala and d-Ala-d-Lac. The intent of the study was to elucidate the role selected key methyl groups may play in the improvement of the in vitro antimicrobial profile of the tetra methyl ether derivative of vancomycin aglycon against vancomycin-resistant Enterococci faecalis previously reported. In these studies, methodology for selective derivatization of the A-, B-, and D-ring was developed that defines the relative reactivity of the four phenols of vancomycin aglycon, providing a foundation for future efforts for site-directed modification of the vancomycin aglycon core.
Co-reporter:James P. Lajiness ; William M. Robertson ; Irene Dunwiddie ; Melinda A. Broward ; George A. Vielhauer ; Scott J. Weir
Journal of Medicinal Chemistry 2010 Volume 53(Issue 21) pp:7731-7738
Publication Date(Web):October 13, 2010
DOI:10.1021/jm1010397
A series of N-acyl O-amino derivatives of seco-CBI-indole2 are reported and examined as prototypical members of a unique class of reductively activated (cleaved) prodrugs of the duocarmycin and CC-1065 family of antitumor agents. These prodrugs were designed to be potentially preferentially activated in hypoxic tumor environments which carry an intrinsically higher concentration of “reducing” nucleophiles (e.g., thiols) capable of activating such derivatives by nucleophilic cleavage of a weak N−O bond. A remarkable range of stabilities and a resulting direct correlation with in vitro/in vivo biological potencies was observed for these prodrugs, even enlisting subtle variations in the electronic and steric environment around the weak N−O bond. An in vivo evaluation of several of the prodrugs demonstrates that some approach the potency and exceed the efficacy of the free drug itself (CBI-indole2), suggesting the prodrugs may offer an additional advantage related to a controlled or targeted release.
Co-reporter:William M. Robertson, David B. Kastrinsky, Inkyu Hwang, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 9) pp:2722-2725
Publication Date(Web):1 May 2010
DOI:10.1016/j.bmcl.2010.03.078
The synthesis and evaluation of a key series of analogs of duocarmycin SA, bearing a single substituent at the C5′ position of the DNA binding subunit, are described.
Co-reporter:Cyrine Ezzili, Katerina Otrubova, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 20) pp:5959-5968
Publication Date(Web):15 October 2010
DOI:10.1016/j.bmcl.2010.08.048
Key studies leading to the discovery and definition of the role of endogenous fatty acid amide signaling molecules are summarized.Key studies leading to the discovery and role of endogenous fatty acid amide signaling molecules are summarized.
Co-reporter:Annie Tam, Hiroaki Gotoh, William M. Robertson, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 22) pp:6408-6410
Publication Date(Web):15 November 2010
DOI:10.1016/j.bmcl.2010.09.091
The examination of the catharanthine C16 substituent effects on the Fe(III)-promoted biomimetic coupling reaction with vindoline is detailed, confirming the importance of the presence of a C16 electron-withdrawing substituent, and establishing an unanticipated unique role (>10-fold) that the C16 methyl ester plays in the expression of the natural product properties. Thus, replacement of the vinblastine C16′ methyl ester with an ethyl ester (10-fold), a cyano group (100-fold), an aldehyde (100-fold), a hydroxymethyl group (1000-fold) or a primary carboxamide (>1000-fold) led to surprisingly large reductions in cytotoxic activity.
Co-reporter:Christopher P. Burke, Mark R. Swingle, Richard E. Honkanen, and Dale L. Boger
The Journal of Organic Chemistry 2010 Volume 75(Issue 22) pp:7505-7513
Publication Date(Web):July 29, 2010
DOI:10.1021/jo1010203
Full details of the total synthesis of phostriecin (2), the assignment of its relative and absolute stereochemistry, and the resultant structural reassignment of the natural product previously represented as sultriecin (1), a phosphate versus sulfate monoester, are detailed. Studies with authentic material confirmed that phostriecin, but not sultriecin, is an effective and selective inhibitor of protein phosphatase 2A (PP2A) defining a mechanism of action responsible for its antitumor activity. The extension of the studies to the synthesis and evaluation of a series of key synthetic analogues is disclosed that highlights the importance of the natural product phosphate monoester (vs sulfate or free alcohol, both inactive and >250-fold), the α,β-unsaturated lactone (12-fold), and the hydrophobic Z,Z,E-triene tail (C12−C22, ca. 200-fold) including the unique importance of its unsaturation (50-fold, and no longer PP2A selective).
Co-reporter:Joie Garfunkle ; F. Scott Kimball ; John D. Trzupek ; Shinobu Takizawa ; Hiroyuki Shimamura ; Masaki Tomishima
Journal of the American Chemical Society 2009 Volume 131(Issue 44) pp:16036-16038
Publication Date(Web):October 19, 2009
DOI:10.1021/ja907193b
The first total synthesis of chloropeptin II (1, complestatin) is disclosed. Key elements of the approach include the use of an intramolecular Larock indole synthesis for the initial macrocyclization, adopting conditions that permit utilization of a 2-bromoaniline, incorporating a terminal alkyne substituent (−SiEt3) that sterically dictates the indole cyclization regioselectivity, and benefiting from an aniline protecting group (−Ac) that enhances the atropdiastereoselectivity and diminishes the strained indole reactivity toward subsequent electrophilic reagents. Not only did this key reaction provide the fully functionalized right-hand ring system of 1 in superb conversion (89%) and good atropdiastereoselectivity (4:1 R:S), but it also represents the first reported example of what will prove to be a useful Larock macrocyclization strategy. Subsequent introduction of the left-hand ring system enlisting an aromatic nucleophilic substitution reaction for macrocyclization with biaryl ether formation completed the assemblage of the core bicyclic structure of 1. Intrinsic in the design of the approach and by virtue of the single-step acid-catalyzed conversion of chloropeptin II (1) to chloropeptin I (2), the route also provides a total synthesis of 2.
Co-reporter:Ryan C. Clark, Sang Yeul Lee, Mark Searcey and Dale L. Boger
Natural Product Reports 2009 vol. 26(Issue 4) pp:465-477
Publication Date(Web):03 Feb 2009
DOI:10.1039/B821676B
Covering: up to December 2008
Co-reporter:Christine M. Crane
Journal of Medicinal Chemistry 2009 Volume 52(Issue 5) pp:1471-1476
Publication Date(Web):February 11, 2009
DOI:10.1021/jm801549b
The synthesis and biological evaluation of a series of vancomycin aglycon analogues bearing alternative residue 1 N-methyl-d-amino acids are described. The analogues were prepared to define whether H-bonding d-amino acids could improve the affinity for the model ligands N,N′-Ac2-l-Lys-d-Ala-d-Ala (2) and N,N′-Ac2-l-Lys-d-Ala-d-Lac (3) and improve antimicrobial activity against vancomycin-sensitive or vancomycin-resistant bacteria. Additionally, a series of analogues with appended nucleophiles (hydrazines and amines) on the residue 1 d-amino acids are described that were examined for their ability to react with the C-terminal ester of 3, forming a covalent attachment of l-Lys-d-Ala to the natural product analogues.
Co-reporter:Karen S. MacMillan
Journal of Medicinal Chemistry 2009 Volume 52(Issue 19) pp:5771-5780
Publication Date(Web):July 29, 2009
DOI:10.1021/jm9006214
Co-reporter:Karl E. Hauschild, James S. Stover, Dale L. Boger, Aseem Z. Ansari
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 14) pp:3779-3782
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmcl.2009.04.097
Determining the sequence specifity of DNA binding molecules is a non-trivial task. Here we describe the development of a platform for assaying the sequence specificity of DNA ligands using label free detection on high density DNA microarrays. This is achieved by combining Cognate Site Identification (CSI) with Fluorescence Intercalation Displacement (FID) to create CSI–FID. We use the well-studied small molecule DNA ligand netropsin to develop this high throughput platform. Analysis of the DNA binding properties of protein- and small molecule-based libraries with CSI–FID will advance the development of genome-anchored molecules for therapeutic purposes.
Co-reporter:Xiao Fang, Joonwoo Nam, Dongwoo Shin, Yosup Rew, Dale L. Boger, Suzanne Walker
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 21) pp:6189-6191
Publication Date(Web):1 November 2009
DOI:10.1016/j.bmcl.2009.09.001
Ramoplanin is a potent lipoglycodepsipeptide antibiotic that is active against a wide range of Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococcus (VRE). It acts as an inhibitor of peptidoglycan (PG) biosynthesis that disrupts glycan chain polymerization by binding and sequestering Lipid II, a PG precursor. Herein, we report the functional antimicrobial activity (MIC, S. aureus) and fundamental biochemical assessments against a peptidoglycan glycosyltransferase (Escherichia coli PBP1b) of a set of key alanine scan analogues of ramoplanin that provide insight into the importance and role of each of its individual amino acid residues.
Co-reporter:Jin Shi, James S. Stover, Landon R. Whitby, Peter K. Vogt, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 21) pp:6038-6041
Publication Date(Web):1 November 2009
DOI:10.1016/j.bmcl.2009.09.044
The preparation and evaluation of a series of inhibitors of Myc/Max dimerization and Myc-induced cell transformation are described providing mycmycin-1 (3) and mycmycin-2 (4).
Co-reporter:Karen S. MacMillan, James P. Lajiness, Carlota Lopez Cara, Romeo Romagnoli, William M. Robertson, Inkyu Hwang, Pier Giovanni Baraldi, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 24) pp:6962-6965
Publication Date(Web):15 December 2009
DOI:10.1016/j.bmcl.2009.10.063
The design, synthesis, and preliminary evaluation of methyl 1,2,8,8a-tetrahydrocyclopropa[c]thieno[3,2-e]indol-4-one-6-carboxylate (CTI) derivatives are detailed representing a single atom change (N to S) embedded in the duocarmycin SA alkylation subunit.
Co-reporter:Landon R. Whitby, Andrew M. Lee, Stefan Kunz, Michael B.A. Oldstone, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 14) pp:3771-3774
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmcl.2009.04.098
The comparative characterization of a series of 4-acyl-1,6-dialkylpiperazin-2-ones as potent cell entry inhibitors of the hemorrhagic fever arenavirus Lassa (LASV) is disclosed. The resolution and examination of the individual enantiomers of the prototypical LASV cell entry inhibitor 3 (16G8) is reported and the more potent (–)-enantiomer was found to be 15-fold more active than the corresponding (+)-enantiomer. The absolute configuration of (–)-3 was established by asymmetric synthesis of the active inhibitor (–)-(S)-3 (lassamycin-1). A limited deletion scan of lassamycin-1 defined key structural features required of the prototypical inhibitors.
Co-reporter:Carla M. Gauss, Akiyuki Hamasaki, Jay P. Parrish, Karen S. MacMillan, Thomas J. Rayl, Inkyu Hwang, Dale L. Boger
