Co-reporter:Yannick Geboes;Frank De Proft
Acta Crystallographica Section B 2017 Volume 73(Issue 2) pp:168-178
Publication Date(Web):2017/04/01
DOI:10.1107/S2052520617001354
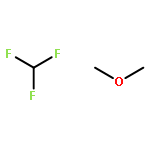
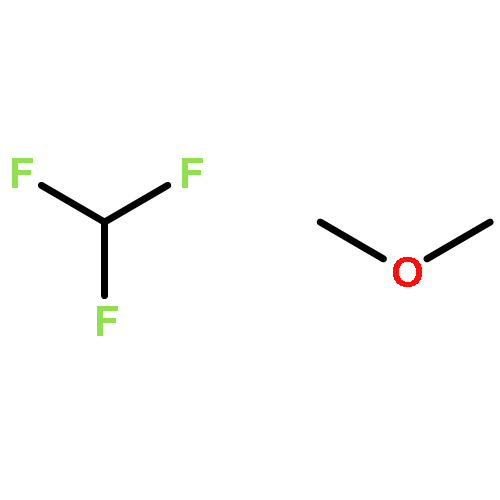
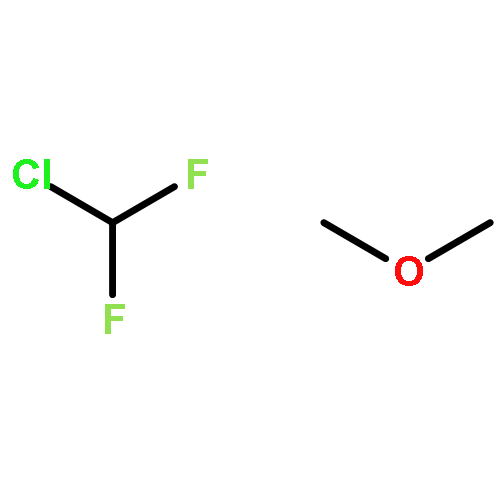
To rationalize the driving factors in the competition of halogen bonding and hydrogen bonding, the complexes of the combined halogen-/hydrogen-bond donor difluoroiodomethane with the Lewis bases trimethylphosphine, dimethyl sulfide and chloromethane are studied. For all Lewis bases, ab initio calculations lead to halogen- and hydrogen-bonded complexes. Fourier transform–IR experiments involving solutions of mixtures of difluoroiodomethane with trimethylphosphine(-d9) or dimethyl sulfide(-d6) in liquid krypton confirm the coexistence of a halogen-bonded and hydrogen-bonded complex. Also for solutions containing chloromethane, evidence of the formation of binary associations is found, but no definitive assignment of the multiple complex bands could be made. Using van't Hoff plots, the experimental complexation enthalpies for the halogen- and hydrogen-bonded complex of difluoroiodomethane with trimethylphosphine are determined to be −15.4 (4) and −10.5 (3) kJ mol−1, respectively, while for the halogen- and hydrogen-bonded complexes with dimethyl sulfide, the values are −11.3 (5) and −7.7 (6) kJ mol−1, respectively. The experimental observation that for both trimethylphospine and dimethyl sulfide the halogen-bonded complex is more stable than the hydrogen-bonded complex supports the finding that softer Lewis bases tend to favor iodine halogen bonding over hydrogen bonding.
Co-reporter:Liene I. De Beuckeleer, Wouter A. Herrebout
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2017 Volume 171() pp:60-71
Publication Date(Web):15 January 2017
DOI:10.1016/j.saa.2016.07.036
•Association of dimethyl ether, acetone, CF3Br and CF3I is observed in the infrared spectra of cryosolutions.•Fitting of polynomials to the recorded data allows us to resolve spectral regions with overlapping absorbance bands.•Fitting of polynomials yields accurate information on monomer, dimer, and trimer species present in the solutions.•The least-squares fitting method shows an added value towards other methods often used.Acetone molecules dissolved in liquid krypton are inclined to self-associate into dimers. This behavior affects its use as a prototype Lewis base in studies of weak intermolecular interactions. In this study infrared spectra of mixed solutions of dimethyl ether and CF3X and of acetone and CF3X (with X = I or Br) dissolved in liquid argon and liquid krypton are recorded at constant temperature. The dataset for dimethyl ether is used to validate a numerical method based on least-squares fitting of a model including contributions of both monomers and a heterodimer with 1:1 stoichiometry. The resulting monomer and dimer spectra show excellent agreement with previous studies found in literature. The analysis of the dataset for acetone requires an extension of the model with contributions for the acetone homodimer and for (acetone)2·CF3X and acetone·(CF3X)2 trimers. The results show that many signals for acetone·CF3I and (acetone)2·CF3I are observed, while only a few bands due to acetone·(CF3I)2 occur. The use of numerical approaches adjusted to the specificities of a mixture of two compounds allows to reliably resolve overlapping spectra of monomers and heterocomplexes and characterizing heterocomplex features that could not be deduced using earlier methods. To support the assignments made, ab initio calculations predicting geometries, relative stabilities and harmonic vibrational frequencies for the species envisaged are performed.