Tetrahedron 2009 65(33) pp: 6591-6599
Publication Date(Web):
DOI:10.1016/j.tet.2009.02.065
Co-reporter:Ryan C. Clark, Sang Yeul Lee, Inkyu Hwang, Mark Searcey, Dale L. Boger
Tetrahedron Letters 2009 50(26) pp: 3151-3153
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.01.081
Co-reporter:Sang Yeul Lee, Dale L. Boger
Tetrahedron 2009 65(16) pp: 3281-3284
Publication Date(Web):
DOI:10.1016/j.tet.2008.09.029
Co-reporter:Mark S. Tichenor and Dale L. Boger
Natural Product Reports 2008 vol. 25(Issue 2) pp:220-226
Publication Date(Web):06 Nov 2007
DOI:10.1039/B705665F
Covering: up to 2007
Co-reporter:F. Scott Kimball ; F. Anthony Romero ; Cyrine Ezzili ; Joie Garfunkle ; Thomas J. Rayl ; Dustin G. Hochstatter ; Inkyu Hwang
Journal of Medicinal Chemistry 2008 Volume 51(Issue 4) pp:937-947
Publication Date(Web):February 5, 2008
DOI:10.1021/jm701210y
A series of α-ketooxazoles containing conformational constraints in the flexible C2 acyl side chain of 2 (OL-135) and representative oxazole C5 substituents were prepared and examined as inhibitors of fatty acid amide hydrolase (FAAH). Exceptionally potent and selective FAAH inhibitors emerged from the series (e.g., 6, Ki = 200 and 260 pM for rat and rhFAAH). With simple and small C5 oxazole substituents, each series bearing a biphenylethyl, phenoxyphenethyl, or (phenoxymethyl)phenethyl C2 side chain was found to follow a well-defined linear relationship between −log Ki and Hammett σp of a magnitude (ρ = 2.7–3.0) that indicates that the substituent electronic effect dominates, confirming its fundamental importance to the series and further establishing its predictive value. Just as significantly, the nature of the C5 oxazole substituent substantially impacts the selectivity of the inhibitors whereas the effect of the C2 acyl chain was more subtle but still significant even in the small series examined. Combination of these independent features, which display generalized trends across a range of inhibitor series, simultaneously improves FAAH potency and selectivity and can provide exquisitely selective and potent FAAH inhibitors.
Co-reporter:Joie Garfunkle ; Cyrine Ezzili ; Thomas J. Rayl ; Dustin G. Hochstatter ; Inkyu Hwang
Journal of Medicinal Chemistry 2008 Volume 51(Issue 15) pp:4392-4403
Publication Date(Web):July 16, 2008
DOI:10.1021/jm800136b
The synthesis and evaluation of a refined series of α-ketoheterocycles based on the oxazole 2 (OL-135) incorporating systematic changes in the central heterocycle bearing a key set of added substituents are described. The nature of the central heterocycle, even within the systematic and minor perturbations explored herein, significantly influenced the inhibitor activity: 1,3,4-oxadiazoles and 1,2,4-oxadiazoles 9 > tetrazoles, the isomeric 1,2,4-oxadiazoles 10, 1,3,4-thiadiazoles > oxazoles including 2 > 1,2-diazines > thiazoles > 1,3,4-triazoles. Most evident in these trends is the observation that introduction of an additional heteroatom at position 4 (oxazole numbering, N > O > CH) substantially increases activity that may be attributed to a reduced destabilizing steric interaction at the FAAH active site. Added heterocycle substituents displaying well-defined trends may be utilized to enhance the inhibitor potency and, more significantly, to enhance the inhibitor selectivity. These trends, exemplified herein, emerge from both enhancements in the FAAH activity and simultaneous disruption of binding affinity for competitive off-target enzymes.
Co-reporter:Jessica K. DeMartino ; Inkyu Hwang ; Stephen Connelly ; Ian A. Wilson
Journal of Medicinal Chemistry 2008 Volume 51(Issue 17) pp:5441-5448
Publication Date(Web):August 8, 2008
DOI:10.1021/jm800555h
Glycinamide ribonucleotide transformylase (GAR Tfase) catalyzes the first of two formyl transfer steps in the de novo purine biosynthetic pathway that require folate cofactors and has emerged as a productive target for antineoplastic therapeutic intervention. The asymmetric synthesis and evaluation of the two diastereomers of 10-methylthio-DDACTHF (10R-3 and 10S-3) and related analogues as potential inhibitors of GAR Tfase are reported. This work, which defines the importance of the C10 stereochemistry for this class of inhibitors of GAR Tfase, revealed that both diastereomers are potent inhibitors of rhGAR Tfase (10R-3 Ki = 210 nM, 10S-3 Ki = 180 nM) that exhibit effective cell growth inhibition (CCRF-CEM IC50 = 80 and 50 nM, respectively), which is dependent on intracellular polyglutamation by folylpolyglutamate synthetase (FPGS) but not intracellular transport by the reduced folate carrier.
Co-reporter:Gregory I. Elliott, Juraj Velcicky, Hayato Ishikawa, YongKai Li,Dale L. Boger
Angewandte Chemie International Edition 2006 45(4) pp:620-622
Publication Date(Web):
DOI:10.1002/anie.200503024
Co-reporter:Gregory I. Elliott Dr.;Juraj Velcicky Dr.;Hayato Ishikawa Dr.;YongKai Li Dr.
Angewandte Chemie 2006 Volume 118(Issue 4) pp:
Publication Date(Web):15 DEC 2005
DOI:10.1002/ange.200503024
Per Tandem zum Pentacyclus: Eine intramolekulare Tandem-Kaskade aus [4+2]- und [3+2]-Cycloaddition überführt das 1,3,4-Oxadiazol (Z)-1 in das pentacyclische Gerüst 2 von (−)- und ent-(+)-Vindorosin. Dabei werden alle erforderlichen Substituenten und Funktionalitäten in einem einzigen Schritt eingeführt, in dem drei neue Ringe und vier C-C-Bindungen gebildet und alle sechs Schlüssel-Stereozentren erzeugt werden.
Co-reporter:Heng Cheng, Inkyu Hwang, Youhoon Chong, Ali Tavassoli, Michael E. Webb, Yan Zhang, Ian A. Wilson, Stephen J. Benkovic, Dale L. Boger
Bioorganic & Medicinal Chemistry 2005 Volume 13(Issue 10) pp:3593-3599
Publication Date(Web):16 May 2005
DOI:10.1016/j.bmc.2004.11.049
The synthesis and evaluation of N-{4-[5-(2,4-diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-2-(2,2,2-trifluoroacetyl)pentyl]benzoyl}-l-glutamic acid (2) as an inhibitor of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide ribonucleotide transformylase (AICAR Tfase) are reported. The inhibitor 2 was prepared in a convergent synthesis involving C-alkylation of methyl 4-(4,4,4-trifluoro-3-dimethylhydrazonobutyl)benzoate with 1-chloro-3-iodopropane followed by construction of the pyrimidinone ring. Compound 2 was found to be an effective inhibitor of recombinant human GAR Tfase (Ki = 0.50 μM), whereas it was inactive (Ki > 100 μM) against E. coli GAR Tfase as well as recombinant human AICAR Tfase. Compound 2 exhibited modest, purine-sensitive growth inhibitory activity against the CCRF-CEM cell line (IC50 = 6.0 μM).
Co-reporter:Youhoon Chong, Inkyu Hwang, Ali Tavassoli, Yan Zhang, Ian A. Wilson, Stephen J. Benkovic, Dale L. Boger
Bioorganic & Medicinal Chemistry 2005 Volume 13(Issue 10) pp:3587-3592
Publication Date(Web):16 May 2005
DOI:10.1016/j.bmc.2004.11.050
Structurally-related, but non-polyglutamylatable, derivatives of 10-CF3CO-DDACTHF (1), which incorporate l-glutamine (2) and l-isoglutamine (3) in place of l-glutamate, were prepared and evaluated as inhibitors of recombinant human (rh) GAR Tfase. While the l-glutamate α-carboxamide derivative 3 was much less effective as a rhGAR Tfase inhibitor (Ki = 4.8 μM) and inactive in cellular functional assays, the γ-carboxamide derivative 2 was found to be a potent and selective rhGAR Tfase inhibitor (Ki = 0.056 μM) being only 4-fold less potent than 1 (Ki = 0.015 μM). Moreover, 2 was effective in cellular functional assays exhibiting purine sensitive cytotoxic activity (IC50 = 300 nM, CCRF-CEM) only 20-fold less potent than 1 (IC50 = 16 nM), consistent with inhibition of de novo purine biosynthesis via selective inhibition of GAR Tfase. Like 1, 2 is transported into the cell by the reduced folate carrier. Unlike 1, the functional activity of 2 is not dependent upon FPGS polyglutamylation.
Co-reporter:Heng Cheng, Youhoon Chong, Inkyu Hwang, Ali Tavassoli, Yan Zhang, Ian A. Wilson, Stephen J. Benkovic, Dale L. Boger
Bioorganic & Medicinal Chemistry 2005 Volume 13(Issue 10) pp:3577-3585
Publication Date(Web):16 May 2005
DOI:10.1016/j.bmc.2004.12.004
The synthesis and evaluation of 10-methanesulfonyl-DDACTHF (1), 10-methanesulfonyl-5-DACTHF (2), and 10-methylthio-DDACTHF (3) as potential inhibitors of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide ribonucleotide transformylase (AICAR Tfase) are reported. The compounds 10-methanesulfonyl-DDACTHF (1, Ki = 0.23 μM), 10-methanesulfonyl-5-DACTHF (2, Ki = 0.58 μM), and 10-methylthio-DDACTHF (3, Ki = 0.25 μM) were found to be selective and potent inhibitors of recombinant human GAR Tfase. Of these, 3 exhibited exceptionally potent, purine sensitive growth inhibition activity (3, IC50 = 100 nM) against the CCRF–CEM cell line being 3-fold more potent than Lometrexol and 30-fold more potent than the parent, unsubstituted DDACTHF, whereas 1 and 2 exhibited more modest growth inhibition activity (1, IC50 = 1.0 μM and 2, IC50 = 2.0 μM).
Co-reporter:Yosup Rew;Dongwoo Shin
PNAS 2004 Volume 101 (Issue 33 ) pp:11977-11979
Publication Date(Web):2004-08-17
DOI:10.1073/pnas.0401419101
Ramoplanin is a potent antibiotic, first disclosed in 1984, that acts by inhibiting bacterial cell-wall biosynthesis. The
original ramoplanin complex was shown to consist of a mixture of three closely related compounds, ramoplanin A1–A3, of which
ramoplanin A2 is the most abundant. The structure of ramoplanin A2 was unambiguously established first through a series of
extensive spectroscopic studies, allowing complete stereochemical assignments and subsequently providing a minor reassignment
of the side-chain double-bond stereochemistry and, most recently, through total synthesis of authentic material. Here we report
the total syntheses of the aglycons of the minor components of the ramoplanin complex, A1 and A3, which unambiguously establish
their structure and provide an expected structural revision for the lipid side-chain double-bond stereochemistry.
Co-reporter:Thomas H Marsilje, Michael P Hedrick, Joel Desharnais, Ali Tavassoli, Yan Zhang, Ian A Wilson, Stephen J Benkovic, Dale L Boger
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 20) pp:4487-4501
Publication Date(Web):October 2003
DOI:10.1016/S0968-0896(03)00456-5
A series of simplified α-keto heterocycle, trifluoromethyl ketone, and formyl substituted folate analogues lacking the benzoylglutamate subunit were prepared and examined as potential inhibitors of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide transformylase (AICAR Tfase).A novel series of potential inhibitors of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide transformylase (AICAR Tfase) that incorporate an electrophilic carbonyl group is reported.
Co-reporter:Thomas H Marsilje, Michael P Hedrick, Joel Desharnais, Kevin Capps, Ali Tavassoli, Yan Zhang, Ian A Wilson, Stephen J Benkovic, Dale L Boger
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 20) pp:4503-4509
Publication Date(Web):October 2003
DOI:10.1016/S0968-0896(03)00457-7
The design and synthesis of 10-(2-benzoxazolcarbonyl)-DDACTHF (1) as an inhibitor of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide transformylase (AICAR Tfase) are reported. Ketone 1 and the corresponding alcohol 13 were evaluated for inhibition of GAR Tfase and AICAR Tfase and the former was found to be a potent inhibitor of recombinant human (rh) GAR Tfase (Ki=600 nM).The design and synthesis of 10-(2-benzoxazolcarbonyl)-DDACTHF as an inhibitor of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide transformylase (AICAR Tfase) are reported.
Co-reporter:Joel Desharnais, Inkyu Hwang, Yan Zhang, Ali Tavassoli, Justin Baboval, Stephen J Benkovic, Ian A Wilson, Dale L Boger
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 20) pp:4511-4521
Publication Date(Web):October 2003
DOI:10.1016/S0968-0896(03)00458-9
The synthesis and evaluation of analogues and key derivatives of 10-CF3CO-DDACTHF as inhibitors of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide transformylase (AICAR Tfase) are reported. Polyglutamate analogues of 1 were evaluated as inhibitors of Escherichia coli and recombinant human (rh) GAR Tfase, and AICAR Tfase. Although the pentaglutamate 6 was found to be the most active inhibitor of the series tested against rhGAR Tfase (Ki=0.004 μM), little distinction between the mono–pentaglutamate derivatives was observed (Ki=0.02–0.004 μM), suggesting that the principal role of the required polyglutamation of 1 is intracellular retention. In contrast, 1 and its defined polyglutamates 3–6 were much less inactive when tested against rhAICAR Tfase (Ki=65–0.120 μM) and very selective (≥100-fold) for rh versus E. coli GAR Tfase. Additional key analogues of 1 were examined (7 and 8) and found to be much less active (1000-fold) highlighting the exceptional characteristics of 1.The synthesis and evaluation of analogues and key derivatives of 10-CF3CO-DDACTHF (1) as inhibitors of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide transformylase (AICAR Tfase) are reported.
Co-reporter:Dale L. Boger ;Joel Desharnais;Kevin Capps
Angewandte Chemie 2003 Volume 115(Issue 35) pp:
Publication Date(Web):15 SEP 2003
DOI:10.1002/ange.200300574
Hochdurchsatzsynthese und -screening von Verbindungsbibliotheken haben große Erwartungen für die Wirkstoffsuche geweckt. Für die Herstellung solcher Bibliotheken an der Festphase und in Lösung sind leistungsfähige Methoden entwickelt worden. Die Synthesestrategie wird schon lange nicht mehr nach allgemeinen Erwägungen ausgewählt, sondern mit Blick auf die spezielle Fragestellung. In diesem Aufsatz beschreiben wir Unterschiede zwischen den beiden Methoden, die bei der Entwicklung neuer Programme helfen sollen. Als Beispiel beschreiben wir unsere Versuche, mithilfe von Lösungstechniken Modulatoren für zelluläre Signale durch Beeinflussung von Protein-Protein- oder Protein-DNA-Wechselwirkungen zu identifizieren. Das Vorurteil, niedermolekulare Substanzen seien für therapeutische Intervention an solchen Zielstrukturen ungeeignet, konnten wir beim Screening unserer Bibliotheken gegen eine repräsentative Auswahl extrazellulärer und intrazellulärer Targets widerlegen: Mithilfe zahlreicher Testmethoden identifizierten wir die ersten niedermolekularen Modulatoren für Protein-Protein-Wechselwirkungen und erarbeiteten ein allgemeines Verfahren für derartige Untersuchungen.
Co-reporter:Dale L. Boger ;Joel Desharnais;Kevin Capps
Angewandte Chemie International Edition 2003 Volume 42(Issue 35) pp:
Publication Date(Web):15 SEP 2003
DOI:10.1002/anie.200300574
The high-throughput synthesis and screening of compound libraries hold tremendous promise for drug discovery and powerful methods for both solid-phase and solution-phase library preparation have been introduced. The question of which approach (solution-phase versus solid-phase) is best for the preparation of chemical libraries has been replaced by which approach is most appropriate for a particular target or screen. Herein we highlight distinctions in the two approaches that might serve as useful considerations at the onset of new programs. This is followed by a more personal account of our own focus on solution-phase techniques for the preparation of libraries designed to modulate cellular signaling by targeting protein–protein or protein–DNA interactions. The screening of our libraries against a prototypical set of extracellular and intracellular targets, using a wide range of assay formats, provided the first small-molecule modulators of the protein–protein interactions studied, and a generalized approach for conducting such studies.
Co-reporter:Thomas H. Marsilje, Marc A. Labroli, Michael P. Hedrick, Qing Jin, Joel Desharnais, Stephen J. Baker, Lata T. Gooljarsingh, Joseph Ramcharan, Ali Tavassoli, Yan Zhang, Ian A. Wilson, G.Peter Beardsley, Stephen J. Benkovic, Dale L. Boger
Bioorganic & Medicinal Chemistry 2002 Volume 10(Issue 8) pp:2739-2749
Publication Date(Web):August 2002
DOI:10.1016/S0968-0896(02)00102-5
The synthesis of 10-formyl-DDACTHF (3) as a potential inhibitor of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide ribonucleotide transformylase (AICAR Tfase) is reported. Aldehyde 3, the corresponding γ- and α-pentaglutamates 21 and 25 and related agents were evaluated for inhibition of folate-dependent enzymes including GAR Tfase and AICAR Tfase. The inhibitors were found to exhibit potent cytotoxic activity (CCRF-CEM IC50 for 3=60 nM) that exceeded their enzyme inhibition potency [Ki (3)=6 and 1 μM for Escherichia coli GAR and human AICAR Tfase, respectively]. Cytotoxicity rescue by medium purines, but not pyrimidines, indicated that the potent cytotoxic activity is derived from selective purine biosynthesis inhibition and rescue by AICAR monophosphate established that the activity is derived preferentially from GAR versus AICAR Tfase inhibition. The potent cytotoxic compounds including aldehyde 3 lost activity against CCRF-CEM cell lines deficient in the reduced folate carrier (CCRF-CEM/MTX) or folylpolyglutamate synthase (CCRF-CEM/FPGS−) establishing that their potent activity requires both reduced folate carrier transport and polyglutamation. Unexpectedly, the pentaglutamates displayed surprisingly similar Ki's versus E. coli GAR Tfase and only modestly enhanced Ki's versus human AICAR Tfase. On the surface this initially suggested that the potent cytotoxic activity of 3 and related compounds might be due simply to preferential intracellular accumulation of the inhibitors derived from effective transport and polyglutamation (i.e., ca. 100-fold higher intracellular concentrations). However, a subsequent examination of the inhibitors against recombinant human GAR Tfase revealed they and the corresponding γ-pentaglutamates were unexpectedly much more potent against the human versus E. coli enzyme (Ki for 3, 14 nM against rhGAR Tfase versus 6 μM against E. coli GAR Tfase) which also accounts for their exceptional cytotoxic potency.The synthesis and evaluation of 10-formyl-DDACTHF (3) as a potential inhibitor of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide ribonucleotide transformylase (AICAR Tfase) are reported.
Co-reporter:Craig R. Woods, Nicolas Faucher, Bernd Eschgfaller, Kenneth W. Bair, Dale L. Boger
Bioorganic & Medicinal Chemistry Letters 2002 Volume 12(Issue 18) pp:2647-2650
Publication Date(Web):16 September 2002
DOI:10.1016/S0960-894X(02)00467-5
A series of saturated heterocyclic analogues of distamycin were prepared and examined. A fluorescent intercalator displacement (FID) assay conducted on p[dA]–p[dT] DNA to obtain C50 values and a hairpin deoxyoligonucleotide containing an A/T-rich binding site was used to evaluate DNA binding affinity. It is observed that saturated heterocycles greatly reduce the DNA binding relative to distamycin.A series of saturated heterocyclic analogues of distamycin was prepared and DNA binding affinity evaluated with a fluorescent intercalator displacement (FID) assay.
Co-reporter:Dale L. Boger, Marc A. Labroli, Thomas H. Marsilje, Qing Jin, Michael P. Hedrick, Stephen J. Baker, Jae Hoon Shim, Stephen J. Benkovic
Bioorganic & Medicinal Chemistry 2000 Volume 8(Issue 5) pp:1075-1086
Publication Date(Web):May 2000
DOI:10.1016/S0968-0896(00)00051-1
The synthesis and evaluation of a series of conformationally restricted analogues of 10-formyl-tetrahydrofolate as potential inhibitors of glycinamide ribonucleotide transformylase (GAR Tfase) or aminoimidazole carboxamide transformylase (AICAR Tfase) are reported.
Co-reporter:Dale L. Boger, Thomas H. Marsilje, René A. Castro, Michael P. Hedrick, Qing Jin, Stephen J. Baker, Jae Hoon Shim, Stephen J. Benkovic
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 13) pp:1471-1475
Publication Date(Web):3 July 2000
DOI:10.1016/S0960-894X(00)00271-7
The examination results of a novel series of potential inhibitors of glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazole carboxamide transformylase (AICAR Tfase) are reported. These agents incorporate an electrophilic fluoronitrophenyl group that can potentially react with an active site nucleophile or the substrate GAR/AICAR amine via nucleophilic aromatic substitution.
Co-reporter:Dale L Boger, Robert A Fecik, Jean E Patterson, Hiroshi Miyauchi, Matthew P Patricelli, Benjamin F Cravatt
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 23) pp:2613-2616
Publication Date(Web):4 December 2000
DOI:10.1016/S0960-894X(00)00528-X
Fatty acid amide hydrolase (FAAH), also referred to as oleamide hydrolase and anandamide amidohydrolase, is a serine hydrolase responsible for the degradation of endogenous oleamide and anandamide, fatty acid amides that function as chemical messengers. FAAH hydrolyzes a range of fatty acid amides, and the present study examines the relative rates of hydrolysis of a variety of natural and unnatural fatty acid primary amide substrates using pure recombinant rat FAAH.
Co-reporter:Dale L. Boger, Yan Chen, Carolyn A. Foster
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 15) pp:1741-1744
Publication Date(Web):7 August 2000
DOI:10.1016/S0960-894X(00)00336-X
The aza analogue of the cyclic heptadepsipeptide HUN-7293 (1), which is a potent naturally occurring inhibitor of inducible cell adhesion molecule expression, and its C23 (MLEU3 C2) epimer were prepared via solution-phase synthesis. Biological evaluations of these two compounds as inhibitors of cell adhesion molecules expression are detailed.
Co-reporter:Joel Goldberg;Shigeki Satoh;Yves Ambroise;Steven B. Cohen;Peter K. Vogt;Dale L. Boger
Helvetica Chimica Acta 2000 Volume 83(Issue 8) pp:1825-1845
Publication Date(Web):15 AUG 2000
DOI:10.1002/1522-2675(20000809)83:8<1825::AID-HLCA1825>3.0.CO;2-4