Co-reporter:Yannick Geboes, Frank De Proft, Wouter A. Herrebout
Chemical Physics 2016 Volume 476() pp:1-8
Publication Date(Web):12 September 2016
DOI:10.1016/j.chemphys.2016.07.014
In this theoretical and experimental study, the ability of carbonyl fluoride (COF2) and carbonyl chloride fluoride (COFCl) to form noncovalent interactions with the Lewis base dimethyl ether (DME) is assessed. From ab initio calculations, two stable complexes are found for COF2·DME, both formed through a lone pair⋯π interaction. FTIR measurements on liquefied noble gas solutions, supported by ab initio calculations, statistical thermodynamical calculations and Monte Carle Free Energy Perturbation calculations, show that a 1:1 lone pair⋯π bonded complex is found in solution, with an experimental complexation enthalpy of −14.5(3) kJ mol−1. For COFCl·DME three lone pair⋯π complexes, as well as a Cl⋯O halogen bonded complex, are found from ab initio calculations. Experimentally, clear complex bands for 1:1 lone pair⋯π complexes are observed, with an experimental complexation enthalpy of −11.4(2) kJ mol−1. Furthermore, indications of the presence of a small amount of the halogen bonded complex are also observed.
Co-reporter:Yannick Geboes, Frank De Proft, Wouter A. Herrebout
Chemical Physics Letters 2016 Volume 647() pp:26-30
Publication Date(Web):March 2016
DOI:10.1016/j.cplett.2016.01.029
Highlights
- •
Cryosolutions of C6F6 with dimethyl ether or trimethylamine are studied with FTIR.
- •
Complex bands are assigned to lone pair···π complex using ab initio calculations.
- •
Assignment of observed monomer and complex bands is given.
- •
Using Van ‘t Hoff plots, complex enthalpies of both complexes are determined.
Co-reporter:Liene I. De Beuckeleer, Wouter A. Herrebout
Journal of Molecular Structure 2016 Volume 1118() pp:34-41
Publication Date(Web):15 August 2016
DOI:10.1016/j.molstruc.2016.03.089
•Self-association of NH3 and ND3 is observed in the infrared spectra of cryosolutions with liquid xenon.•Least-squares fitting of polynomials to the data allows us to resolve spectral regions with overlapping absorbance bands.•Least-squares fitting of polynomials yields accurate information on monomer and oligomer species present in the solutions.•The least-squares fitting method shows an added value towards other methods often used.In this study we report on the analysis of isothermal spectra of NH3 and ND3 solutions in liquid xenon at 203 K using newly developed and validated least-squares approaches to investigate the its self-associating behavior. For both species we observe clear dimer bands in the spectral area of the ν1+ν4, ν3+ν4 and ν1+ν2, ν3+ν2 combination bands. The analysis of the N−D stretching area, allows us to characterize clear contributions of dimers and trimers. The analysis of the NH stretching area is hampered by the occurrence of a time dependent band due to solid water traces during the experiments. For NH3 we also performed an investigation of the NH bending region, ν2, which demonstrated a small dimer absorption band. These obtained results compare well with literature data.
Co-reporter:Liene I. De Beuckeleer, Wouter A. Herrebout
Journal of Molecular Structure 2016 Volume 1108() pp:71-79
Publication Date(Web):15 March 2016
DOI:10.1016/j.molstruc.2015.11.067
•Self-association of pyrrole is observed in the infrared spectra of cryosolutions with liquid xenon.•Fitting of polynomials to the recorded data allows us to resolve spectral regions with overlapping absorbance bands.•Fitting of polynomials yields accurate information on monomer, dimer, trimer and tetramer species present in the solutions.•The least-squares fitting method shows an added value towards other methods often used.The self-associating behavior of pyrrole in liquid xenon was investigated by analyzing a data set of 185–113 infrared spectra obtained for different concentrations recorded at a constant temperature of 203 K. Analysis of the data using a recently developed least-squares approach allows the vibrational spectra of the monomer and of the different oligomers to be isolated. Apart from the monomer transitions, intense absorption bands originating from pyrrole trimers are observed in almost every spectral region including regions for which no data have yet been reported. Apart from these bands, weak features proving the presence of pyrrole dimer and pyrrole tetramer in the solutions are also reported. The weak character of the dimer bands observed and the low concentrations of these species deduced are explained by the fact that the cryosolutions studied are in chemical equilibrium and by the fact that due to strong cooperative effect present in the trimer, the complexation equilibria are strongly shifted towards the latter species, thereby strongly reducing the equilibrium concentrations of dimer and tetramer.