The development of a solution-phase approach to the rapid, parallel synthesis of highly functionalized piperazinones in only four steps starting from N-Boc-iminodiacetic acid is detailed. The efforts represent the extension of the solution-phase synthesis of combinatorial libraries from N-Boc-iminodiacetic acid to non-amide-based libraries where simple liquid-liquid extractions are employed to purify all reaction products. This methodology was applied to the synthesis of a diverse 150-member library with substituents in three positions of the piperazinone core. Screening results from a luciferase reporter assay indicate that a number of library members are novel repressors of LEF-1/β-catenin-mediated transcription, and may be effective agents against colorectal tumors. Two secondary libraries (100 members each) designed from these lead structures were synthesized and screened, providing additional active agents and insight into key structure-activity relationships in the series. These compounds represent only the second class of small molecules which repress transcription of reporter genes containing LEF-1 responsive elements, and the first group not based on DNA minor-groove-binding agents.
Co-reporter:Dale L. Boger;Haruhiko Sato;Aaron E. Lerner;Michael P. Hedrick;Robert A. Fecik;Hiroshi Miyauchi;Gordon D. Wilkie;Bryce J. Austin;Matthew P. Patricelli;Benjamin F. Cravatt
PNAS 2000 Volume 97 (Issue 10 ) pp:5044-5049
Publication Date(Web):2000-05-09
DOI:10.1073/pnas.97.10.5044
The development of exceptionally potent inhibitors of fatty acid amide hydrolase (FAAH), the enzyme responsible for the degradation
of oleamide (an endogenous sleep-inducing lipid), and anandamide (an endogenous ligand for cannabinoid receptors) is detailed.
The inhibitors may serve as useful tools to clarify the role of endogenous oleamide and anandamide and may prove to be useful
therapeutic agents for the treatment of sleep disorders or pain. The combination of several features—an optimal C12–C8 chain
length, π-unsaturation introduction at the corresponding arachidonoyl Δ8,9/Δ11,12 and oleoyl Δ9,10 location, and an α-keto N4 oxazolopyridine with incorporation of a second weakly basic nitrogen provided FAAH inhibitors
with Kis that drop below 200 pM and are 102–103 times more potent than the corresponding trifluoromethyl ketones.
Co-reporter:Dale L. Boger;Mark W. Ledeboer;Masaharu Kume;Qing Jin
Angewandte Chemie International Edition 1999 Volume 38(Issue 16) pp:
Publication Date(Web):6 AUG 1999
DOI:10.1002/(SICI)1521-3773(19990816)38:16<2424::AID-ANIE2424>3.0.CO;2-9
The relative and absolute stereochemistry of the naturally occurring potent HIV reverse transcriptase (RT) inhibitors 1 and 2, quinoxapeptin A and B, were established by total synthesis. Their synthetic precursor 3 (dubbed quinoxapeptin C) was found to be a more potent HIV-1 RT inhibitor and to lack the potent cytotoxic activity characteristic of 1 and 2.
Co-reporter:Dale L. Boger;Mark W. Ledeboer;Masaharu Kume;Qing Jin
Angewandte Chemie 1999 Volume 111(Issue 16) pp:
Publication Date(Web):6 AUG 1999
DOI:10.1002/(SICI)1521-3757(19990816)111:16<2533::AID-ANGE2533>3.0.CO;2-I
Die relative und die absolute Konfiguration der natürlich vorkommenden, wirksamen Inhibitoren der Reversen Transkriptase (RT) von HIV, Chinoxapeptin A und B (1 bzw. 2), wurden durch Totalsynthese bestimmt. Die Synthesevorstufe 3 (Chinoxapeptin C) erwies sich als noch wirksamerer HIV-1-RT-Inhibitor, ohne die für 1 und 2 charakteristische, hohe Cytotoxizität.
Co-reporter:Dale L. Boger;Hui Cai
Angewandte Chemie International Edition 1999 Volume 38(Issue 4) pp:
Publication Date(Web):24 FEB 1999
DOI:10.1002/(SICI)1521-3773(19990215)38:4<448::AID-ANIE448>3.0.CO;2-W
The subtleties of the structure and function of bleomycin A2 (1), a clinically employed antitumor agent that derives its biological properties through the sequence-selective cleavage of DNA in a process that is dependent on the metal ion and O2, have been unraveled in a stepwise manner. Systematic modifications in the structure of 1 enabled many of the subtle functional roles of the individual subunits and their substituents in the efficiency, selectivity, and preference (double strand versus single strand) of DNA cleavage to be elucidated.
Co-reporter:Dale L. Boger;Hui Cai
Angewandte Chemie 1999 Volume 111(Issue 4) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19990215)111:4<470::AID-ANGE470>3.0.CO;2-M
Stückweise entschlüsselt wurden die Feinheiten der Struktur und Funktion von Bleomycin A21, einem klinisch eingesetzten Antitumormittel, das seine biologische Wirkung über die Metallionen- und O2-abhängige, sequenzselektive Spaltung von DNA ausübt. Durch systematisches Modifizieren der Struktur von 1 war es möglich, die subtilen funktionellen Rollen der einzelnen Molekülteile und ihrer Substituenten für Effizienz, Selektivität und Präferenz (Doppelstrang- gegenüber Einzelstrang-DNA) der DNA-Spaltung aufzuklären.
Co-reporter: Dale L. Boger; Douglas S. Johnson
Angewandte Chemie 1996 Volume 108(Issue 13‐14) pp:
Publication Date(Web):31 JAN 2006
DOI:10.1002/ange.19961081306
Wir fassen hier Arbeiten zusammen, die sich mit den DNA-Alkylierungs-Eigenschaften einer Klasse wirksamer Cytostatica/Antibiotica, zu denen CC–1065 und die Duocarmycine zählen, befaßten, sowie Untersuchungen, die für diese Verbindungen die elementaren Zusammenhänge zwischen Struktur, funktioneller Reaktivität und biologischem Verhalten ermittelten. Neben dem Studium der Naturstoffe selbst haben Untersuchungen mit strukturell stark veränderten, synthetischen Substraten und den nicht in der Natur vorkommenden („nichtnatürlichen”) Enantiomeren der Naturstoffe und verwandter Analoga dazu beigetragen, die strukturelle Grundlage für die sequenzselektive Alkylierung von Duplex-DNA zu bestimmen, und den Schlüssel zum Verständnis der elementaren Zusammenhänge zwischen chemischer Struktur, funktioneller Reaktivität und biologischem Verhalten geliefert. Die charakteristische DNA-Alkylierung erfolgt durch reversible, stereoelektronisch gesteuerte Adenin-N3-Addition an das am wenigsten substituierte Kohlenstoffatom des aktivierten Antibioticum-Cyclopropanrings innerhalb AT-reicher Bindungsstellen in der kleinen Furche. Sowohl die natürlichen als auch die nichtnatürlichen Enantiomere alkylieren DNA, und in beiden Fällen wird die Sequenzselektivität durch die bevorzugte nichtkovalente Bindung der Substrate an AT-reiche Regionen innerhalb der schmaleren und tieferen kleinen Furche und durch die Zugänglichkeit der Alkylierungsstelle beim Eindringen in diese Furche gesteuert und dominiert. Unter den elementaren Beziehungen zwischen Struktur und Eigenschaften fällt besonders der direkte Zusammenhang zwischen chemischer oder funktioneller Stabilität und biologischer Wirksamkeit auf. In kurzer Zeit wurden vereinfachte, auf Naturstoff-Leitbildern basierende und leicht zugängliche synthetische Substrate mit einer vergleichbaren und außergewöhnlichen biologischen Wirksamkeit (IC50 = 1–50 pM) und einer verbesserten Wirkung entwickelt. Die bisher definierten elementaren Zusammenhänge könnten den Grundstein für künftige Entwicklungen und Fortschritte legen.
Co-reporter:Yoshikazu Sasaki ; Daisuke Kato
Journal of the American Chemical Society () pp:
Publication Date(Web):September 1, 2010
DOI:10.1021/ja106284s
Concise asymmetric total syntheses of vindoline (1) and vindorosine (2) are detailed based on a unique intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles inspired by the natural product structures. A chiral substituent on the tether linking the dienophile and oxadiazole was used to control the facial selectivity of the initiating Diels−Alder reaction and set the absolute stereochemistry of the remaining six stereocenters in the cascade cycloadduct. This key reaction introduced three rings and four C−C bonds central to the pentacyclic ring system setting all six stereocenters and introducing essentially all the functionality found in the natural products in a single step. Implementation of the approach for the synthesis of 1 and 2 required the development of a ring expansion reaction to provide a 6-membered ring suitably functionalized for introduction of the Δ6,7-double bond found in the core structure of the natural products. Two unique approaches were developed that defined our use of a protected hydroxymethyl group as the substituent that controls the stereochemical course of the cycloaddition cascade. In the course of these studies, several analogues of vindoline were prepared containing deep-seated structural changes presently accessible only by total synthesis. These analogues, bearing key modifications at C6−C8, were incorporated into vinblastine analogues and used to probe the unusual importance (100-fold) and define the potential role of the vinblastine Δ6,7-double bond.
Co-reporter:Shouliang Yang, Kuppusamy Sankar, Colin K. Skepper, Timothy J. Barker, John C. Lukesh III, Daniel M. Brody, Manuela M. Brütsch and Dale L. Boger
Chemical Science (2010-Present) 2017 - vol. 8(Issue 2) pp:NaN1569-1569
Publication Date(Web):2016/11/03
DOI:10.1039/C6SC04146A
The total synthesis and evaluation of a key systematic series of vinblastines that incorporate the first deep-seated changes to the substituent at C4 are detailed. The synthetic approach features an expanded and redefined scope of a 1,3,4-oxadiazole [4 + 2]/[3 + 2] cycloaddition cascade in which electronically mismatched electron-deficient trisubstituted alkenes and unactivated trisubstituted alkenes were found to productively initiate the cycloaddition cascade with tethered electron-deficient 1,3,4-oxadiazoles. Such cycloaddition cascades were used to directly introduce altered C4 substituents, providing the basis for concise total syntheses of a series of C4 modified vindolines and their subsequent single-step incorporation into the corresponding synthetic vinblastines in routes as short as 8–12 steps. Evaluation of the synthetic vinblastines revealed a surprisingly large impact and role of the C4 substituent on activity even though it was previously not thought to intimately interact with the biological target tubulin. Only the introduction of a C4 methyl ester, a constitutional isomer of vinblastine in which the carbonyl carbon and ester oxygen of the C4 acetate are transposed, provided a synthetic vinblastine that matched the potency of the natural product. In contrast, even introduction of a C4 acetamide or N-methyl carboxamide, which incorporate single heavy atom exchanges (amide NH for ester oxygen) in vinblastine or the C4 methyl ester, provided compounds that were ≥10-fold less active than vinblastine. Other C4 acetate replacements, including a C4 amine, carboxylic acid, hydroxymethyl or acetoxymethyl group, led to even greater reductions in potency. Even replacement of the C4 acetoxy group or its equally active C4 methyl ester with an ethyl or isopropyl ester led to 10-fold or more reductions in activity. These remarkable trends in activity, which correlate with relative tubulin binding affinities, retrospectively may be ascribed to the role the substituent serves as a H-bond acceptor for α-tubulin Lys336 and Asn329 side chains at a site less tolerant of a H-bond donor, placing the methyl group of the C4 acetate or C4 methyl ester in a spatially restricted and well-defined hydrophobic half pocket created by a surrounding well-ordered loop. This remarkable impact of the C4 substituent, its stringency, and even the magnitude of its effect are extraordinary, and indicate that its presence was selected in Nature to enhance the effects of vinblastine and related natural products.
.jpg)











![SILANE, [4-(2-BROMOETHYL)PHENOXY]TRIS(1-METHYLETHYL)-](http://img.cochemist.com/ccimg/613300/613233-76-4.png)
![SILANE, [4-(2-BROMOETHYL)PHENOXY]TRIS(1-METHYLETHYL)-](http://img.cochemist.com/ccimg/613300/613233-76-4_b.png)
![Phosphonium, triphenyl[[4-(phenylmethoxy)phenyl]methyl]-, bromide](http://img.cochemist.com/ccimg/383200/383189-06-8.png)
![Phosphonium, triphenyl[[4-(phenylmethoxy)phenyl]methyl]-, bromide](http://img.cochemist.com/ccimg/383200/383189-06-8_b.png)


















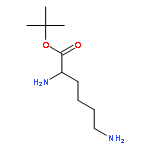

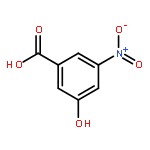
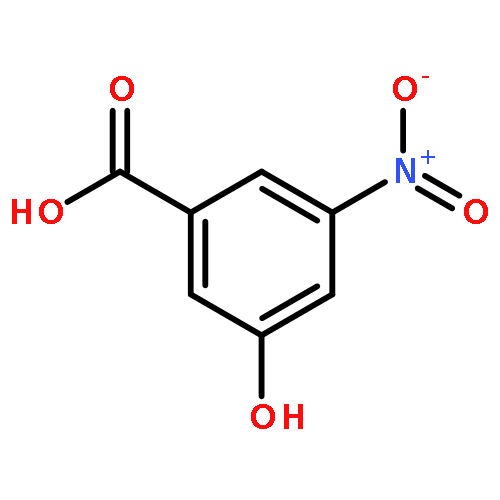
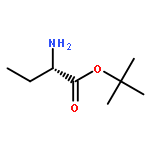
![5-HYDROXY-4-OXO-5-(2-OXOPROPYL)-1H-PYRROLO[2,3-F]QUINOLINE-2,7,9-TRICARBOXYLIC ACID](http://img.cochemist.com/ccimg/73100/73030-04-3.png)
![5-HYDROXY-4-OXO-5-(2-OXOPROPYL)-1H-PYRROLO[2,3-F]QUINOLINE-2,7,9-TRICARBOXYLIC ACID](http://img.cochemist.com/ccimg/73100/73030-04-3_b.png)
![4,5-Dioxo-4,5-dihydro-1H-pyrrolo[2,3-f]quinoline-2,7,9-tricarboxylic acid](/data/chemimg/654800/72909-34-3.png)
![4,5-Dioxo-4,5-dihydro-1H-pyrrolo[2,3-f]quinoline-2,7,9-tricarboxylic acid](/data/chemimg/654800/72909-34-3_b.png)
![BENZ[H]ISOQUINOLINE, 5,6-DIHYDRO-](http://img.cochemist.com/ccimg/67600/67545-18-0.png)
![BENZ[H]ISOQUINOLINE, 5,6-DIHYDRO-](http://img.cochemist.com/ccimg/67600/67545-18-0_b.png)









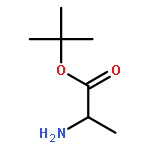
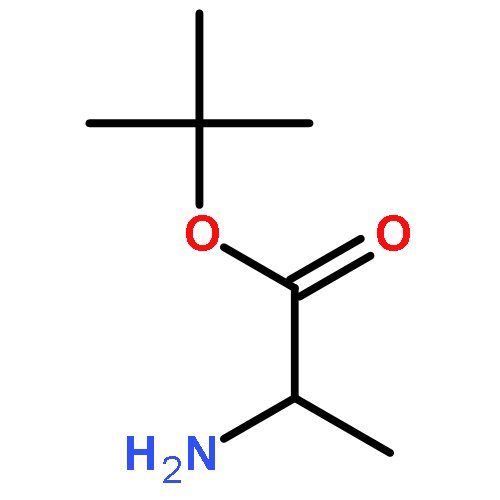

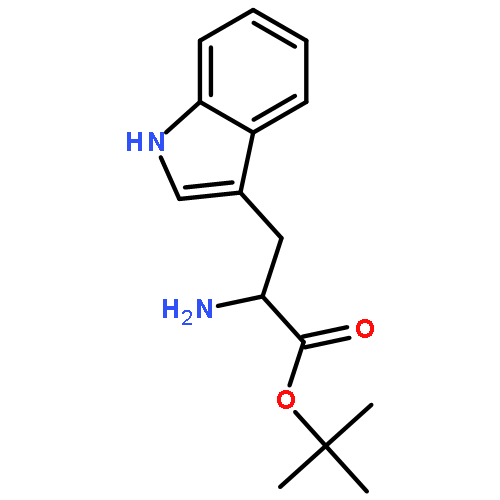
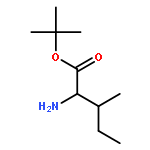


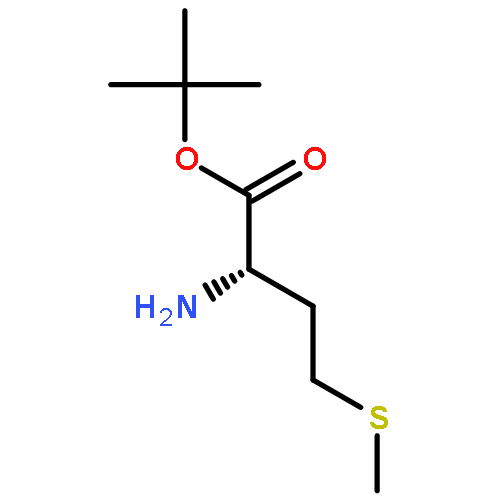


![7-Azabicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/300/279-40-3.png)
![7-Azabicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/300/279-40-3_b.png)











![Benzene, [(2-azidopropoxy)methyl]-](http://img.cochemist.com/ccimg/858400/858344-64-6.png)
![Benzene, [(2-azidopropoxy)methyl]-](http://img.cochemist.com/ccimg/858400/858344-64-6_b.png)


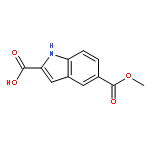
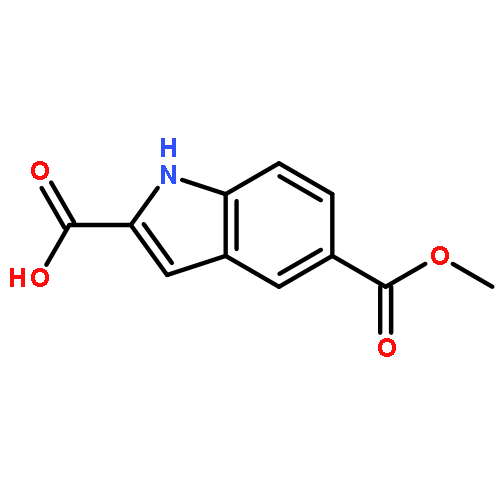
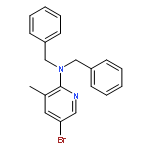
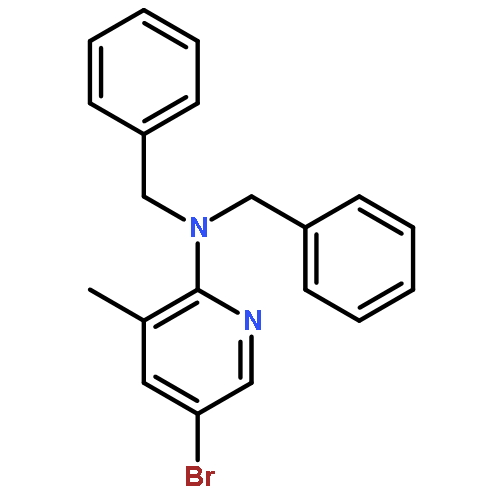
![Hexanoic acid, 5-[[(2S)-2,6-bis(acetylamino)-1-oxohexyl]amino]-2-methyl-4-oxo-, (2S,5R)-](/data/chemimg/127500/593278-45-6.png)
![Hexanoic acid, 5-[[(2S)-2,6-bis(acetylamino)-1-oxohexyl]amino]-2-methyl-4-oxo-, (2S,5R)-](/data/chemimg/127500/593278-45-6_b.png)


![L-Glutamic acid,N-[4-[4-(2,6-diamino-1,4-dihydro-4-oxo-5-pyrimidinyl)-1-(trifluoroacetyl)butyl]benzoyl]-](http://img.cochemist.com/ccimg/553700/553681-09-7.png)
![L-Glutamic acid,N-[4-[4-(2,6-diamino-1,4-dihydro-4-oxo-5-pyrimidinyl)-1-(trifluoroacetyl)butyl]benzoyl]-](http://img.cochemist.com/ccimg/553700/553681-09-7_b.png)