Co-reporter:Shi Qiu;Kourosch Abbaspour Tehrani;Sergey Sergeyev;Patrick Bultinck;Wouter Herrebout;Benoit Mathieu
Chirality 2016 Volume 28( Issue 3) pp:215-225
Publication Date(Web):
DOI:10.1002/chir.22558
Abstract
The stereochemistry of all four stereoisomers of brivaracetam was determined using vibrational circular dichroism (VCD) spectroscopy. By comparing experimentally obtained VCD spectra and computationally simulated ones, the absolute configurations can be confidently assigned without prior knowledge of their relative stereochemistry. Neither the corrected mean absolute errors analysis of the nuclear magnetic resonance (NMR) data, nor the matching of experimental and calculated infrared spectra allowed the diastereoisomers to be distinguished. VCD spectroscopy itself suffices to establish the absolute configurations of all diastereoisomers. The relative stereochemistry could also be statistically confirmed by matching experimental and computed NMR spectra using the CP3 algorithm. The combination of VCD and NMR is recommended for molecules bearing more than one chiral center, as the relative configurations obtained from NMR serve as an independent check for those established with VCD. Analysis of the calculated VCD spectra reveals that the localized NH2 scissoring mode at around 1600 cm-1 is characteristic for intramolecular hydrogen bonding, while the orientation of the ethyl group is reflected by the delocalized modes between 1150 and 1050 cm-1. Chirality 28:215–225, 2016. © 2016 Wiley Periodicals, Inc.
Co-reporter:Liene I. De Beuckeleer and Wouter A. Herrebout
The Journal of Physical Chemistry A 2016 Volume 120(Issue 6) pp:884-894
Publication Date(Web):January 25, 2016
DOI:10.1021/acs.jpca.5b10405
Acetone molecules are inclined to self-associate through dipole–dipole interactions because of their large dipole moment. Infrared spectroscopy of compounds dissolved in liquid noble gases supported by high level ab initio calculations allows investigating the self-associating behavior and determining the thermodynamical properties. In this study, infrared spectra of various concentrations of acetone dissolved in liquid krypton are recorded at constant temperature. Overlapping monomer and dimer spectra are separated by analyzing the obtained data sets with numerical methods based on least-squares fitting. Although acetone is known to self-associate, only a few spectral features have been presented in literature before. In this study, the application of new numerical approaches succeeds in resolving overlapping spectra and allows observing isolated acetone dimer absorption bands for the complete mid infrared spectrum. By use of data sets of spectra recorded at temperatures between 134 and 142 K, the experimental standard dimerization enthalpy was determined to be −10.8 kJ mol–1. MP2/aug-cc-pVDZ calculations predicted a stacked and planar dimer geometry of which the stacked geometry is more stable. Combining MP2 energies and single point corrections involving CCSD(T) calculations and complete basis set extrapolations based on the MP2/aug-cc-pVDZ equilibrium geometry lead to complexation energy of −28.4 kJ mol–1 for the stacked geometry and −15.1 kJ mol–1 for the planar geometry. The corresponding values for the complexation enthalpies in solution, obtained by combining these values with corrections for thermal and solvent influences are −13.7 and −5.8 kJ mol–1.