![Hexanoic acid, 4-[(phenylmethoxy)methylene]-, (4Z)-](http://img.cochemist.com/ccimg/473800/473742-41-5.png)
![Hexanoic acid, 4-[(phenylmethoxy)methylene]-, (4Z)-](http://img.cochemist.com/ccimg/473800/473742-41-5_b.png)
![HEXANOIC ACID, 4-[(PHENYLMETHOXY)METHYLENE]-, (4E)-](http://img.cochemist.com/ccimg/473800/473742-40-4.png)
![HEXANOIC ACID, 4-[(PHENYLMETHOXY)METHYLENE]-, (4E)-](http://img.cochemist.com/ccimg/473800/473742-40-4_b.png)




![Benzaldehyde, 3,4,5-tris[2-(2-methoxyethoxy)ethoxy]-](http://img.cochemist.com/ccimg/422300/422267-98-9.png)
![Benzaldehyde, 3,4,5-tris[2-(2-methoxyethoxy)ethoxy]-](http://img.cochemist.com/ccimg/422300/422267-98-9_b.png)









![5-tert-butoxycarbonylamino-benzo[b]thiophene-2-carboxylic Acid](http://img.cochemist.com/ccimg/292100/292068-77-0.png)
![5-tert-butoxycarbonylamino-benzo[b]thiophene-2-carboxylic Acid](http://img.cochemist.com/ccimg/292100/292068-77-0_b.png)
![2-Thiophenecarboxylic acid, 4-[[(1,1-dimethylethoxy)carbonyl]amino]-](/data/chemimg/127100/292068-69-0.png)
![2-Thiophenecarboxylic acid, 4-[[(1,1-dimethylethoxy)carbonyl]amino]-](/data/chemimg/127100/292068-69-0_b.png)

![5H-Cyclopenta[b]pyridine-2-carboxylic acid, 6,7-dihydro-, methyl ester](http://img.cochemist.com/ccimg/221200/221137-08-2.png)
![5H-Cyclopenta[b]pyridine-2-carboxylic acid, 6,7-dihydro-, methyl ester](http://img.cochemist.com/ccimg/221200/221137-08-2_b.png)
![L-Alanine, N-[(1,1-dimethylethoxy)carbonyl]-N-methyl-D-leucyl-(βR)-4-fluoro-β-hydroxy-3-nitro-D-phenylalanyl-3-cyano-](/data/chemimg/127500/207390-83-8.png)
![L-Alanine, N-[(1,1-dimethylethoxy)carbonyl]-N-methyl-D-leucyl-(βR)-4-fluoro-β-hydroxy-3-nitro-D-phenylalanyl-3-cyano-](/data/chemimg/127500/207390-83-8_b.png)










![N-[5-[2-[2-Amino-4-oxo-4,6,7,8-tetrahydro-3H-pyrimido[5,4-b][1,4]thiazin-6(S)-yl]ethyl]thien-2-ylcarbonyl]-L-glutamic acid](http://img.cochemist.com/ccimg/177600/177575-17-6.png)
![N-[5-[2-[2-Amino-4-oxo-4,6,7,8-tetrahydro-3H-pyrimido[5,4-b][1,4]thiazin-6(S)-yl]ethyl]thien-2-ylcarbonyl]-L-glutamic acid](http://img.cochemist.com/ccimg/177600/177575-17-6_b.png)


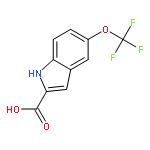
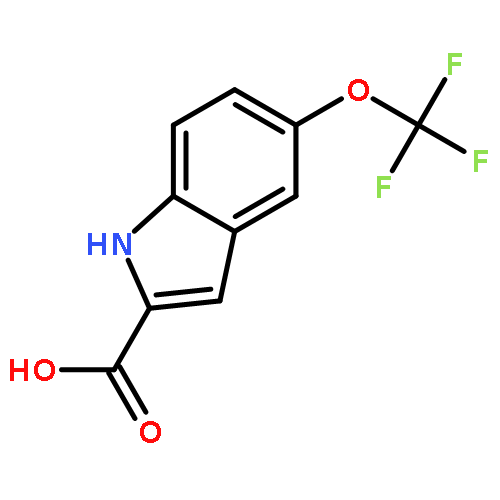






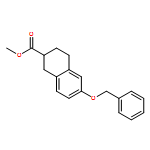
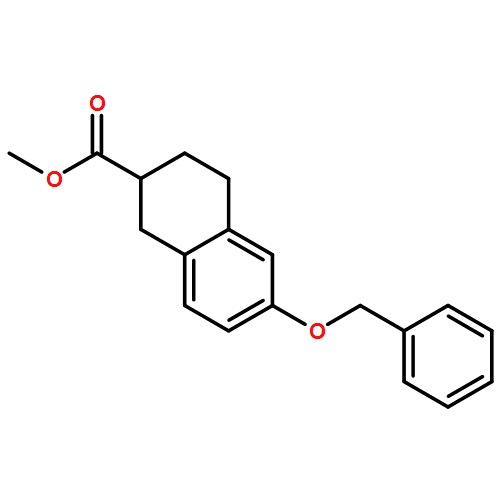
![Vancomycin,22-O-(3-amino-2,3,6-trideoxy-3-C-methyl-a-L-arabino-hexopyranosyl)-N3''-[(4'-chloro[1,1'-biphenyl]-4-yl)methyl]-,(4''R)-](http://img.cochemist.com/ccimg/171100/171099-57-3.png)
![Vancomycin,22-O-(3-amino-2,3,6-trideoxy-3-C-methyl-a-L-arabino-hexopyranosyl)-N3''-[(4'-chloro[1,1'-biphenyl]-4-yl)methyl]-,(4''R)-](http://img.cochemist.com/ccimg/171100/171099-57-3_b.png)


![2-Hexenal, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/163300/163277-83-6.png)
![2-Hexenal, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/163300/163277-83-6_b.png)

![3-Bromo-6,7-dihydro-5H-cyclopenta[b]pyridine](http://img.cochemist.com/ccimg/158400/158331-18-1.png)
![3-Bromo-6,7-dihydro-5H-cyclopenta[b]pyridine](http://img.cochemist.com/ccimg/158400/158331-18-1_b.png)
![Ethanamine, 2-[[tris(1-methylethyl)silyl]oxy]-](/data/chemimg/128100/158198-43-7.png)
![Ethanamine, 2-[[tris(1-methylethyl)silyl]oxy]-](/data/chemimg/128100/158198-43-7_b.png)








![L-Alanine, 1-[(1,1-dimethylethoxy)carbonyl]-L-histidyl-, 1,1-dimethylethyl ester](/data/chemimg/1095900/138899-48-6.png)
![L-Alanine, 1-[(1,1-dimethylethoxy)carbonyl]-L-histidyl-, 1,1-dimethylethyl ester](/data/chemimg/1095900/138899-48-6_b.png)




![Propanoic acid, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, methyl ester,(2S)-](http://img.cochemist.com/ccimg/133000/132969-71-2.png)
![Propanoic acid, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, methyl ester,(2S)-](http://img.cochemist.com/ccimg/133000/132969-71-2_b.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, (2S)-](http://img.cochemist.com/ccimg/133000/132969-60-9.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, (2S)-](http://img.cochemist.com/ccimg/133000/132969-60-9_b.png)



![L-Glutamic acid,N-[[5-[2-[(6R)-2-amino-3,4,5,6,7,8-hexahydro-4-oxopyrido[2,3-d]pyrimidin-6-yl]ethyl]-2-thienyl]carbonyl]-](http://img.cochemist.com/ccimg/127300/127228-54-0.png)
![L-Glutamic acid,N-[[5-[2-[(6R)-2-amino-3,4,5,6,7,8-hexahydro-4-oxopyrido[2,3-d]pyrimidin-6-yl]ethyl]-2-thienyl]carbonyl]-](http://img.cochemist.com/ccimg/127300/127228-54-0_b.png)


![4,8-Dioxaspiro[2.5]oct-1-ene, 6,6-dimethyl-1-phenyl-](http://img.cochemist.com/ccimg/122800/122762-89-4.png)
![4,8-Dioxaspiro[2.5]oct-1-ene, 6,6-dimethyl-1-phenyl-](http://img.cochemist.com/ccimg/122800/122762-89-4_b.png)
![Carbamic acid, N-[1-bromo-4-(phenylmethoxy)-2-naphthalenyl]-, 1,1-dimethylethyl ester](/data/chemimg/1070000/122745-37-3.png)
![Carbamic acid, N-[1-bromo-4-(phenylmethoxy)-2-naphthalenyl]-, 1,1-dimethylethyl ester](/data/chemimg/1070000/122745-37-3_b.png)
![Vancomycin,44-O-de[2-O-(3-amino-2,3,6-trideoxy-3-C-methyl-a-L-lyxo-hexopyranosyl)-b-D-glucopyranosyl]-19-dechloro-](http://img.cochemist.com/ccimg/119800/119789-44-5.png)
![Vancomycin,44-O-de[2-O-(3-amino-2,3,6-trideoxy-3-C-methyl-a-L-lyxo-hexopyranosyl)-b-D-glucopyranosyl]-19-dechloro-](http://img.cochemist.com/ccimg/119800/119789-44-5_b.png)











![L-Glutamic acid,N-[4-[2-[(6R)-2-amino-3,4,5,6,7,8-hexahydro-4-oxopyrido[2,3-d]pyrimidin-6-yl]ethyl]benzoyl]-](http://img.cochemist.com/ccimg/106500/106400-81-1.png)
![L-Glutamic acid,N-[4-[2-[(6R)-2-amino-3,4,5,6,7,8-hexahydro-4-oxopyrido[2,3-d]pyrimidin-6-yl]ethyl]benzoyl]-](http://img.cochemist.com/ccimg/106500/106400-81-1_b.png)






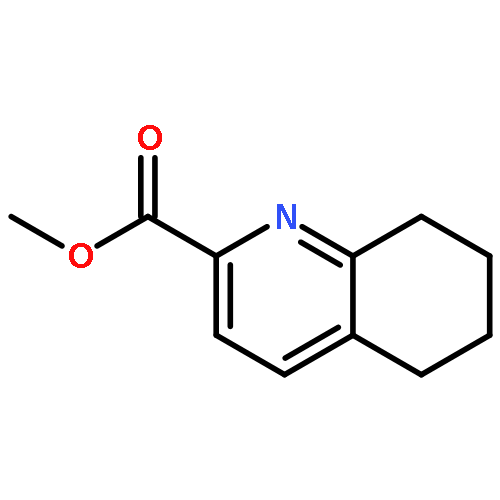

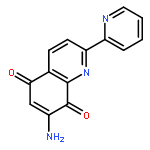

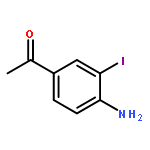

![L-Glutamic acid,N-[4-[2-(2-amino-3,4,5,6,7,8-hexahydro-4-oxopyrido[2,3-d]pyrimidin-6-yl)ethyl]benzoyl]-](http://img.cochemist.com/ccimg/95700/95693-76-8.png)
![L-Glutamic acid,N-[4-[2-(2-amino-3,4,5,6,7,8-hexahydro-4-oxopyrido[2,3-d]pyrimidin-6-yl)ethyl]benzoyl]-](http://img.cochemist.com/ccimg/95700/95693-76-8_b.png)

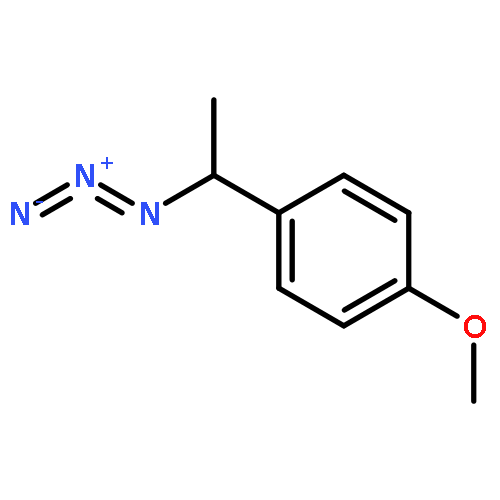
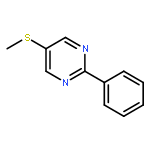

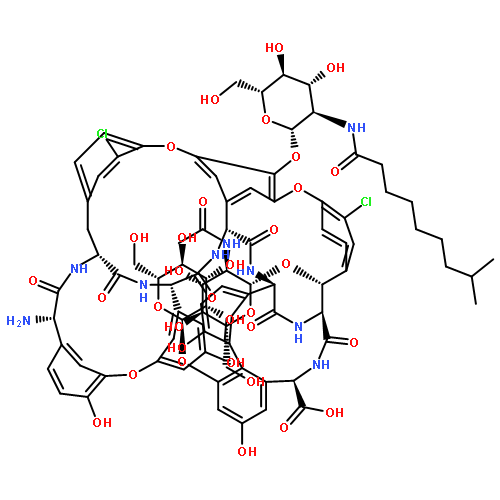
![BENZALDEHYDE, 3,5-DIMETHOXY-4-[2-(2-METHOXYETHOXY)ETHOXY]-](http://img.cochemist.com/ccimg/90600/90521-07-6.png)
![BENZALDEHYDE, 3,5-DIMETHOXY-4-[2-(2-METHOXYETHOXY)ETHOXY]-](http://img.cochemist.com/ccimg/90600/90521-07-6_b.png)



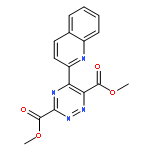



![Acetamide, N-[2,6-bis[[(4-methylphenyl)sulfonyl]oxy]phenyl]-](http://img.cochemist.com/ccimg/88200/88172-87-6.png)
![Acetamide, N-[2,6-bis[[(4-methylphenyl)sulfonyl]oxy]phenyl]-](http://img.cochemist.com/ccimg/88200/88172-87-6_b.png)




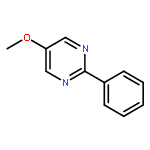

![Piperazine, 1-([1,1'-biphenyl]-4-ylmethyl)-](http://img.cochemist.com/ccimg/84400/84359-51-3.png)
![Piperazine, 1-([1,1'-biphenyl]-4-ylmethyl)-](http://img.cochemist.com/ccimg/84400/84359-51-3_b.png)

![Phosphorane, triphenyl[(phenylmethoxy)methylene]-](http://img.cochemist.com/ccimg/82600/82599-12-0.png)
![Phosphorane, triphenyl[(phenylmethoxy)methylene]-](http://img.cochemist.com/ccimg/82600/82599-12-0_b.png)



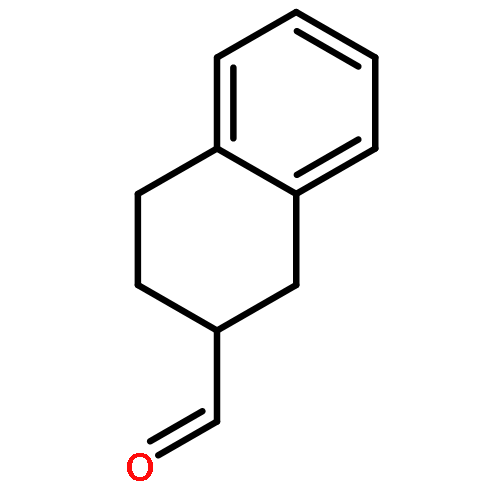


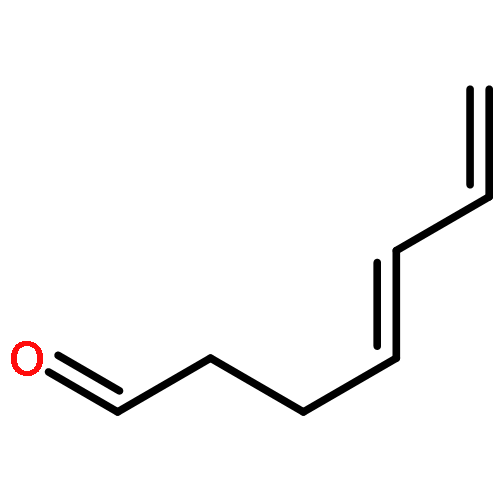

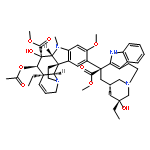
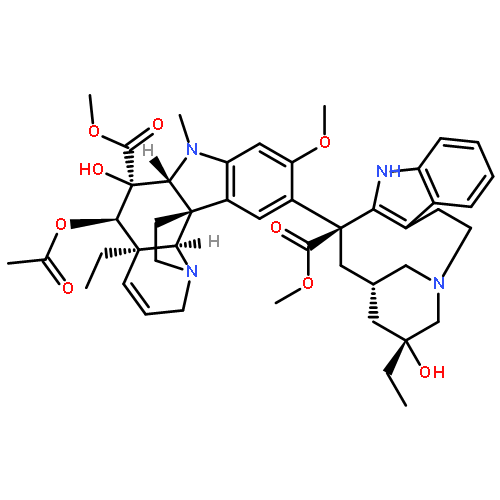



![1-[1-(4-METHOXYPHENYL)ETHENYL]PYRROLIDINE](http://img.cochemist.com/ccimg/73100/73031-42-2.png)
![1-[1-(4-METHOXYPHENYL)ETHENYL]PYRROLIDINE](http://img.cochemist.com/ccimg/73100/73031-42-2_b.png)



![2-Hexyn-1-ol, 6-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/66100/66084-35-3.png)
![2-Hexyn-1-ol, 6-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/66100/66084-35-3_b.png)
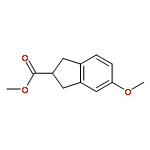







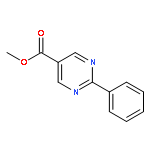





![Ethanol, 2-[2-(2-methoxyethoxy)ethoxy]-, 1-(4-methylbenzenesulfonate)](/data/chemimg/636900/62921-74-8.png)
![Ethanol, 2-[2-(2-methoxyethoxy)ethoxy]-, 1-(4-methylbenzenesulfonate)](/data/chemimg/636900/62921-74-8_b.png)




![PHOSPHONIC ACID, [2-(2-FURANYL)-2-OXOETHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/56000/55984-13-9.png)
![PHOSPHONIC ACID, [2-(2-FURANYL)-2-OXOETHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/56000/55984-13-9_b.png)





![1-benzoyl-1,2,2a,5-tetrahydrobenzo[cd]indol-4(3H)-one](http://img.cochemist.com/ccimg/51900/51867-06-2.png)
![1-benzoyl-1,2,2a,5-tetrahydrobenzo[cd]indol-4(3H)-one](http://img.cochemist.com/ccimg/51900/51867-06-2_b.png)
![Phosphonic acid, [(methylsulfinyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/50800/50746-61-7.png)
![Phosphonic acid, [(methylsulfinyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/50800/50746-61-7_b.png)

![Phosphonic acid, [(methylsulfonyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/40200/40137-11-9.png)
![Phosphonic acid, [(methylsulfonyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/40200/40137-11-9_b.png)

![Cyclopent[b]indole-3-carboxylic acid, 1,2,3,4-tetrahydro-, methyl ester](http://img.cochemist.com/ccimg/35600/35515-63-0.png)
![Cyclopent[b]indole-3-carboxylic acid, 1,2,3,4-tetrahydro-, methyl ester](http://img.cochemist.com/ccimg/35600/35515-63-0_b.png)















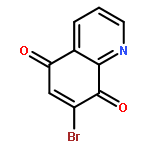








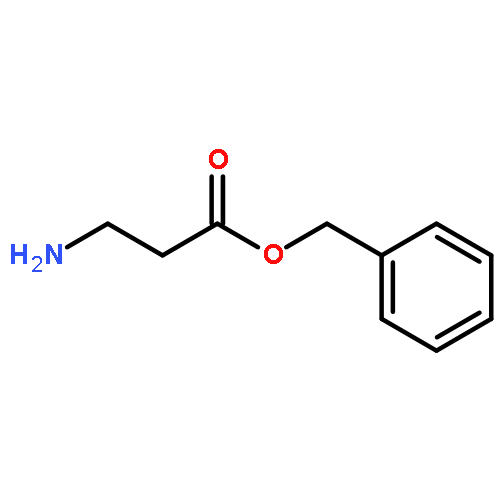



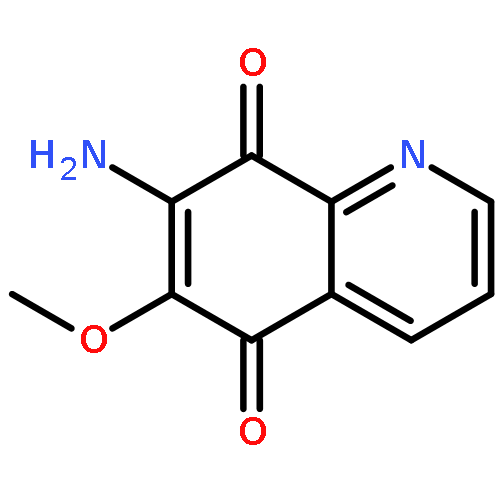
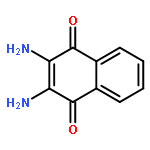

















































![N-[3-[4-(3-acetamidopropylamino)butylamino]propyl]acetamide,dihydrochloride](http://img.cochemist.com/ccimg/900/825-60-5.png)
![N-[3-[4-(3-acetamidopropylamino)butylamino]propyl]acetamide,dihydrochloride](http://img.cochemist.com/ccimg/900/825-60-5_b.png)