Co-reporter:Liene I. De Beuckeleer, Wouter A. Herrebout
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2016 Volume 154() pp:89-97
Publication Date(Web):5 February 2016
DOI:10.1016/j.saa.2015.10.012
•Polynomials are least-squares fitted to the experimental data to study the self-association in cryosolutions.•The appropriate polynomial degree is chosen using the AIC and BIC information criteria.•As a prototype of self-association, HCl is used for validation.•The methodology should allow other self-associating systems to be analyzed with higher accuracy than before.To rationalize the concentration dependent behavior observed for a large spectral data set of HCl recorded in liquid argon, least-squares based numerical methods are developed and validated. In these methods, for each wavenumber a polynomial is used to mimic the relation between monomer concentrations and measured absorbances. Least-squares fitting of higher degree polynomials tends to overfit and thus leads to compensation effects where a contribution due to one species is compensated for by a negative contribution of another. The compensation effects are corrected for by carefully analyzing, using AIC and BIC information criteria, the differences observed between consecutive fittings when the degree of the polynomial model is systematically increased, and by introducing constraints prohibiting negative absorbances to occur for the monomer or for one of the oligomers. The method developed should allow other, more complicated self-associating systems to be analyzed with a much higher accuracy than before.
Co-reporter:Yannick Geboes, Nick Nagels, Balazs Pinter, Frank De Proft, and Wouter A. Herrebout
The Journal of Physical Chemistry A 2015 Volume 119(Issue 11) pp:2502-2516
Publication Date(Web):November 12, 2014
DOI:10.1021/jp5087812
Inspection of the electrostatic potential of C2F3X (X = F, Cl, Br, and I) revealed a second electropositive region in the immediate vicinity of the C═C double bond apart from the σ hole of chlorine, bromine, and iodine, leading to C(sp2)–X···Y halogen bonding, through which complexes stabilized by so-called lone pair···π interactions can be formed. Consequently, the experimental studies for the complexes of dimethyl ether with C2F3X (X = F, Cl, Br, and I) not only allowed one to experimentally characterize and rationalize the effects of hybridization on halogen bonding but, for the first time, also allowed the competition of C–X···Y halogen bonding and lone pair···π interactions to be studied at thermodynamic equilibrium. Analysis of the infrared and Raman spectra reveals that in the cryosolutions of dimethyl ether and C2F3I, solely the halogen-bonded complex is present, whereas C2F3Br and C2F3Cl give rise to a lone pair···π bonded complex as well as a halogen-bonded complex. Mixtures of dimethyl ether with C2F4 solely yield a lone pair···π bonded complex. The experimentally derived complexation enthalpies for the halogen bonded complexes are found to be −14.2(5) kJ mol–1 for C2F3I·DME and −9.3(5) kJ mol–1 for C2F3Br·DME. For the complexes of C2F3Cl with dimethyl ether, no experimental complexation enthalpy could be obtained, whereas the C2F4·DME complex has a complexation enthalpy of −5.5(3) kJ mol–1. The observed trends have been rationalized with the aid of an interaction energy decomposition analysis (EDA) coupled to a Natural Orbital for Chemical Valence (NOCV) analysis and also using the noncovalent interaction index method.
Co-reporter:Yannick Geboes, Frank De Proft, and Wouter A. Herrebout
The Journal of Physical Chemistry A 2015 Volume 119(Issue 22) pp:5597-5606
Publication Date(Web):April 29, 2015
DOI:10.1021/acs.jpca.5b02283
The molecular electrostatic potential surface of unsaturated, locally electron-deficient molecules shows a positive region perpendicular to (a part of) the molecular framework. In recent years it has been shown both theoretically and experimentally that molecules are able to form noncovalent interactions with Lewis bases through this π-hole. When studying unsaturated perfluorohalogenated molecules containing a higher halogen atom, a second electropositive region is also observed near the halogen atom. This region, often denoted as a σ-hole, allows the molecules to interact with Lewis bases and form a halogen bond. To experimentally characterize the competition between both these noncovalent interactions, Fourier transform infrared and Raman spectra of liquefied noble gas solutions containing perfluorohalogenated ethylene derivatives (C2F3X; X = F, Cl, Br, or I) and trimethylamine(-d9) were investigated. Analysis of the spectra shows that in mixed solutions of trimethylamine(-d9) and C2F4 or C2F3Cl lone pair···π complex is present, while evidence for halogen-bonded complex is found in solutions containing trimethylamine(-d9) and C2F3Cl, C2F3Br, or C2F3I. For all species observed, complexation enthalpies were determined, the values varying between −4.9(1) and −24.4 kJ mol–1.