![2-Pyridinecarboxylic acid, 6-[2-(1-oxo-7-phenylheptyl)-5-oxazolyl]-](/data/chemimg/126800/935264-47-4.png)
![2-Pyridinecarboxylic acid, 6-[2-(1-oxo-7-phenylheptyl)-5-oxazolyl]-](/data/chemimg/126800/935264-47-4_b.png)


![Oxazole,2-[1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-7-phenylheptyl]-5-(tributylstannyl)-](http://img.cochemist.com/ccimg/808200/808134-98-7.png)
![Oxazole,2-[1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-7-phenylheptyl]-5-(tributylstannyl)-](http://img.cochemist.com/ccimg/808200/808134-98-7_b.png)


![Phosphonium, triphenyl[(phenylmethoxy)methyl]-, bromide](http://img.cochemist.com/ccimg/473800/473742-39-1.png)
![Phosphonium, triphenyl[(phenylmethoxy)methyl]-, bromide](http://img.cochemist.com/ccimg/473800/473742-39-1_b.png)


![Glycine,N-[2-(3,5-dichloro-4-hydroxyphenyl)-2-oxoacetyl]-D-alanyl-(2R)-2-(3,5-dichloro-4-hydroxyphenyl)glycyl-(2R)-2-(3,4-dihydroxyphenyl)glycyl-(2R)-2-(3,5-dichloro-4-hydroxyphenyl)glycyl-N-methyl-L-tyrosyl-2-(4-hydroxyphenyl)-,cyclic (33®5)-ether, cyclic13,35-(1H-indole-3,7-diyl) deriv., (2R)-](http://img.cochemist.com/ccimg/160300/160219-64-7.png)
![Glycine,N-[2-(3,5-dichloro-4-hydroxyphenyl)-2-oxoacetyl]-D-alanyl-(2R)-2-(3,5-dichloro-4-hydroxyphenyl)glycyl-(2R)-2-(3,4-dihydroxyphenyl)glycyl-(2R)-2-(3,5-dichloro-4-hydroxyphenyl)glycyl-N-methyl-L-tyrosyl-2-(4-hydroxyphenyl)-,cyclic (33®5)-ether, cyclic13,35-(1H-indole-3,7-diyl) deriv., (2R)-](http://img.cochemist.com/ccimg/160300/160219-64-7_b.png)



![1-Propanol, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, (2R)-](http://img.cochemist.com/ccimg/136400/136320-64-4.png)
![1-Propanol, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, (2R)-](http://img.cochemist.com/ccimg/136400/136320-64-4_b.png)


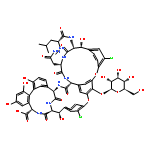

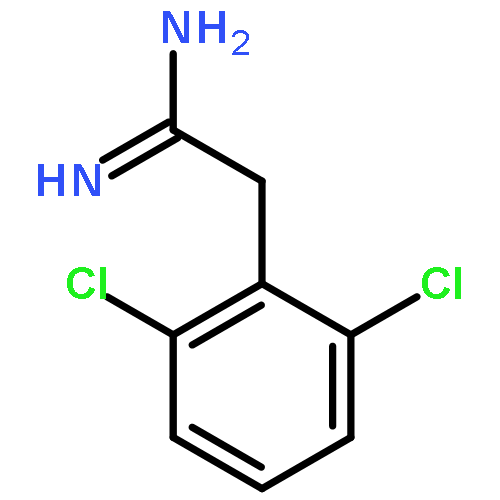


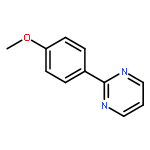
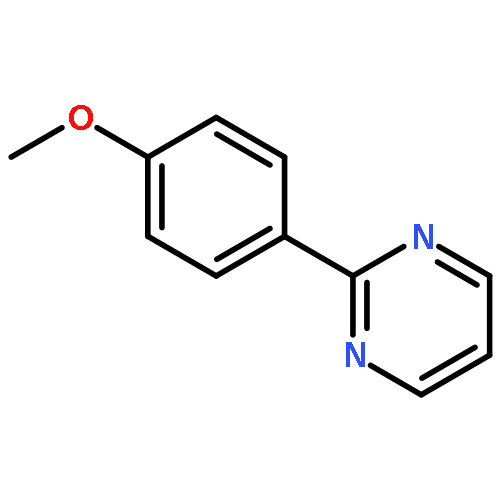


![4,8-Dioxaspiro[2.5]oct-1-ene, 6,6-dimethyl-](http://img.cochemist.com/ccimg/61000/60935-22-0.png)
![4,8-Dioxaspiro[2.5]oct-1-ene, 6,6-dimethyl-](http://img.cochemist.com/ccimg/61000/60935-22-0_b.png)
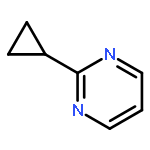
















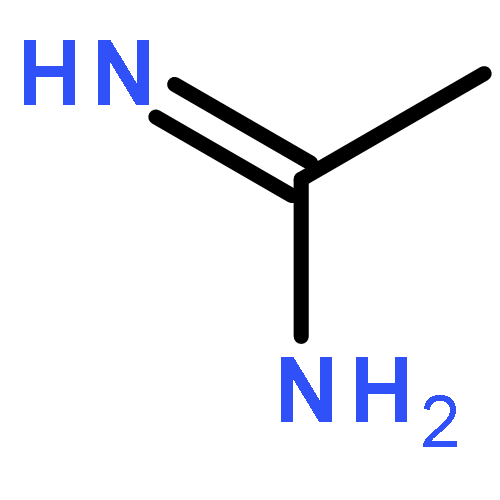




![Glycine,N-[2-(3,5-dichloro-4-hydroxyphenyl)-2-oxoacetyl]-D-alanyl-(2R)-2-(3,5-dichloro-4-hydroxyphenyl)glycyl-(2R)-2-(3,4-dihydroxyphenyl)glycyl-(2R)-2-(3,5-dichloro-4-hydroxyphenyl)glycyl-N-methyl-L-tyrosyl-2-(4-hydroxyphenyl)-,cyclic (33®5)-ether, cyclic13,35-(6R)-1H-indole-3,6-diyl deriv., (2R)-](http://img.cochemist.com/ccimg/69600/69598-75-0.png)
![Glycine,N-[2-(3,5-dichloro-4-hydroxyphenyl)-2-oxoacetyl]-D-alanyl-(2R)-2-(3,5-dichloro-4-hydroxyphenyl)glycyl-(2R)-2-(3,4-dihydroxyphenyl)glycyl-(2R)-2-(3,5-dichloro-4-hydroxyphenyl)glycyl-N-methyl-L-tyrosyl-2-(4-hydroxyphenyl)-,cyclic (33®5)-ether, cyclic13,35-(6R)-1H-indole-3,6-diyl deriv., (2R)-](http://img.cochemist.com/ccimg/69600/69598-75-0_b.png)











![6H,13aH-3a,5a-Ethano-1H-indolizino[8,1-cd]carbazole-5-carboxylicacid, 2,3,4,5,11,12-hexahydro-, methyl ester, (3aR,5R,5aR,10bR,13aS)-](http://img.cochemist.com/ccimg/600/559-51-3.png)
![6H,13aH-3a,5a-Ethano-1H-indolizino[8,1-cd]carbazole-5-carboxylicacid, 2,3,4,5,11,12-hexahydro-, methyl ester, (3aR,5R,5aR,10bR,13aS)-](http://img.cochemist.com/ccimg/600/559-51-3_b.png)







![2H-Pyran-2-one,5,6-dihydro-5-hydroxy-6-[(7Z,9Z,11E)-6-hydroxy-5-methyl-4-(sulfooxy)-1,7,9,11-heptadecatetraen-1-yl]-,sodium salt (1:1)](http://img.cochemist.com/ccimg/131800/131774-59-9.png)
![2H-Pyran-2-one,5,6-dihydro-5-hydroxy-6-[(7Z,9Z,11E)-6-hydroxy-5-methyl-4-(sulfooxy)-1,7,9,11-heptadecatetraen-1-yl]-,sodium salt (1:1)](http://img.cochemist.com/ccimg/131800/131774-59-9_b.png)
![Cyclopropa[c]pyrrolo[3,2-e]indole-6-carboxylicacid,1,2,4,5,6,7,8,8a-octahydro-6-methyl-4,7-dioxo-2-[(5,6,7-trimethoxy-1H-indol-2-yl)carbonyl]-,methyl ester, (6R,7bR,8aS)-](http://img.cochemist.com/ccimg/118300/118292-34-5.png)
![Cyclopropa[c]pyrrolo[3,2-e]indole-6-carboxylicacid,1,2,4,5,6,7,8,8a-octahydro-6-methyl-4,7-dioxo-2-[(5,6,7-trimethoxy-1H-indol-2-yl)carbonyl]-,methyl ester, (6R,7bR,8aS)-](http://img.cochemist.com/ccimg/118300/118292-34-5_b.png)


![Benzo[1,2-b:4,3-b']dipyrrole-3(2H)-carboxamide,7-[[1,6-dihydro-4-hydroxy-5-methoxy-7-[[(7bR,8aS)-4,5,8,8a-tetrahydro-7-methyl-4-oxocyclopropa[c]pyrrolo[3,2-e]indol-2(1H)-yl]carbonyl]benzo[1,2-b:4,3-b']dipyrrol-3(2H)-yl]carbonyl]-1,6-dihydro-4-hydroxy-5-methoxy-](http://img.cochemist.com/ccimg/69900/69866-21-3.png)
![Benzo[1,2-b:4,3-b']dipyrrole-3(2H)-carboxamide,7-[[1,6-dihydro-4-hydroxy-5-methoxy-7-[[(7bR,8aS)-4,5,8,8a-tetrahydro-7-methyl-4-oxocyclopropa[c]pyrrolo[3,2-e]indol-2(1H)-yl]carbonyl]benzo[1,2-b:4,3-b']dipyrrol-3(2H)-yl]carbonyl]-1,6-dihydro-4-hydroxy-5-methoxy-](http://img.cochemist.com/ccimg/69900/69866-21-3_b.png)
![2-[2-(2,6-diacetamidohexanoylamino)propanoylamino]propanoic Acid](http://img.cochemist.com/ccimg/24600/24570-39-6.png)
![2-[2-(2,6-diacetamidohexanoylamino)propanoylamino]propanoic Acid](http://img.cochemist.com/ccimg/24600/24570-39-6_b.png)



![1H-Indole-2-carboxylic acid, 5-[(1H-indol-2-ylcarbonyl)amino]-](http://img.cochemist.com/ccimg/101200/101134-91-2.png)
![1H-Indole-2-carboxylic acid, 5-[(1H-indol-2-ylcarbonyl)amino]-](http://img.cochemist.com/ccimg/101200/101134-91-2_b.png)