Co-reporter:Dr. Nick Nagels;Yannick Geboes;Dr. Balazs Pinter;Dr. Frank DeProft;Dr. Wouter A. Herrebout
Chemistry - A European Journal 2014 Volume 20( Issue 27) pp:8433-8443
Publication Date(Web):
DOI:10.1002/chem.201402116
Abstract
Insight into the key factors driving the competition of halogen and hydrogen bonds is obtained by studying the affinity of the Lewis bases trimethylamine (TMA), dimethyl ether (DME), and methyl fluoride (MF) towards difluoroiodomethane (CHF2I). Analysis of the infrared and Raman spectra of solutions in liquid krypton containing mixtures of TMA and CHF2I and of DME and CHF2I reveals that for these Lewis bases hydrogen and halogen-bonded complexes appear simultaneously. In contrast, only a hydrogen-bonded complex is formed for the mixtures of CHF2I and MF. The complexation enthalpies for the CH⋅⋅⋅Y hydrogen-bonded complexes with TMA, DME, and MF are determined to be −14.7(2), −10.5(5) and −5.1(6) kJ mol−1, respectively. The values for the CI⋅⋅⋅Y halogen-bonded isomers are −19.0(3) kJ mol−1 for TMA and −9.9(8) kJ mol−1 for DME. Generalization of the observed trends suggests that, at least for the bases studied here, softer Lewis bases such as TMA favor halogen bonding, whereas harder bases such as MF show a substantial preference for hydrogen bonding.
Co-reporter:Shi Qiu ; Ewoud De Gussem ; Kourosch Abbaspour Tehrani ; Sergey Sergeyev ; Patrick Bultinck ;Wouter Herrebout
Journal of Medicinal Chemistry 2013 Volume 56(Issue 21) pp:8903-8914
Publication Date(Web):October 11, 2013
DOI:10.1021/jm401407w
The stereochemistry of all four stereoisomers of tadalafil is determined using vibrational circular dichroism (VCD), electronic circular dichroism (ECD), and optical rotatory dispersion (ORD) spectroscopy. By comparing experimentally obtained VCD spectra to computationally simulated ones, the absolute configuration of the enantiomeric pair (6R, 12aR)/(6S, 12aS) can be confidently assigned without prior knowledge of their relative stereochemistry. IR and NMR spectra are used to aid the assignment of the relative stereochemistry. The IR and VCD difference spectra further confirm the assignment of all stereoisomers. ECD and ORD spectra are used to investigate the complementarity of the three chiroptical techniques. VCD spectroscopy itself is found to have the ability to identify diastereoisomers, and simultaneous use of these chiroptical spectroscopic methods and NMR chemical shifts aids in increasing the reliability of stereochemistry assignment of diastereoisomers.
Co-reporter:Dieter Hauchecorne and Wouter A. Herrebout
The Journal of Physical Chemistry A 2013 Volume 117(Issue 45) pp:11548-11557
Publication Date(Web):October 15, 2013
DOI:10.1021/jp4077323
Using FTIR and Raman spectroscopy, we investigated the formation of halogen bonded complexes of the trifluorohalomethanes CF3Cl, CF3Br, and CF3I with the halomethanes CH3F and CH3Cl and the haloethanes C2H5F and C2H5Cl dissolved in liquid krypton. For CF3Br and CF3I, evidence was found for the formation of C–X···F and C–X···Cl halogen bonded 1:1 complexes. Using spectra recorded at different temperatures, we determined the complexation enthalpies for the complexes to be −7.0(3) kJ mol–1 for CF3Br·CH3F, −7.6(1) kJ mol–1 for CF3I·CH3F, −5.9(2) kJ mol–1 for CF3Br·CH3Cl, −8.3(3) kJ mol–1 for CF3I·CH3Cl, −7.1(1) kJ mol–1 for CF3Br·C2H5F, −8.7(2) kJ mol–1 for CF3I·C2H5F, −6.5(2) kJ mol–1 for CF3Br·C2H5Cl, and −8.8(3) kJ mol–1 for CF3I·C2H5Cl. For all halogen bonded complexes with a fluorine-electron donor, a blue shift ranging from +0.6 to +1.5 cm–1 was observed for the C–X stretching mode. The results from the cyrospectroscopic study are compared with ab initio calculations at the MP2/aug-cc-pVDZ(-PP) level.
Co-reporter:F. Passareli, A. N. L. Batista, A. J. Cavalheiro, W. A. Herrebout and J. M. Batista Junior
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 45) pp:NaN30906-30906
Publication Date(Web):2016/10/21
DOI:10.1039/C6CP07171F
A simple IR and VCD spectra–structure relationship is proposed for the determination of the relative and absolute configurations of polyhydroxylated molecules using a series of styryl–pyrones as model compounds. Spectral signatures identified for free molecules and acetonide derivatives may be used for stereochemical assignments of related molecules without the aid of quantum-chemical calculations.